Lipoprotein Lipase Inhibitor, Nordihydroguaiaretic Acid, Aggravates Metabolic Phenotypes and Alters HDL Particle Size in the Western Diet-Fed db/db Mice



Abstract
:1. Introduction
2. Results
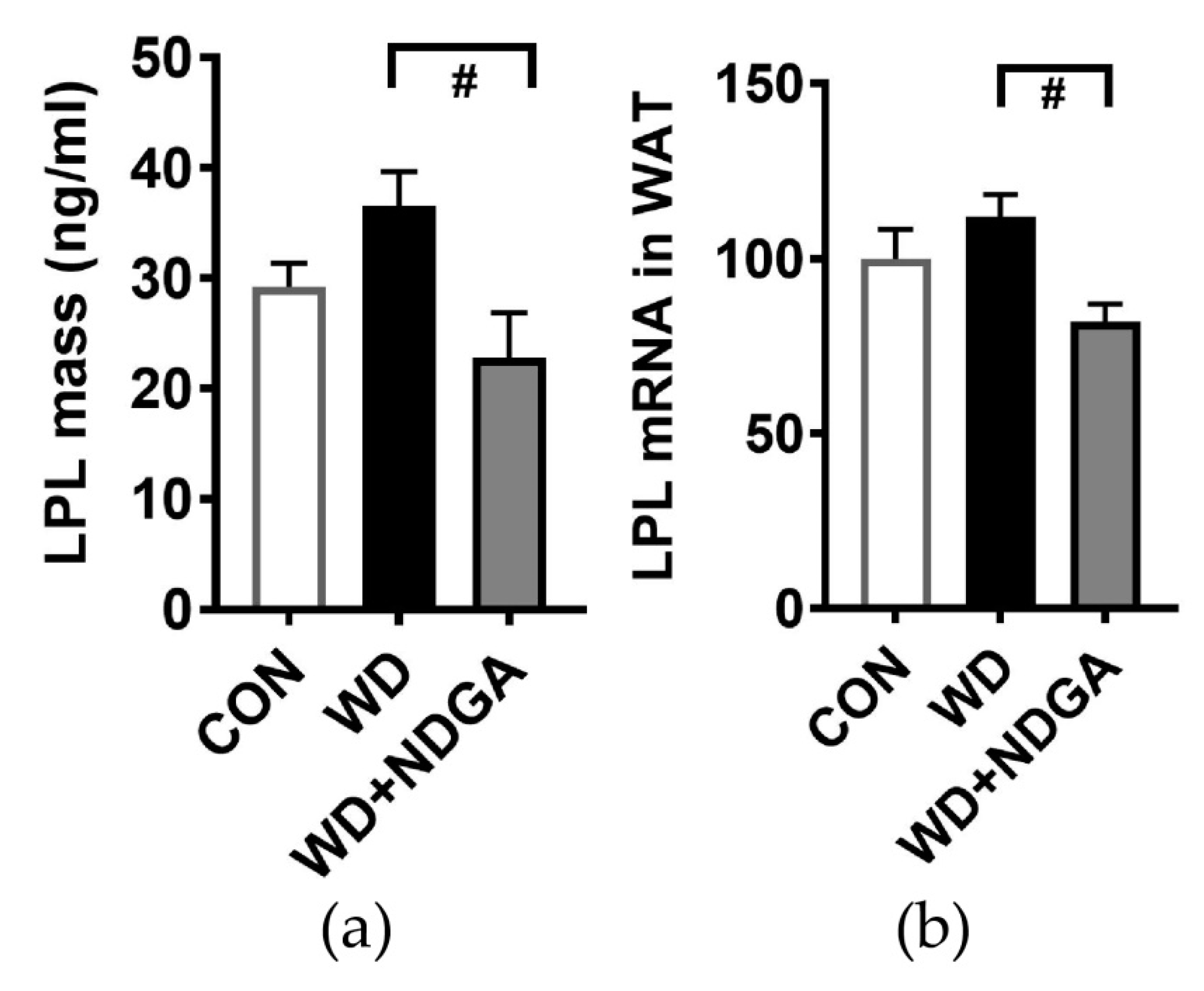
2.1. LPL Inhibition by NDGA
2.2. Changes of Metabolic Parameters, Glucose and Insulin Levels by LPL Inhibitor, NDGA in db/db Mice
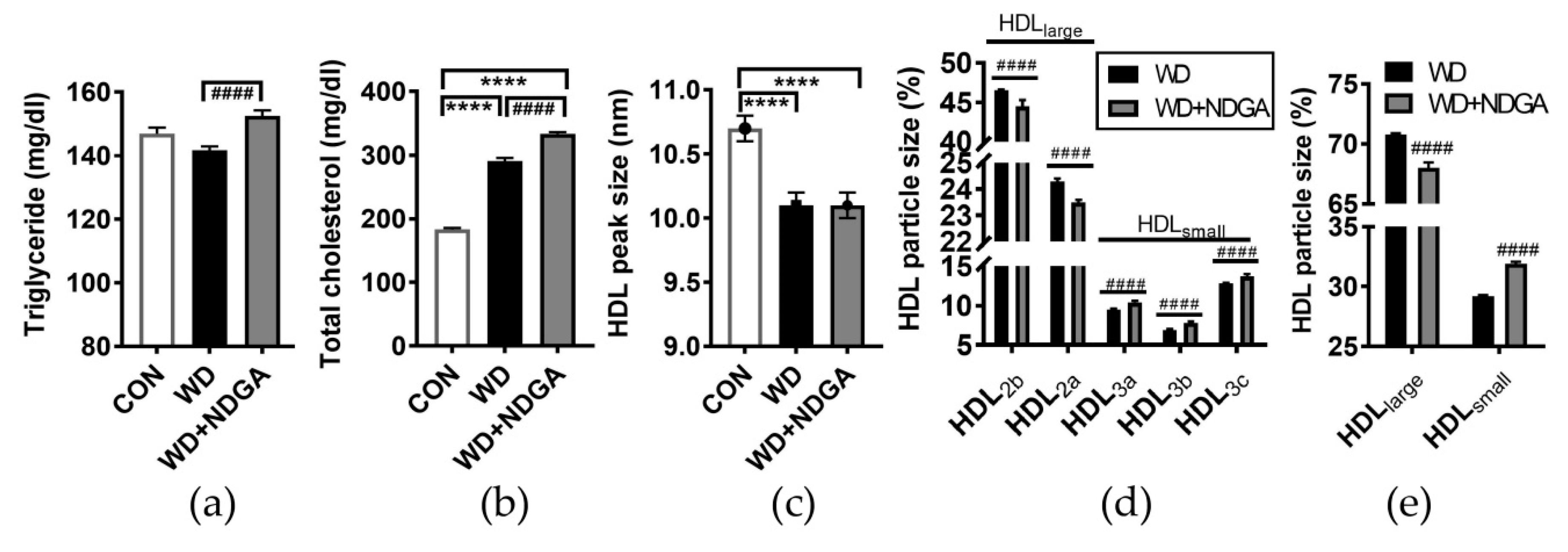
2.3. Changes of Lipid Profiles and HDL Particle Size by the LPL Inhibitor, NDGA, in db/db Mice
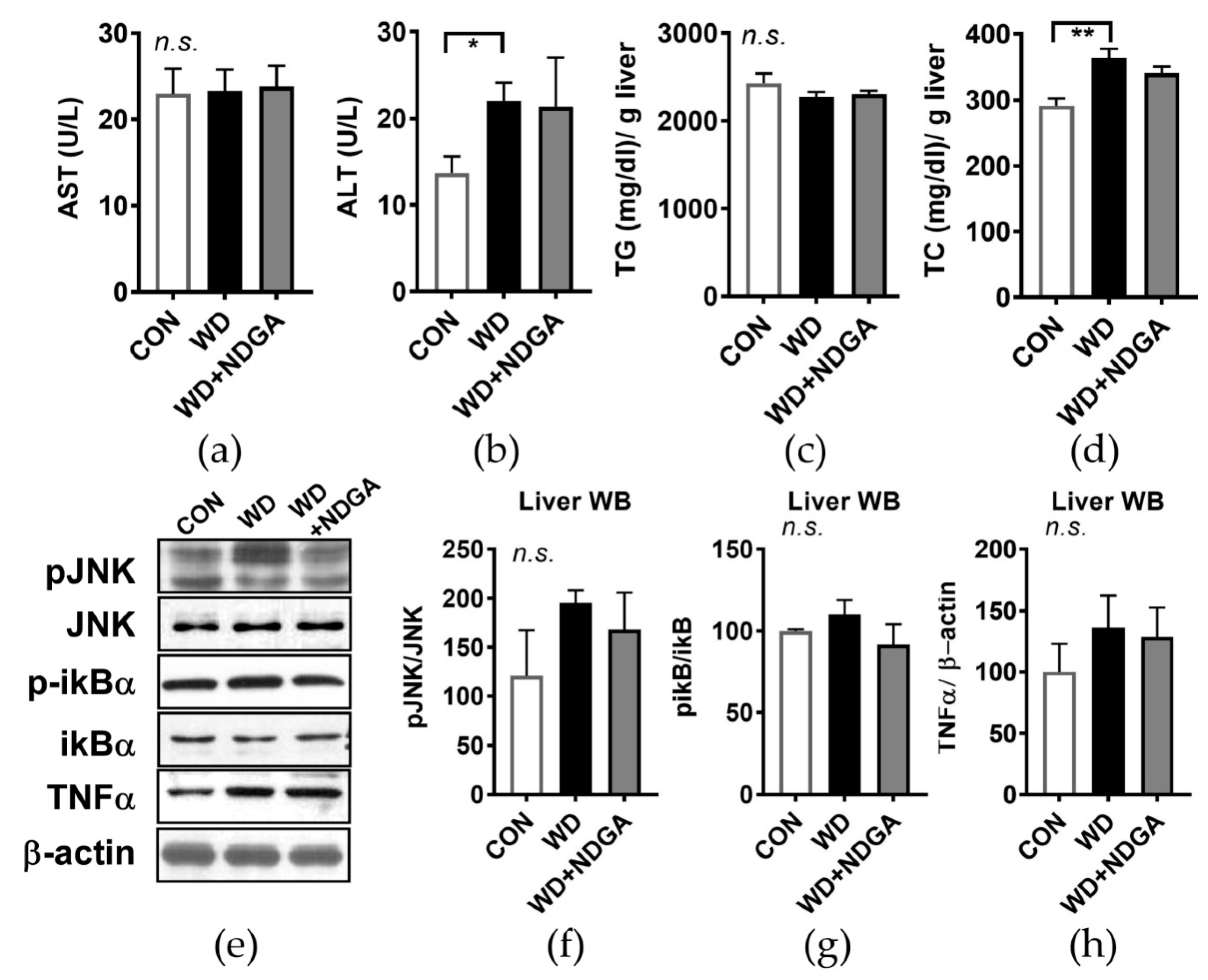
2.4. Hepatic Stress Levels by the LPL Inhibitor, NDGA, in db/db Mice
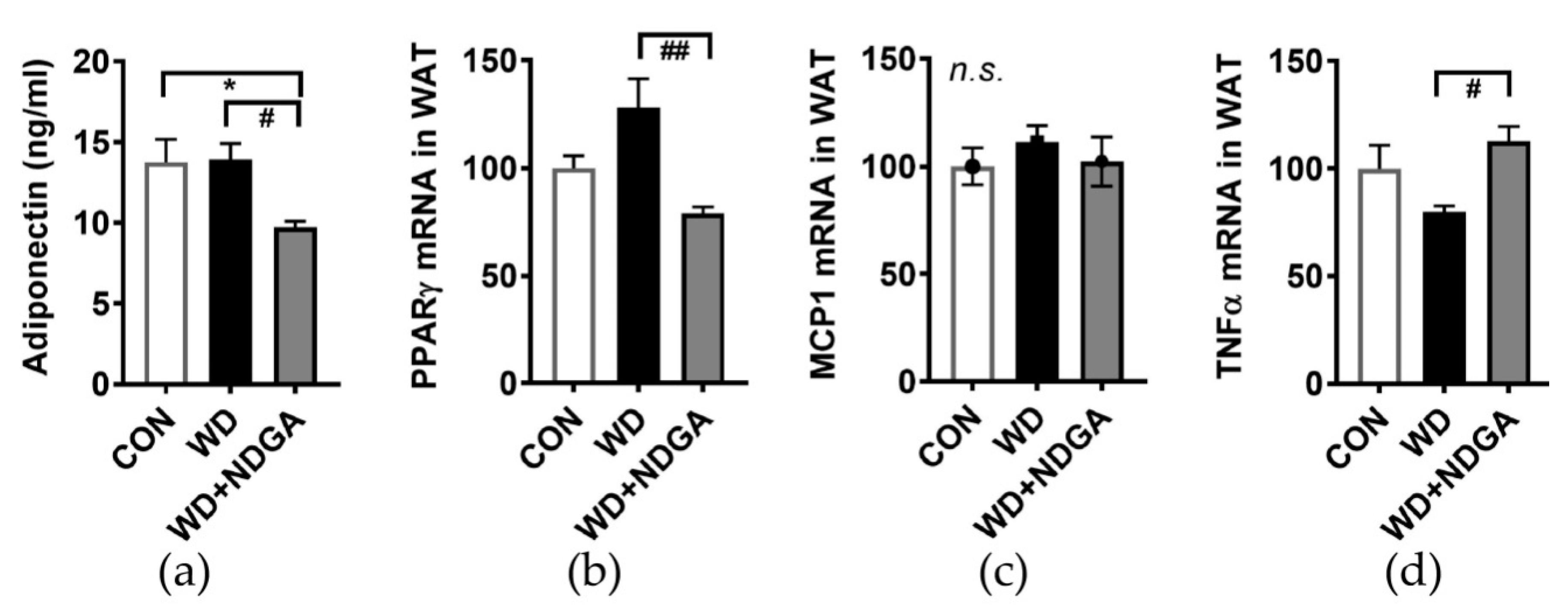
2.5. LPL Inhibition Alters Inflammation-Relation Makers in Epididymal Fat
3. Discussion
4. Materials and Methods
4.1. Care of Animals and Experimental Groups
4.2. Blood and Tissue Collection
4.3. Measurements of Biochemical Parameters
4.4. HDL Particle Size Phenotyping
4.5. Preheparin LPL Mass
4.6. Gene Expression Analysis (Quantitative PCR)
4.7. Western Blotting Analysis
4.8. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ABCA1 | ATP-binding cassette (ABC), sub-family A, member 1 |
| ABCG1 | ABC, sub-family G, member 1 |
| ALT | Alanine aminotransferase |
| AST | Aspartate aminotransferase |
| CETP | Cholesteryl ester transfer protein |
| CON | Control, AIN76-A diet |
| CVD | Cardiovascular diseases |
| ER | Endoplasmic reticulum |
| HDL | High-density lipoprotein |
| IkB | Inhibitor kappa B |
| JNK | C-Jun N-terminal kinases |
| LDL | Low-density lipoprotein |
| LPL | Lipoprotein lipase |
| MCP1 | Monocyte chemoattractant protein-1 |
| MetSyn | Metabolic syndrome |
| NDGA | Nordihydroguaiaretic acid |
| PPARg | Peroxisome proliferator-activated receptor gamma |
| RCT | Reverse cholesterol transport |
| SR-B1 | Scavenger receptor class B, member 1 |
| TC | Total cholesterol |
| TG | Triglyceride |
| TNFα | Tumor necrosis factor alpha |
| WD | Western diet |
References
- Yusuf, S.; Hawken, S.; Ounpuu, S.; Dans, T.; Avezum, A.; Lanas, F.; McQueen, M.; Budaj, A.; Pais, P.; Varigos, J.; et al. Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): Case-control study. Lancet 2004, 364, 937–952. [Google Scholar] [CrossRef]
- Walldius, G.; Jungner, I.; Holme, I.; Aastveit, A.H.; Kolar, W.; Steiner, E. High apolipoprotein B, low apolipoprotein A-I, and improvement in the prediction of fatal myocardial infarction (AMORIS study): A prospective study. Lancet 2001, 358, 2026–2033. [Google Scholar] [CrossRef]
- Law, M.R.; Wald, N.J. Risk factor thresholds: Their existence under scrutiny. BMJ 2002, 324, 1570–1576. [Google Scholar] [CrossRef] [PubMed]
- Akosah, K.O.; Schaper, A.; Cogbill, C.; Schoenfeld, P. Preventing myocardial infarction in the young adult in the first place: How do the National Cholesterol Education Panel III guidelines perform? J. Am. Coll. Cardiol. 2003, 41, 1475–1479. [Google Scholar] [CrossRef]
- Hero, C.; Svensson, A.M.; Gidlund, P.; Gudbjornsdottir, S.; Eliasson, B.; Eeg-Olofsson, K. LDL cholesterol is not a good marker of cardiovascular risk in Type 1 diabetes. Diabet. Med. 2016, 33, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Studer, M.; Briel, M.; Leimenstoll, B.; Glass, T.R.; Bucher, H.C. Effect of different antilipidemic agents and diets on mortality: A systematic review. Arch. Intern. Med. 2005, 165, 725–730. [Google Scholar] [CrossRef] [PubMed]
- Lewis, G.F.; Rader, D.J. New insights into the regulation of HDL metabolism and reverse cholesterol transport. Circ. Res. 2005, 96, 1221–1232. [Google Scholar] [CrossRef]
- Emerging Risk Factors, C.; Di Angelantonio, E.; Gao, P.; Pennells, L.; Kaptoge, S.; Caslake, M.; Thompson, A.; Butterworth, A.S.; Sarwar, N.; Wormser, D.; et al. Lipid-related markers and cardiovascular disease prediction. JAMA 2012, 307, 2499–2506. [Google Scholar] [CrossRef]
- Group, A.S.; Ginsberg, H.N.; Elam, M.B.; Lovato, L.C.; Crouse, J.R., 3rd; Leiter, L.A.; Linz, P.; Friedewald, W.T.; Buse, J.B.; Gerstein, H.C.; et al. Effects of combination lipid therapy in type 2 diabetes mellitus. N. Engl. J. Med. 2010, 362, 1563–1574. [Google Scholar] [CrossRef]
- Investigators, A.-H.; Boden, W.E.; Probstfield, J.L.; Anderson, T.; Chaitman, B.R.; Desvignes-Nickens, P.; Koprowicz, K.; McBride, R.; Teo, K.; Weintraub, W.; et al. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N. Engl. J. Med. 2011, 365, 2255–2267. [Google Scholar] [CrossRef]
- Asztalos, B.F.; Tani, M.; Schaefer, E.J. Metabolic and functional relevance of HDL subspecies. Curr. Opin. Lipidol. 2011, 22, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Krauss, R.M. Lipoprotein subfractions and cardiovascular disease risk. Curr. Opin. Lipidol. 2010, 21, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Kastelein, J.J. Refocusing on use of cholesteryl ester transfer protein inhibitors. Am. J. Cardiol. 2007, 100, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Krishna, R.; Anderson, M.S.; Bergman, A.J.; Jin, B.; Fallon, M.; Cote, J.; Rosko, K.; Chavez-Eng, C.; Lutz, R.; Bloomfield, D.M.; et al. Effect of the cholesteryl ester transfer protein inhibitor, anacetrapib, on lipoproteins in patients with dyslipidaemia and on 24-h ambulatory blood pressure in healthy individuals: Two double-blind, randomised placebo-controlled phase I studies. Lancet 2007, 370, 1907–1914. [Google Scholar] [CrossRef]
- Bloomfield, D.; Carlson, G.L.; Sapre, A.; Tribble, D.; McKenney, J.M.; Littlejohn, T.W., 3rd; Sisk, C.M.; Mitchel, Y.; Pasternak, R.C. Efficacy and safety of the cholesteryl ester transfer protein inhibitor anacetrapib as monotherapy and coadministered with atorvastatin in dyslipidemic patients. Am. Heart J. 2009, 157, 352–360.e2. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, S.J.; Brewer, H.B.; Kastelein, J.J.; Krueger, K.A.; Wang, M.D.; Shao, M.; Hu, B.; McErlean, E.; Nissen, S.E. Effects of the CETP inhibitor evacetrapib administered as monotherapy or in combination with statins on HDL and LDL cholesterol: a randomized controlled trial. JAMA 2011, 306, 2099–2109. [Google Scholar] [CrossRef] [PubMed]
- Ballantyne, C.M.; Miller, M.; Niesor, E.J.; Burgess, T.; Kallend, D.; Stein, E.A. Effect of dalcetrapib plus pravastatin on lipoprotein metabolism and high-density lipoprotein composition and function in dyslipidemic patients: Results of a phase IIb dose-ranging study. Am. Heart J. 2012, 163, 515–521.e3. [Google Scholar] [CrossRef]
- Hovingh, G.K.; Kastelein, J.J.; van Deventer, S.J.; Round, P.; Ford, J.; Saleheen, D.; Rader, D.J.; Brewer, H.B.; Barter, P.J. Cholesterol ester transfer protein inhibition by TA-8995 in patients with mild dyslipidaemia (TULIP): A randomised, double-blind, placebo-controlled phase 2 trial. Lancet 2015, 386, 452–460. [Google Scholar] [CrossRef]
- Sheridan, C. CETP inhibitors boost ‘good’ cholesterol to no avail. Nat. Biotechnol. 2016, 34, 5–6. [Google Scholar] [CrossRef]
- Group, H.T.R.C.; Bowman, L.; Hopewell, J.C.; Chen, F.; Wallendszus, K.; Stevens, W.; Collins, R.; Wiviott, S.D.; Cannon, C.P.; Braunwald, E.; et al. Effects of Anacetrapib in Patients with Atherosclerotic Vascular Disease. N. Engl. J. Med. 2017, 377, 1217–1227. [Google Scholar] [CrossRef]
- Schwartz, G.G.; Olsson, A.G.; Abt, M.; Ballantyne, C.M.; Barter, P.J.; Brumm, J.; Chaitman, B.R.; Holme, I.M.; Kallend, D.; Leiter, L.A.; et al. Effects of dalcetrapib in patients with a recent acute coronary syndrome. N. Engl. J. Med. 2012, 367, 2089–2099. [Google Scholar] [CrossRef] [PubMed]
- Sprecher, D.L.; Harris, B.V.; Stein, E.A.; Bellet, P.S.; Keilson, L.M.; Simbartl, L.A. Higher triglycerides, lower high-density lipoprotein cholesterol, and higher systolic blood pressure in lipoprotein lipase-deficient heterozygotes. A preliminary report. Circulation. 1996, 94, 3239–3245. [Google Scholar] [CrossRef] [PubMed]
- Paglialunga, S.; Julien, P.; Tahiri, Y.; Cadelis, F.; Bergeron, J.; Gaudet, D.; Cianflone, K. Lipoprotein lipase deficiency is associated with elevated acylation stimulating protein plasma levels. J. Lipid Res. 2009, 50, 1109–1119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, R.C.; Castellani, L.W.; Hosseini, M.; Ben-Zeev, O.; Mao, H.Z.; Weinstein, M.M.; Jung, D.Y.; Jun, J.Y.; Kim, J.K.; Lusis, A.J.; et al. Early hepatic insulin resistance precedes the onset of diabetes in obese C57BLKS-db/db mice. Diabetes 2010, 59, 1616–1625. [Google Scholar] [CrossRef] [PubMed]
- Burke, S.J.; Batdorf, H.M.; Burk, D.H.; Noland, R.C.; Eder, A.E.; Boulos, M.S.; Karlstad, M.D.; Collier, J.J. db/db Mice Exhibit Features of Human Type 2 Diabetes That Are Not Present in Weight-Matched C57BL/6J Mice Fed a Western Diet. J. Diabetes Res. 2017, 2017, 8503754. [Google Scholar] [CrossRef] [PubMed]
- Lagos, K.G.; Filippatos, T.D.; Tsimihodimos, V.; Gazi, I.F.; Rizos, C.; Tselepis, A.D.; Mikhailidis, D.P.; Elisaf, M.S. Alterations in the high density lipoprotein phenotype and HDL-associated enzymes in subjects with metabolic syndrome. Lipids 2009, 44, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Zeljkovic, A.; Vekic, J.; Spasojevic-Kalimanovska, V.; Jelic-Ivanovic, Z.; Bogavac-Stanojevic, N.; Gulan, B.; Spasic, S. LDL and HDL subclasses in acute ischemic stroke: prediction of risk and short-term mortality. Atherosclerosis 2010, 210, 548–554. [Google Scholar] [CrossRef] [PubMed]
- Guo, B.; Li, Z. Endoplasmic reticulum stress in hepatic steatosis and inflammatory bowel diseases. Front. Genet. 2014, 5, 242. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Eckel, R.H. Lipoprotein lipase: from gene to obesity. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E271–E288. [Google Scholar] [CrossRef] [Green Version]
- Trak-Smayra, V.; Paradis, V.; Massart, J.; Nasser, S.; Jebara, V.; Fromenty, B. Pathology of the liver in obese and diabetic ob/ob and db/db mice fed a standard or high-calorie diet. Int. J. Exp. Pathol. 2011, 92, 413–421. [Google Scholar] [CrossRef]
- Laperrousaz, E.; Moulle, V.S.; Denis, R.G.; Kassis, N.; Berland, C.; Colsch, B.; Fioramonti, X.; Philippe, E.; Lacombe, A.; Vanacker, C.; et al. Lipoprotein lipase in hypothalamus is a key regulator of body weight gain and glucose homeostasis in mice. Diabetologia 2017, 60, 1314–1324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Arcos, I.; Hiyama, Y.; Drosatos, K.; Bharadwaj, K.G.; Hu, Y.; Son, N.H.; O’Byrne, S.M.; Chang, C.L.; Deckelbaum, R.J.; Takahashi, M.; et al. Adipose-specific lipoprotein lipase deficiency more profoundly affects brown than white fat biology. J. Biol. Chem. 2013, 288, 14046–14058. [Google Scholar] [CrossRef] [PubMed]
- Walton, R.G.; Zhu, B.; Unal, R.; Spencer, M.; Sunkara, M.; Morris, A.J.; Charnigo, R.; Katz, W.S.; Daugherty, A.; Howatt, D.A.; et al. Increasing adipocyte lipoprotein lipase improves glucose metabolism in high fat diet-induced obesity. J. Biol. Chem. 2015, 290, 11547–11556. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, T.; Takami, N.; Misumi, Y.; Ikehara, Y. Nordihydroguaiaretic acid blocks protein transport in the secretory pathway causing redistribution of Golgi proteins into the endoplasmic reticulum. J. Biol. Chem. 1998, 273, 3068–3075. [Google Scholar] [CrossRef] [PubMed]
- Murase, T.; Cattran, D.C.; Rubenstein, B.; Steiner, G. Inhibition of lipoprotein lipase by uremic plasma, a possible cause of hypertriglyceridemia. Metabolism 1975, 24, 1279–1286. [Google Scholar] [CrossRef]
- Berbee, J.F.; van der Hoogt, C.C.; Sundararaman, D.; Havekes, L.M.; Rensen, P.C. Severe hypertriglyceridemia in human APOC1 transgenic mice is caused by apoC-I-induced inhibition of LPL. J. Lipid Res. 2005, 46, 297–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansson, J.; Olsson, A.G.; Bergstrand, L.; Elinder, L.S.; Nilsson, S.; Erikson, U.; Molgaard, J.; Holme, I.; Walldius, G. Lowering of HDL2b by probucol partly explains the failure of the drug to affect femoral atherosclerosis in subjects with hypercholesterolemia. A Probucol Quantitative Regression Swedish Trial (PQRST) Report. Arterioscler. Thromb. Vasc. Biol. 1995, 15, 1049–1056. [Google Scholar] [CrossRef]
- Watanabe, H.; Soderlund, S.; Soro-Paavonen, A.; Hiukka, A.; Leinonen, E.; Alagona, C.; Salonen, R.; Tuomainen, T.P.; Ehnholm, C.; Jauhiainen, M.; et al. Decreased high-density lipoprotein (HDL) particle size, prebeta-, and large HDL subspecies concentration in Finnish low-HDL families: relationship with intima-media thickness. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 897–902. [Google Scholar] [CrossRef]
- Stefanovic, A.; Kotur-Stevuljevic, J.; Spasic, S.; Vekic, J.; Zeljkovic, A.; Spasojevic-Kalimanovska, V.; Jelic-Ivanovic, Z. HDL 2 particles are associated with hyperglycaemia, lower PON1 activity and oxidative stress in type 2 diabetes mellitus patients. Clin. Biochem. 2010, 43, 1230–1235. [Google Scholar] [CrossRef]
- Lee, M.; Jang, Y.; Kim, K.; Cho, H.; Jee, S.H.; Park, Y.; Kim, M.K. Relationship between HDL3 subclasses and waist circumferences on the prevalence of metabolic syndrome: KMSRI-Seoul Study. Atherosclerosis 2010, 213, 288–293. [Google Scholar] [CrossRef]
- de Souza, J.A.; Vindis, C.; Hansel, B.; Negre-Salvayre, A.; Therond, P.; Serrano, C.V., Jr.; Chantepie, S.; Salvayre, R.; Bruckert, E.; Chapman, M.J.; et al. Metabolic syndrome features small, apolipoprotein A-I-poor, triglyceride-rich HDL3 particles with defective anti-apoptotic activity. Atherosclerosis 2008, 197, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Kontush, A.; Chantepie, S.; Chapman, M.J. Small, dense HDL particles exert potent protection of atherogenic LDL against oxidative stress. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1881–1888. [Google Scholar] [CrossRef] [PubMed]
- Sakuma, N.; Yoshikawa, M.; Hibino, T.; Ohte, N.; Kamiya, T.; Kunimatsu, M.; Kimura, G.; Inoue, M. HDL3 exerts a more powerful antiperoxidative and protective effect against peroxidative modification of LDL than HDL2 does. J. Nutr. Sci. Vitaminol. (Tokyo) 2002, 48, 278–282. [Google Scholar] [CrossRef] [PubMed]
- Camps, L.; Reina, M.; Llobera, M.; Bengtsson-Olivecrona, G.; Olivecrona, T.; Vilaro, S. Lipoprotein lipase in lungs, spleen, and liver: synthesis and distribution. J. Lipid Res. 1991, 32, 1877–1888. [Google Scholar] [PubMed]
- Liu, G.; Xu, J.N.; Liu, D.; Ding, Q.; Liu, M.N.; Chen, R.; Fan, M.; Zhang, Y.; Zheng, C.; Zou, D.J.; et al. Regulation of plasma lipid homeostasis by hepatic lipoprotein lipase in adult mice. J. Lipid. Res. 2016, 57, 1155–1161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharyya, S.; Dudeja, P.K.; Tobacman, J.K. Tumor necrosis factor alpha-induced inflammation is increased but apoptosis is inhibited by common food additive carrageenan. J. Biol. Chem. 2010, 285, 39511–39522. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AIN76-A Diet (CON) | Western Diet (WD) | 0.1% NDGA Mixed Western Diet (WD + NDGA) | |
|---|---|---|---|
| Ingredient | grams/ Kg | grams/ Kg | grams/ Kg |
| Casein | 200 | 195 | 195 |
| DL-Methionine | 3 | 3 | 3 |
| Cornstarch | 150 | 150 | 150 |
| Sucrose | 499.99 | 341.46 | 340.46 |
| Cellulose | 50 | 50 | 50 |
| Milk Fat | 50 (corn-oil) | 210 | 210 |
| Cholesterol | 1.5 | 1.5 | |
| Vitamin Mix #310035 | 12 | 10 | 10 |
| Mineral Mix #200000 | 35 | 35 | 35 |
| Calcium Carbonate | 4 | 4 | |
| Ethoxyquin | 0.01 | 0.04 | 0.04 |
| NDGA | 1 |
| Gene | Forward/Reverse | Sequence (5′-3′) |
|---|---|---|
| mPPARγ | Forward | aac tgc agg gtg aaa ctc tgg gag att ctc c |
| Reverse | gga ttc agc aac cat tgg gtc agc tct | |
| mFAT/CD36 | Forward | tac ctg gga gtt ggc gag |
| Reverse | ttg cca cgt cat ctg ggt tt | |
| mTNFα | Forward | ctg agg tca atc tgc cca agt ac |
| Reverse | ctt cac aga gca atg act cca aag | |
| mβ-actin | Forward | cct cta tgc caa cac agt gc |
| Reverse | gta ctc ctg ctt gct gat cc |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, I.; Park, M.; Yang, S.J.; Lee, M. Lipoprotein Lipase Inhibitor, Nordihydroguaiaretic Acid, Aggravates Metabolic Phenotypes and Alters HDL Particle Size in the Western Diet-Fed db/db Mice. Int. J. Mol. Sci. 2019, 20, 3057. https://doi.org/10.3390/ijms20123057
Kang I, Park M, Yang SJ, Lee M. Lipoprotein Lipase Inhibitor, Nordihydroguaiaretic Acid, Aggravates Metabolic Phenotypes and Alters HDL Particle Size in the Western Diet-Fed db/db Mice. International Journal of Molecular Sciences. 2019; 20(12):3057. https://doi.org/10.3390/ijms20123057
Chicago/Turabian StyleKang, Inhae, Miyoung Park, Soo Jin Yang, and Myoungsook Lee. 2019. "Lipoprotein Lipase Inhibitor, Nordihydroguaiaretic Acid, Aggravates Metabolic Phenotypes and Alters HDL Particle Size in the Western Diet-Fed db/db Mice" International Journal of Molecular Sciences 20, no. 12: 3057. https://doi.org/10.3390/ijms20123057
APA StyleKang, I., Park, M., Yang, S. J., & Lee, M. (2019). Lipoprotein Lipase Inhibitor, Nordihydroguaiaretic Acid, Aggravates Metabolic Phenotypes and Alters HDL Particle Size in the Western Diet-Fed db/db Mice. International Journal of Molecular Sciences, 20(12), 3057. https://doi.org/10.3390/ijms20123057