1. Introduction
Diabetic cardiomyopathy was first described in 1972 [
1] and is characterized by the existence of ventricular dysfunction in the absence of other cardiac risk factors, such as coronary artery disease, hypertension, and significant valvular disease, in individuals with diabetes mellitus. In the first asymptomatic stage, diabetic cardiomyopathy includes a hidden subclinical period characterised by structural and functional abnormalities, including left ventricular hypertrophy and myocardial fibrosis, increased myocardial stiffness, and subclinical diastolic dysfunction. Subsequently, these abnormalities may evolve to heart failure with preserved ejection fraction (HFpEF) [
2]. More pronounced systolic dysfunction may be accompanied by heart failure with reduced ejection fraction (HFrEF) [
2].
Mechanisms leading to left ventricular impairment in type 2 diabetes are systemic changes. Not surprisingly, the right ventricle is also affected in patients with diabetic cardiomyopathy, as demonstrated by right ventricular remodelling and impaired systolic and diastolic function in men with type 2 diabetes, in a similar manner as changes in left ventricular dimension and left ventricular function [
3]. Moreover, structural alterations occur predominantly in the right chambers of the heart during the early phase of experimental diabetes in rats [
4].
Key metabolic abnormalities in type 2 diabetes mellitus are hyperglycemia, hyperinsulinemia, systemic insulin resistance, and impaired cardiac insulin metabolic signalling. Hyperglycemia, insulin resistance, and hyperinsulinemia induce metabolic alterations that lead to mitochondrial dysfunction, oxidative stress, advanced glycation end products (AGEs), impaired mitochondria Ca
2+ handling, inflammation, activation of the renin–angiotensin–aldosterone system, endoplasmic reticulum stress, impaired myocardial microcirculation, and cardiomyocyte death [
5]. A 1% reduction in haemoglobin A1c was associated with a 16% reduction of heart failure incidence in the UK Prospective Diabetes Study [
6].
Type 2 diabetes mellitus impairs the capacity of the myocardium to use glucose as an energy source and fatty acid oxidation is increased in these subjects [
7]. Increased subcellular vesicular recycling of cluster of differentiation 36 (CD36) from endosomes to the plasma membrane increases the rate of cellular uptake of free fatty acids in diabetic hearts [
8]. Diabetic cardiomyopathy is associated with excess cardiac lipid accumulation [
9]. Accumulation of lipid intermediates in the heart may lead to lipotoxicity characterised by cellular dysfunction, cardiomyocyte death, and deterioration of insulin resistance [
9]. Interestingly, deficiency of CD36 rescues lipotoxic cardiomyopathy [
10].
Epidemiological studies implicate added sugars in the development of the metabolic syndrome and type 2 diabetes mellitus [
11,
12,
13]. In westernized cultures, the use of added sweeteners containing fructose (sucrose and high-fructose corn syrup) has increased by approximately 25% over the past three decades [
14]. Fructose constitutes a particular toxic sugar challenge. It emerges that mice that cannot metabolize fructose are healthier when placed on carbohydrate-rich diets [
15,
16,
17]. Fructose consumption may also impact the development of diabetic cardiomyopathy [
5]. High-fructose diets induce cardiomyocyte autophagy, oxidative stress, and impaired insulin metabolic phosphatidylinositol 3-kinase (PI3K)/Akt/endothelial nitric oxide synthase (eNOS) signalling, and interstitial fibrosis [
5].
Pleiotropic effects of high-density lipoproteins (HDL) including its anti-inflammatory, anti-oxidative, and anti-fibrotic properties may exert favourable effects on the myocardium [
18,
19,
20,
21]. We have previously shown that human
apolipoprotein (apo) A-I gene transfer inhibits the development of diabetic cardiomyopathy in rats [
22] and also improves diastolic function in hypercholesterolemic mice [
23]. Furthermore, selective HDL-raising adeno-associated viral serotype 8-mediated human
apo A-I gene transfer prevents heart failure induced by transverse aortic constriction in C57BL/6 low-density lipoprotein receptor-deficient mice [
24]. However, an effect of HDL on established heart failure in a model of obesity and diabetes has never been investigated in an intervention study.
Apo A-I
Milano is an apo A-I mutant resulting from an arginine 173 to cysteine mutation and was discovered in 1980 in a family from Limone sul Garda in Northern Italy [
25,
26]. Heterozygous carriers of this mutant demonstrate apparent longevity [
27] and are characterised by much less atherosclerosis than expected based on their plasma levels of HDL cholesterol (in the lowest 5th percentile (10–30 mg/dL)) [
28]. MDCO-216 is a pharmaceutical product that contains reconstituted HDL comprising highly purified recombinant dimeric apoA-I
Milano complexed with 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylcholine (POPC) [
29]. The objective of this study was to establish a robust murine model of diabetic cardiomyopathy induced by feeding a high-sugar/high-fat (HSHF) diet in C57BL/6N mice and to investigate the effect of intervention with reconstituted HDL
Milano (MDCO-216) on established heart failure in these diabetic mice.
3. Discussion
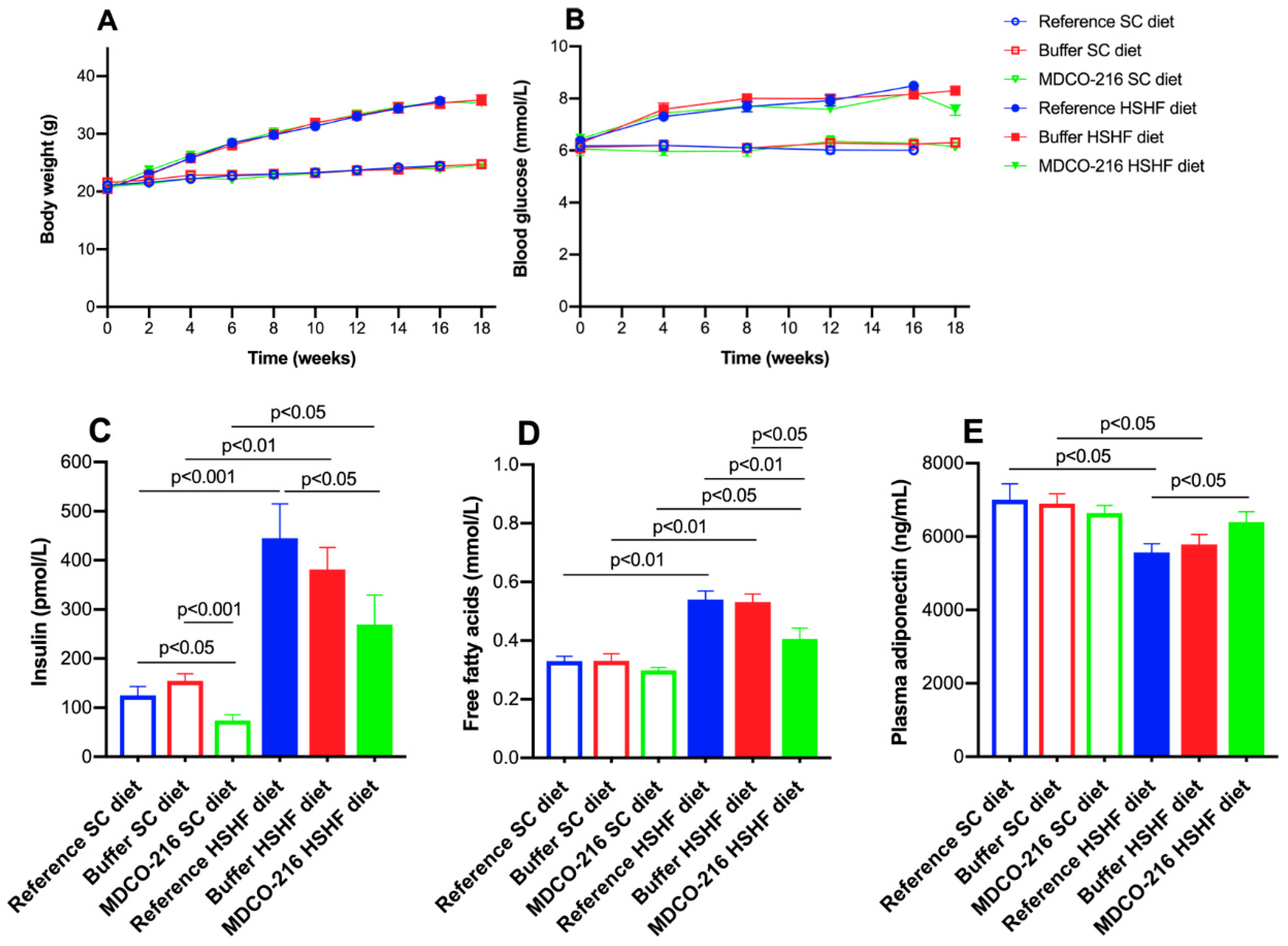
The main findings of the current study are that (1) the HSHF diet induces obesity, type 2 diabetes mellitus, and diabetic cardiomyopathy; (2) the diabetic cardiomyopathy in this model is characterized by cardiac hypertrophy, capillary rarefaction, increased myocardial fibrosis, prominent cardiac dysfunction, and heart failure; (3) intervention with reconstituted HDLMilano in HSHF diet mice reverses heart failure and partially reverses cardiac hypertrophy and pathological remodelling.
Structural and functional alterations and underlying mechanisms leading to diabetic cardiomyopathy in type 2 diabetes mellitus have been mostly investigated in db/db mice, ob/ob mice, Zucker diabetic fatty rats, and in diabetic patients [
5]. Whereas ob/ob mice are leptin-deficient, db/db mice are leptin receptor-deficient [
30]. In Zucker diabetic fatty rats, a mutation in the leptin receptor, OB-R, is associated with leptin resistance and obesity [
31]. Due to single gene mutations that lead to the lack of action by the satiety factor leptin or its cognate receptor, these rodents spontaneously develop severe hyperphagia leading to obesity and manifest some characteristics of type 2 diabetes mellitus [
30]. However, disease-causing genetic mutations in the leptin and leptin receptor are very rare in humans. Moreover, substantial differences exist between these animal models and human type 2 diabetes mellitus [
30]. Considering that the use of added sweeteners containing fructose (sucrose and high-fructose corn syrup) may play a key potentiating role in the development of type 2 diabetes mellitus and associated diabetic cardiomyopathy in humans [
5,
11,
12,
13], an HSHF diet was applied in this study. Important components of this HSHF diet are fructose corn syrup-55 (17.5 weight percentage) and sucrose (17.5 weight percentage).
Previously, it has been demonstrated that short-term feeding of an HSHF diet for 8 weeks in female C57BL/6J starting from the age of 4 weeks induces insulin resistance and diastolic dysfunction as evidenced by echocardiographic analysis [
32]. In the current study, the HSHF diet was initiated at the age of 12 weeks and was maintained for 16 weeks and induced type 2 diabetes mellitus in female C57BL/6N mice. The HSHF diet-induced diabetic cardiomyopathy in female C57BL/6N mice. Cardiomyopathy is defined by the European Society of Cardiology as a myocardial disorder in which the myocardium is both structurally and functionally abnormal, in the absence of coronary artery disease, arterial hypertension, valvular heart disease, and congenital heart disease sufficient to cause the observed abnormality of the heart muscle [
33]. At the structural level, HSHF diet mice were characterized by left ventricular hypertrophy, cardiomyocyte hypertrophy, increased perivascular and interstitial fibrosis, and capillary rarefaction. At the functional level, both systolic and diastolic dysfunction were present. Systolic dysfunctions was evidenced by reduced left ventricular end-systolic elastance (E
es) and decreased preload recruitable stroke work (PRSW), which are parameters of the systolic function that are load-independent. Multiple parameters in the model indicate the presence of diastolic dysfunction in HSHF mice: decreased left ventricular compliance (higher slope of the end-diastolic pressure volume relationship (EDPVR), reduced peak filling rate and impaired left ventricular isovolumetric relaxation. Taken together, the structural and functional data indicate that the HSHF mice represent a
bona fide model of diabetic cardiomyopathy. These abnormalities resulted in a reduced cardiac output and an impaired ventriculo-arterial coupling as evidenced by the significantly increased E
a/E
es ratio in HSHF diet mice. The E
a/E
es ratio or ventriculo-arterial coupling ratio represents a measure of pump efficiency in expelling blood into the vasculature. The most favourable ventriculo-arterial coupling occurs when the E
a/E
es ratio lies in the range 0.5–1.0. However, cardiac dysfunction does not automatically imply the presence of heart failure. The definition of clinical heart failure by the European Society of Cardiology is entirely based on clinical symptoms and signs [
34]. The severe cardiac dysfunction in HSHF diet mice resulted in heart failure as evidenced by the increased wet lung weight and the severely reduced exercise capacity. Taken together, the HSHF diet mice constitute a model of diabetic cardiomyopathy and heart failure.
Intervention with reconstituted HDL
Milano in HSHF diet mice partially reversed cardiac hypertrophy and induced a partial regression of pathological remodelling in HSHF diet mice as evidenced by the increased capillary density and the decreased perivascular fibrosis. The anti-hypertrophic effects of HDL are consistent with prior in vitro and in vivo data. HDL has been shown to downregulate the angiotensin II type 1 receptor [
35,
36] and counteracts mechanical stress-induced autophagy and hypertrophy in cultured cardiomyocytes [
37]. Moreover, continuous infusion of HDL inhibits cardiac hypertrophy in vivo [
36,
37], which may be mediated at least in part via downregulation of the angiotensin II type 1 receptor. We demonstrated that selective HDL-raising gene therapy also exerts anti-hypertrophic effects on the myocardium under conditions of pressure overload [
24]. However, all these prior studies are not the equivalent of clinical intervention in patients with established heart failure since treatment is initiated before the onset of disease. Recently, regression of cardiac hypertrophy induced by administration of reconstituted HDL
Milano was demonstrated in a model of coconut oil-induced HFpEF [
38]. This model was not characterized by diabetes mellitus or by hyperinsulinemia. The increase of capillary density and reduction of perivascular fibrosis reflects the pleiotropic effects of HDL. HDL exert multiple effects on the endothelium [
39] and has potent effects on endothelial progenitor cells [
40,
41,
42,
43]. HDL reduces transforming growth factor-β 1-induced collagen deposition in murine fibroblasts [
44] and decreases transforming growth factor-β1 in the myocardium [
38]. Moreover, HDL has been shown to decrease transforming growth factor-β1-induced endothelial-mesenchymal transition in aortic endothelial cells in vitro [
45]. Gene therapy with an E1E3E4-deleted human
apo A-I gene transfer vectors reduced oxidative stress, inflammation, and myocardial fibrosis in a rat model of diabetic cardiomyopathy [
22]. Adeno-associated viral serotype eight human
apo A-I gene therapy strongly reduced myocardial fibrosis in a model of pressure overload-induced cardiomyopathy following transverse aortic constriction [
24]. Finally, the regression of myocardial fibrosis induced by reconstituted HDL
Milano has recently been demonstrated in a murine model of HFpEF [
38].
Intervention with MDCO-216 resulted in a prominent restoration of cardiac function in HSHF diet mice. Cardiac function at rest was similar in MDCO-216 HSHF diet mice compared to SC diet mice. Several mechanisms may contribute to increased cardiac function. First of all, it can be postulated that HDL has direct electrophysiological effects. HDL may regulate cholesterol distribution between the raft and non-raft membrane fractions [
46]. Microdomain-specific localization of ion channels may affect their function [
47]. Reconstituted HDL containing wild-type apo A-I shortened repolarization of isolated rabbit cardiomyocytes [
48]. Moreover, infusion of reconstituted HDL shortened the heart-rate-corrected QT interval, which represents the duration of the ventricular electrical systole, on surface electrocardiograms in humans [
48]. Direct effects of HDL have been shown in isolated cardiomyocytes in vitro as evidenced by activation of the transcription factor signal transducer and activator of transcription 3 (STAT3) via increased phosphorylation of extracellular signal-regulated kinases (ERK)1/2 [
49] and by augmented phosphorylation of the pro-survival kinase Akt [
22]. Interestingly, Scarb1
−/− mice that are deficient in scavenger receptor class B, type 1 are characterized by more pronounced cardiac hypertrophy, cardiac dysfunction, and heart failure under conditions of pressure overload [
50]. HDL isolated from Scarb1
−/− mice that are deficient in scavenger receptor class B, type 1 is dysfunctional in activating Akt, ERK1/2, and STAT3 in isolated cardiomyocytes [
50]. Finally, the effects of reconstituted HDL
Milano on the cardiac structure may also contribute to improved cardiac function. The increased capillary density and the regression of perivascular fibrosis may enhance cardiac function via improved myocardial microcirculation.
Autophagy is a cellular pathway for lysosomal degradation and recycling of long-lived proteins and organelles, which plays an important role in cardiac homeostasis [
51,
52]. Fructose-induced insulin resistance has been shown to increase autophagy, which may contribute to cardiac pathology [
53]. Since HDL inhibits autophagy in cultured cardiomyocytes [
37], this property may also contribute to the beneficial effects of MDCO-216 in this model of diabetic cardiomyopathy.
HDL modulate glucose metabolism [
19]. HDL exert direct effects on adipose tissue, antagonizes lipolysis and enhances adiponectin expression [
54]. Adiponectin is an adipokine that is downregulated in individuals with obesity-related disorders. HDL also improve peripheral glucose metabolism by phosphorylating AMP-activated protein kinase [
55]. In this study, reconstituted HDL reduced free fatty acids concentrations, glucose levels, and insulin concentrations, and increased plasma adiponectin in HSHF diet mice. Adiponectin has insulin-sensitizing and anti-inflammatory effects [
56]. This adipose-derived plasma protein also influences cardiac remodelling and suppresses pathological cardiac growth [
57]. Taken together, the systemic effects of reconstituted HDL
Milano promote insulin sensitivity and may also contribute to the improvement of cardiac structure and function.
In contrast to adiponectin, levels of several other adipokines including tumour necrosis factor-α (TNF-α) increase in obesity. An inverse relationship exists between TNF-α and adiponectin is observed in humans [
58,
59]. TNF-α plasma levels were significantly decreased in MDCO-216 HSHF diet mice compared to the other HSHF diet groups. Obesity is characterised by chronic systemic inflammation originating from local immune responses in visceral adipose tissue. Infiltration of macrophages into adipose tissue follow the adipocyte-secretion of chemoattractants like TNF-α and free fatty acids [
60]. Inflammation and heart failure are strongly interconnected and may reinforce each other in a mechanism of mutual causality [
61]. TNF-α contributes to cardiac dysfunction and failure [
62]. The cytokine hypothesis of heart failure postulates that heart failure progresses, at least in part, as a consequence of the harmful effects exerted by endogenous cytokine cascades on the heart and the peripheral circulation [
63]. Whereas this hypothesis is essentially unproven, the increased anti-inflammatory potential of HDL and the reduction of TNF-α may have contributed to the beneficial effects of MDCO-216 on cardiac structure and function.
The improved cardiac function in MDCO-216 HSHF diet mice was paralleled by a significant amelioration of exercise capacity compared to the two other HSHF diet groups. The absence of complete normalization of exercise capacity compared to SC diet mice is likely due to the pronounced difference in weight, which requires the development of greater power during running on a 10° incline.
In conclusion, intervention with reconstituted HDLMilano reverses diabetic cardiomyopathy and heart failure in a murine model of type 2 diabetes mellitus.

 ,
, 









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}