Genetic Defects in Mitochondrial Dynamics in Caenorhabditis elegans Impact Ultraviolet C Radiation- and 6-hydroxydopamine-Induced Neurodegeneration

 , ,
, ,  and
and
Abstract
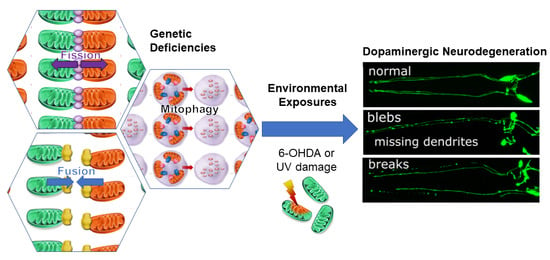
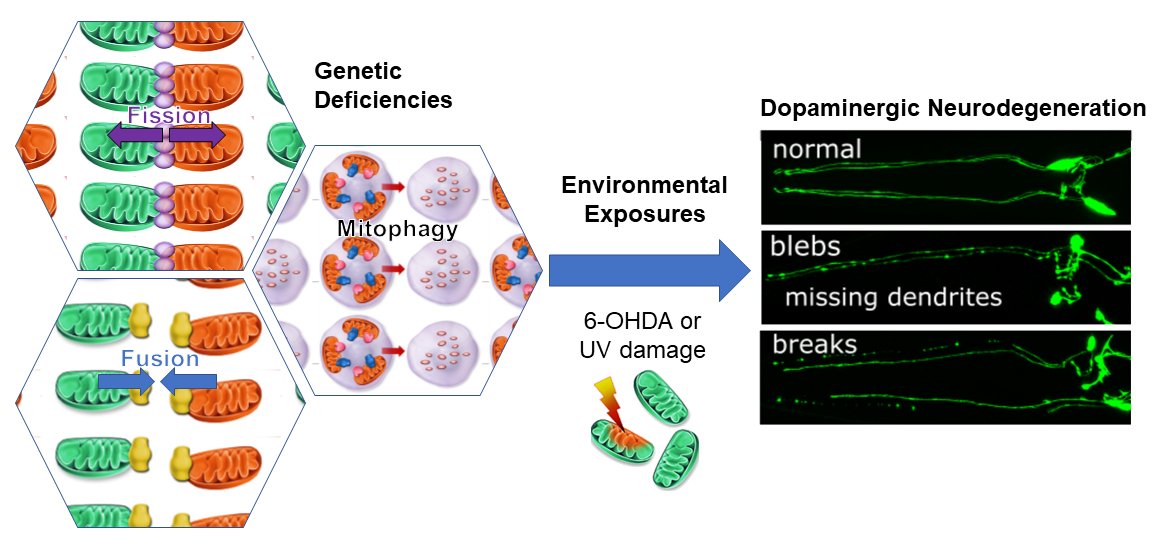
:
1. Introduction
2. Results
2.1. Genetic Deficiencies in Mitochondrial Fission and Fusion Impact Environmentally-Induced Neurodegeneration
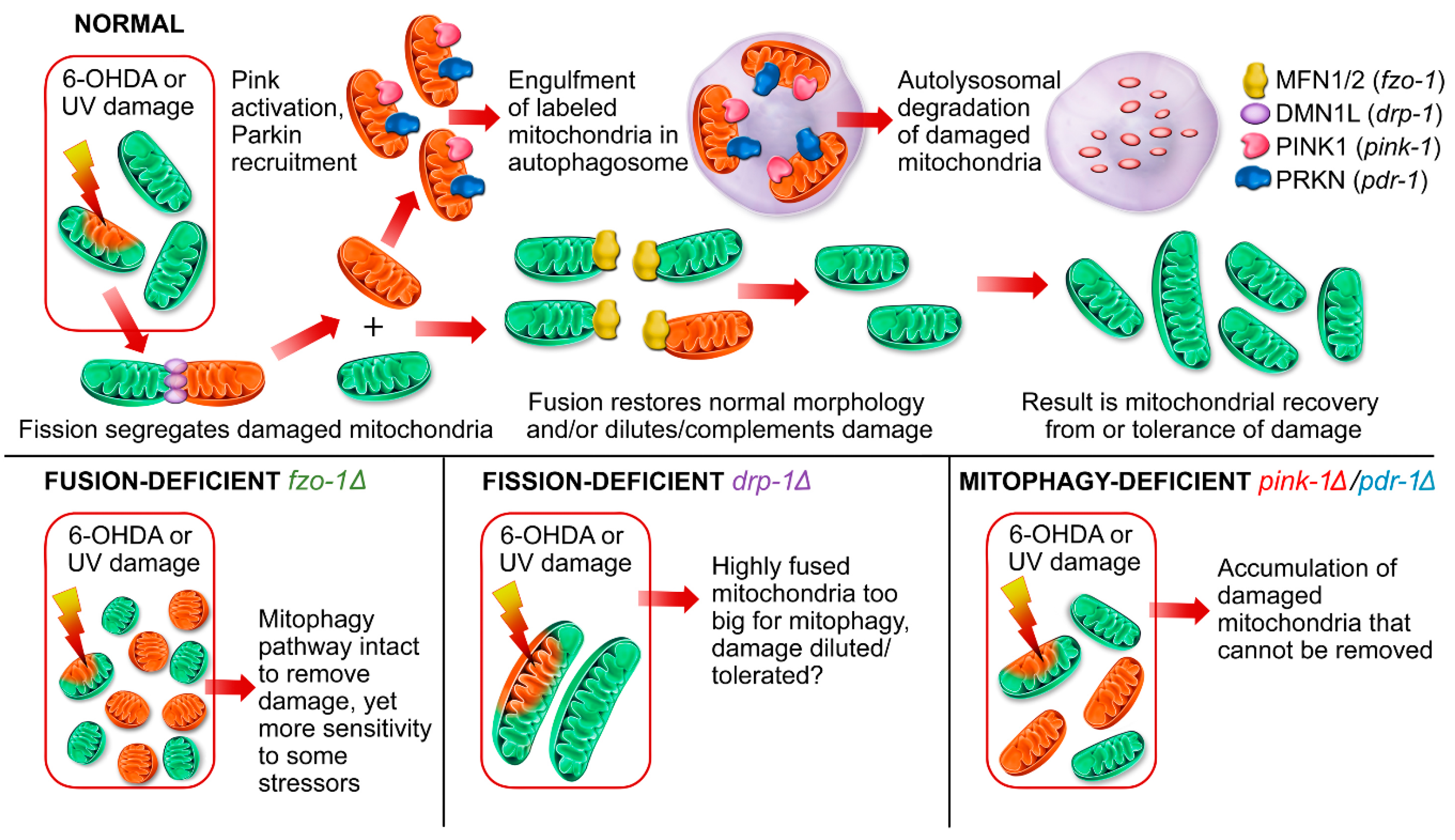
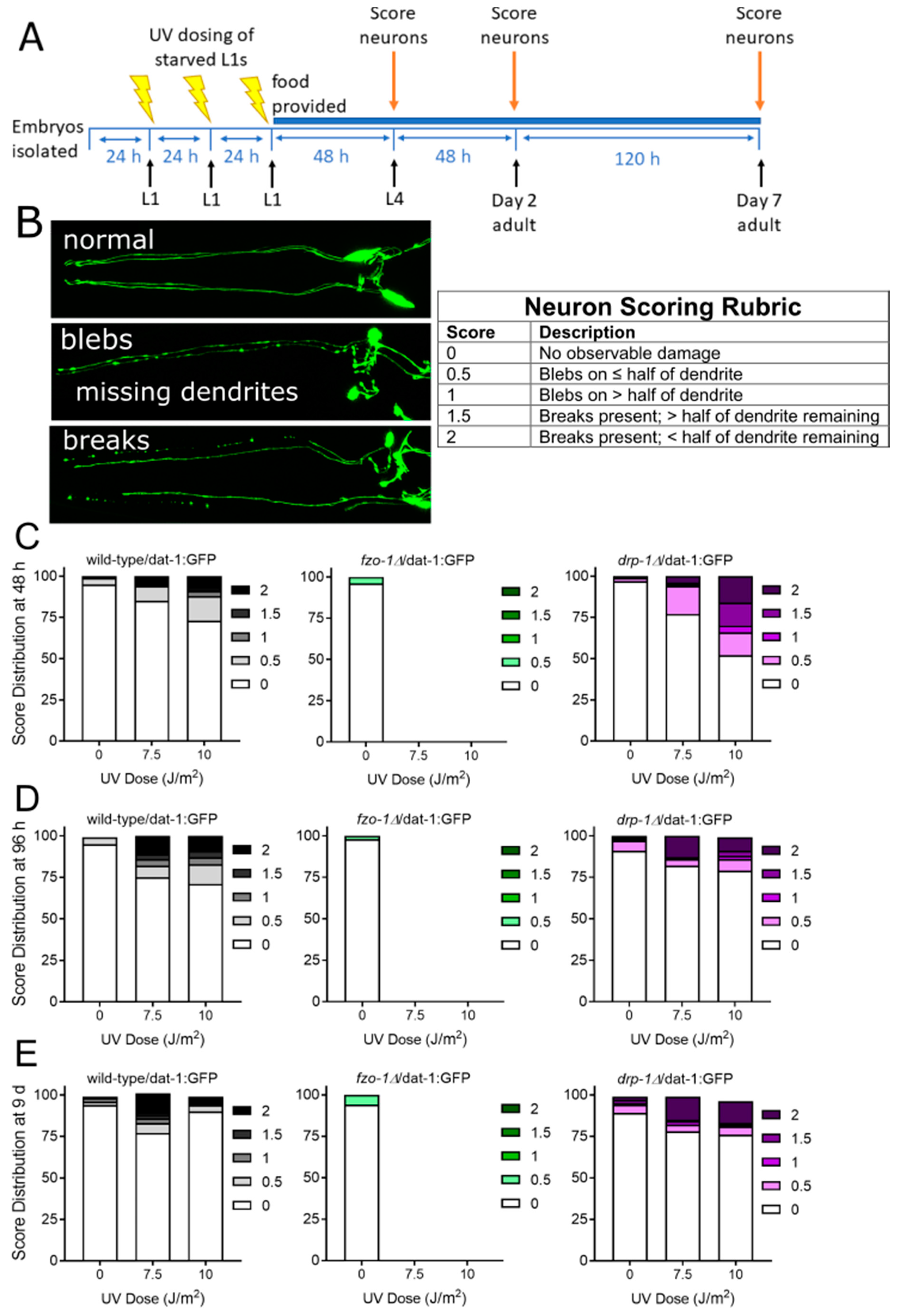
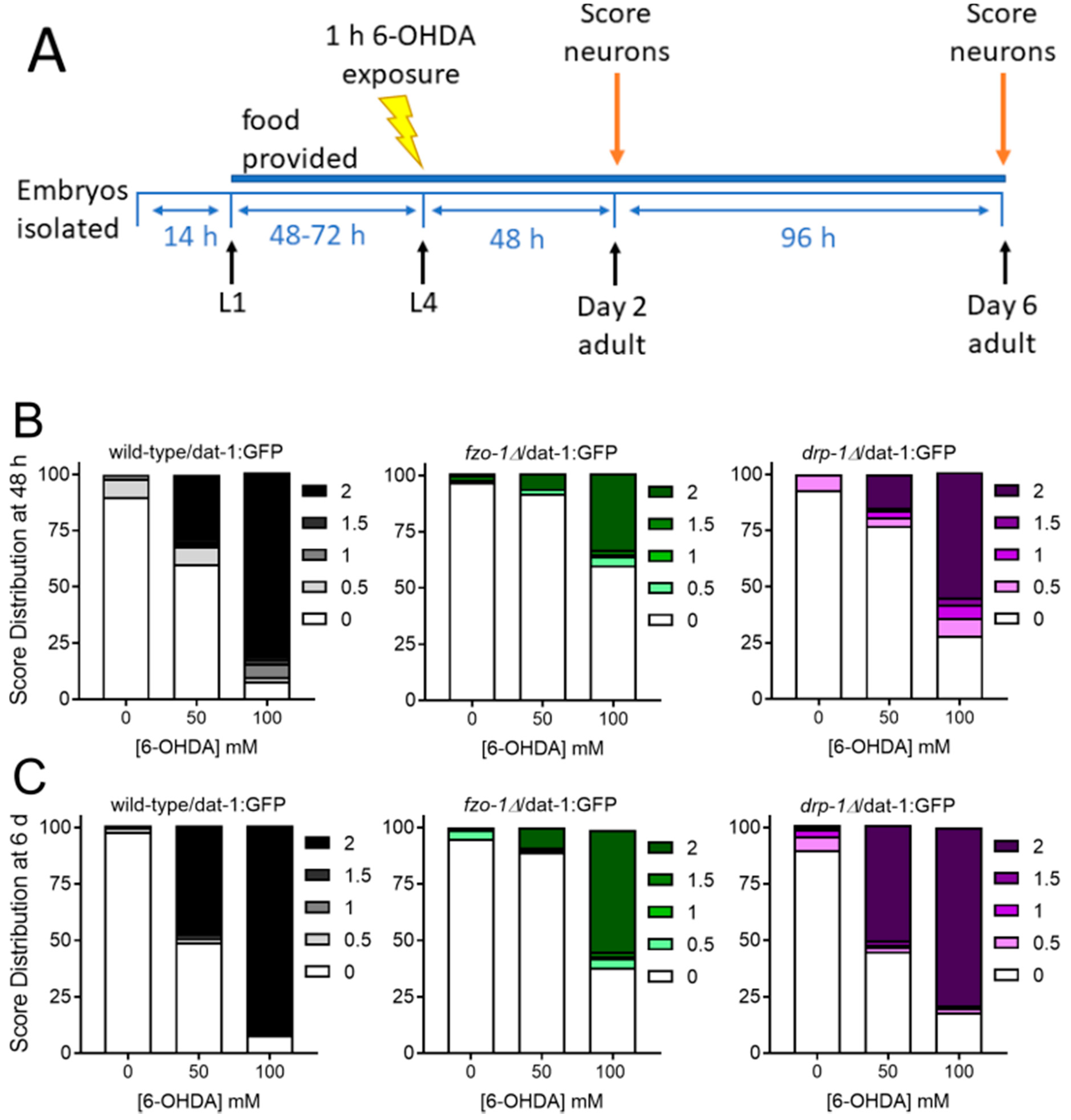
2.1.1. Mitochondrial Fusion Deficiency Sensitized Nematodes to UVC-Induced L1 Larval Arrest and Lethality
2.1.2. Deficiency in Mitochondrial Fission Exacerbates UVC-Induced Neurodegeneration
2.1.3. Mitochondrial Fission and Fusion Mutants are Protected from 6-OHDA-Induced Neurodegeneration
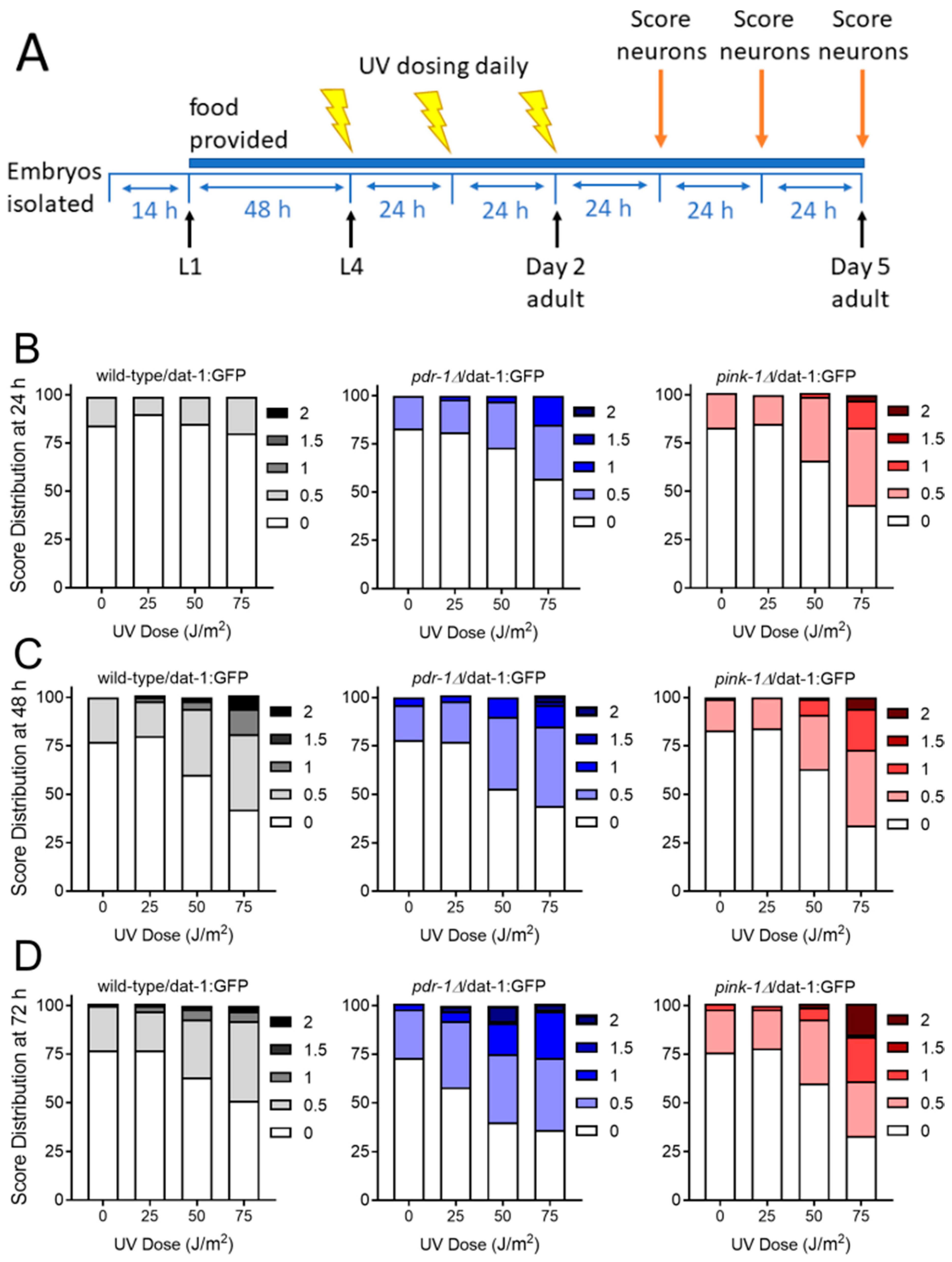
2.2. Animals Lacking Classic Pink1- and Parkin-Mediated Mitophagy are Generally more Sensitive to UVC- and 6-OHDA-Induced Neurodegeneration
2.2.1. Exposure to UVC Caused more Neurodegeneration when Mitophagy was Blocked in pink-1 or pdr-1 Mutants
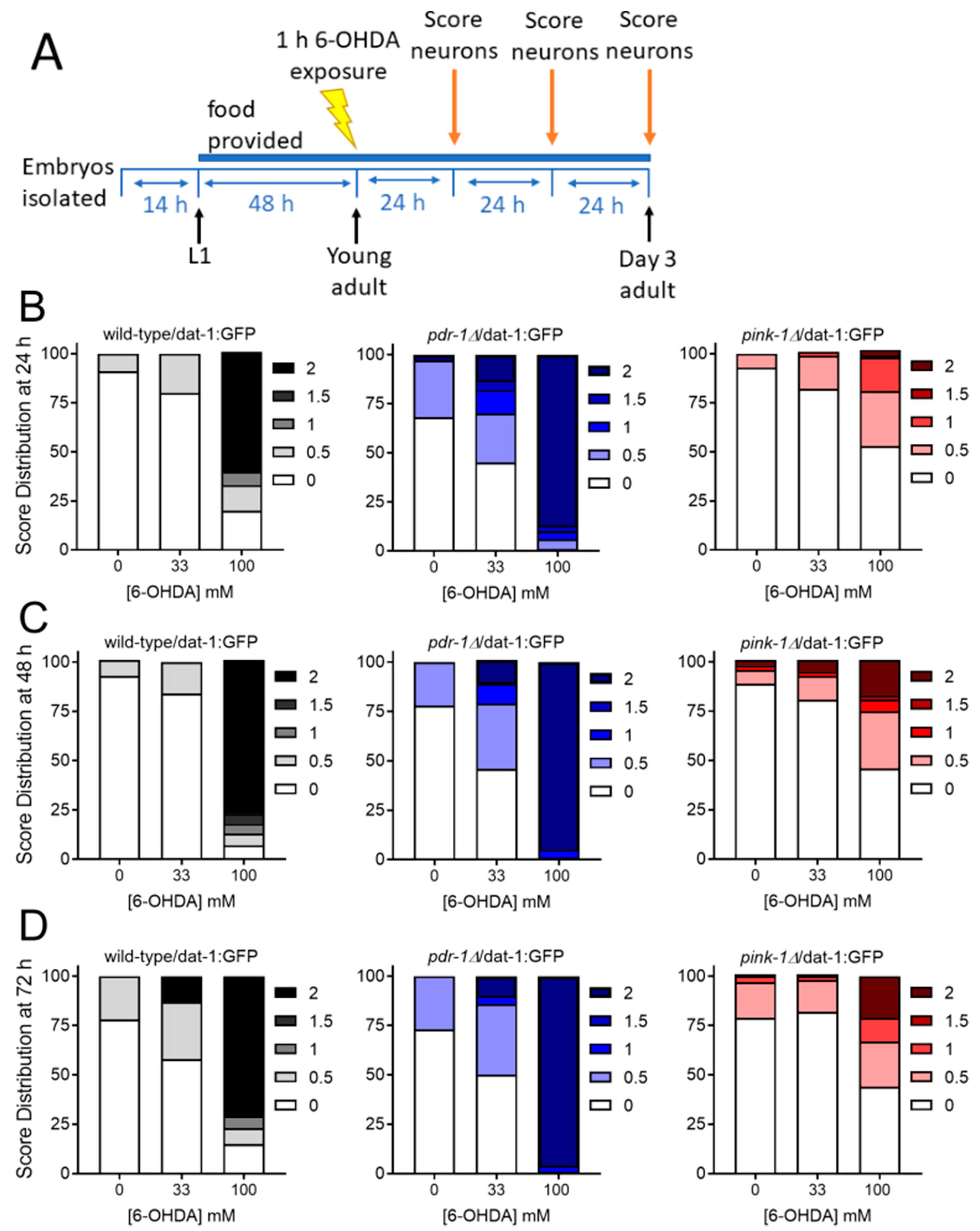
2.2.2. Mutants Deficient in pdr-1 Are More Sensitive to 6-OHDA-Induced Neurodegeneration, but pink-1 Mutants Are Protected
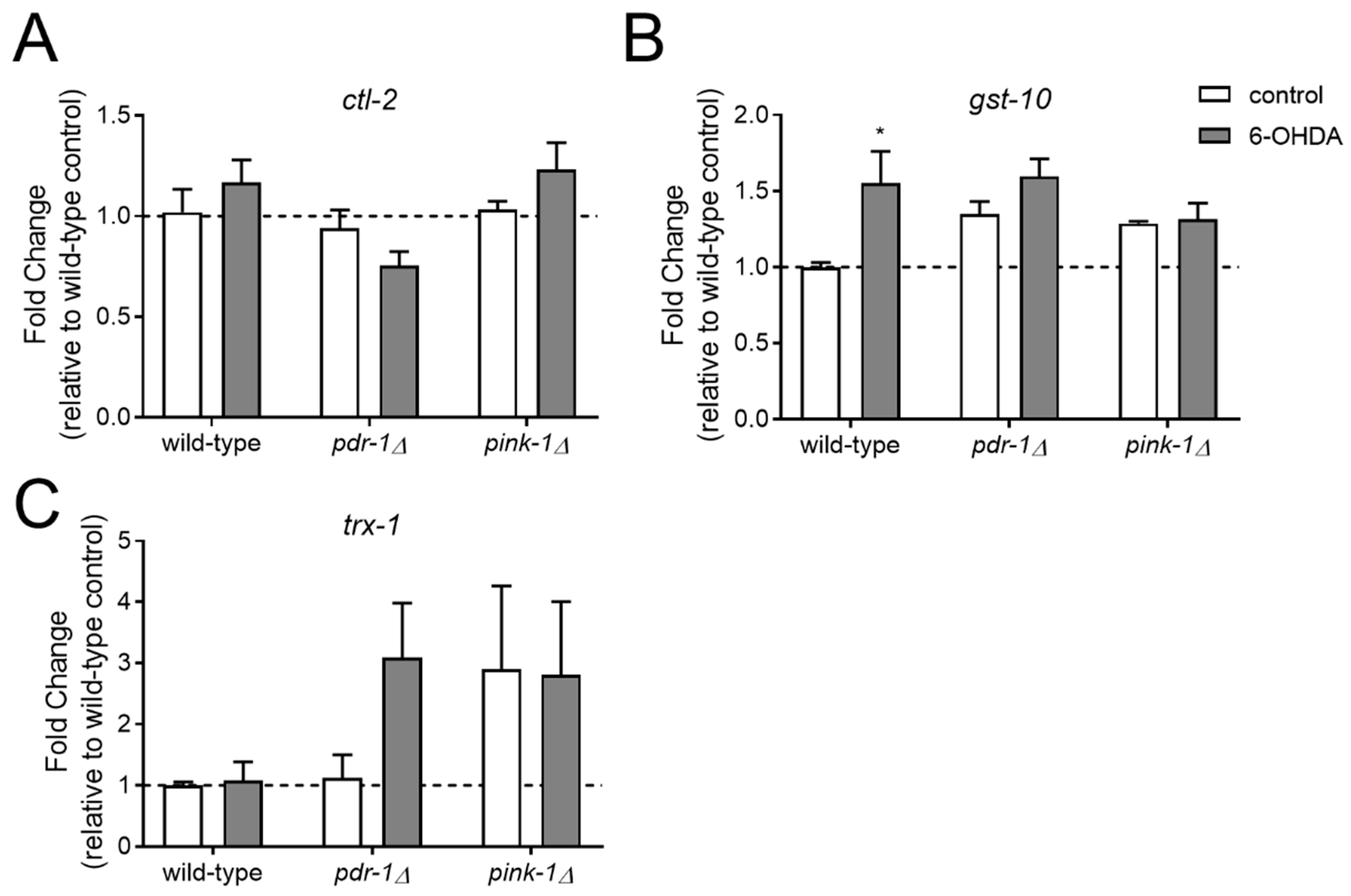
2.3. Resistance to 6-OHDA-Mediated Dopaminergic Neurodegeneration in pink-1 Mutants Is not Mediated by Increased Antioxidant Gene Expression but may Involve Mitochondrial morphology Differences
2.3.1. Pink-1 Mutants do not Differentially Express Antioxidant Genes at Baseline or in Response to 6-OHDA
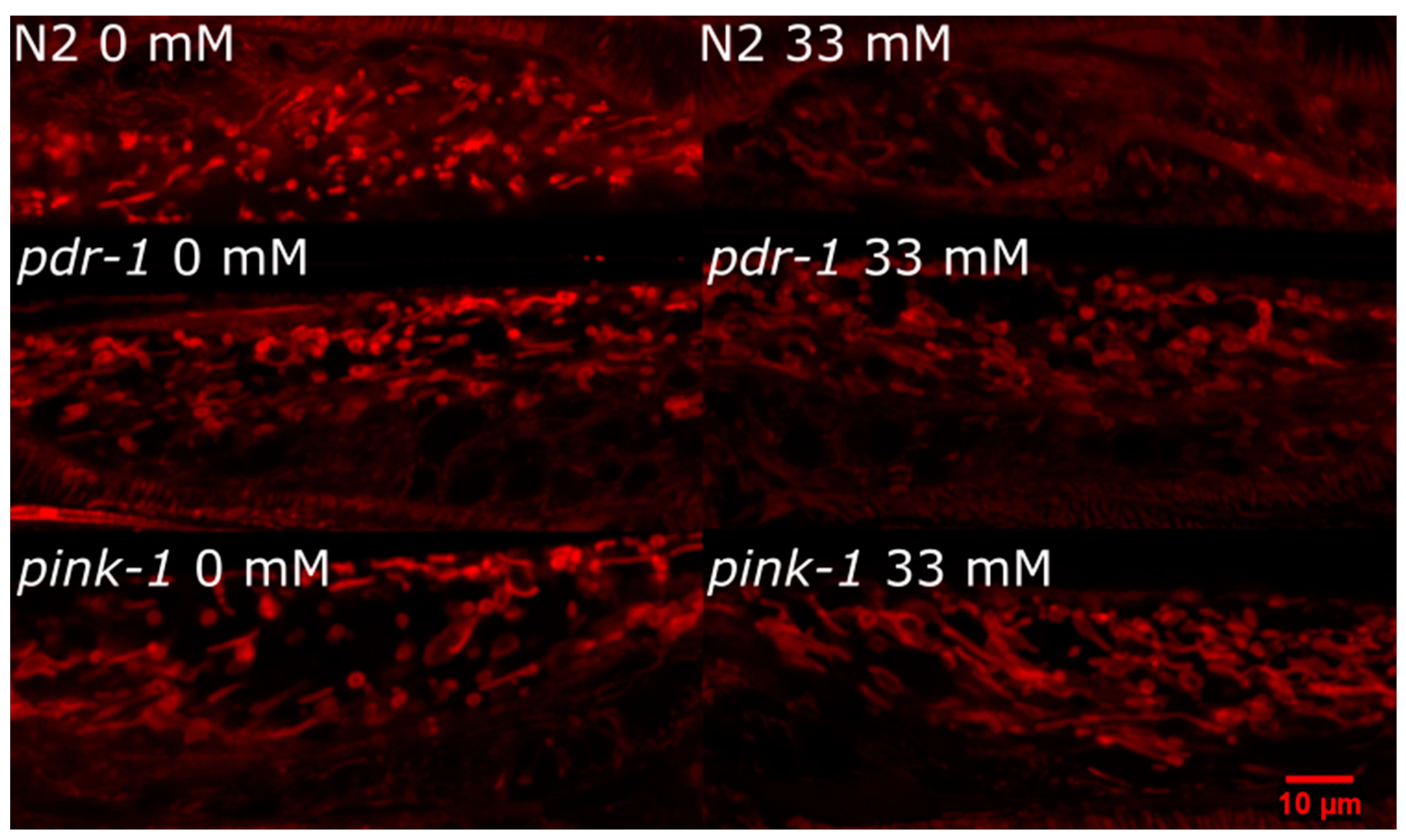
2.3.2. pink-1 Mutant Mitochondrial Morphology Cannot Explain Differential Sensitivity to 6-OHDA
3. Discussion
4. Materials and Methods
4.1. C. elegans Culture
4.2. Toxic Exposures
4.3. Dopaminergic Neurodegeneration Assay
4.4. Targeted Gene Expression Measurements
4.5. Mitochondrial Morphology Evaluation
4.6. Statistical Analyses
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| mtDNA | mitochondrial DNA |
| 6-OHDA | 6-hydroxydopamine |
| UVC | ultraviolet C radiation |
| PD | Parkinson’s Disease |
References
- Cannon, J.R.; Greenamyre, J.T. The role of environmental exposures in neurodegeneration and neurodegenerative diseases. Toxicol. Sci. 2011, 124, 225–250. [Google Scholar] [CrossRef] [PubMed]
- Bronstein, J.; Carvey, P.; Chen, H.; Cory-Slechta, D.; DiMonte, D.; Duda, J.; English, P.; Goldman, S.; Grate, S.; Hansen, J.; et al. Meeting report: Consensus statement-Parkinson’s disease and the environment: Collaborative on health and the environment and Parkinson’s Action Network (CHE PAN) conference 26-28 June 2007. Environ. Health Perspect. 2009, 117, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Meyer, J.N.; Leung, M.C.K.; Rooney, J.P.; Sendoel, A.; Hengartner, M.O.; Kisby, G.E.; Bess, A.S. Mitochondria as a Target of Environmental Toxicants. Toxicol. Sci. 2013. [Google Scholar] [CrossRef] [PubMed]
- Fleming, S.M. Mechanisms of Gene-Environment Interactions in Parkinson’s Disease. Curr. Environ. Health Rep. 2017, 4, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Helley, M.P.; Pinnell, J.; Sportelli, C.; Tieu, K. Mitochondria: A Common Target for Genetic Mutations and Environmental Toxicants in Parkinson’s Disease. Front. Genet. 2017, 8, 177. [Google Scholar] [CrossRef] [PubMed]
- Schapira, A.H. Mitochondria in the aetiology and pathogenesis of Parkinson’s disease. Lancet. Neurol. 2008, 7, 97–109. [Google Scholar] [CrossRef]
- Abou-Sleiman, P.M.; Muqit, M.M.K.; Wood, N.W. Expanding insights of mitochondrial dysfunction in Parkinson’s disease. Nat. Rev. Neurosci. 2006, 7, 207–219. [Google Scholar] [CrossRef]
- Ekstrand, M.I.; Terzioglu, M.; Galter, D.; Zhu, S.; Hofstetter, C.; Lindqvist, E.; Thams, S.; Bergstrand, A.; Hansson, F.S.; Trifunovic, A.; et al. Progressive parkinsonism in mice with respiratory-chain-deficient dopamine neurons. Proc. Natl. Acad. Sci. USA 2007, 104, 1325–1330. [Google Scholar] [CrossRef] [Green Version]
- Sliter, D.A.; Martinez, J.; Hao, L.; Chen, X.; Sun, N.; Fischer, T.D.; Burman, J.L.; Li, Y.; Zhang, Z.; Narendra, D.P.; et al. Parkin and PINK1 mitigate STING-induced inflammation. Nature 2018, 561, 258–262. [Google Scholar] [CrossRef]
- Zimmermann, M.; Reichert, A.S. How to get rid of mitochondria: Crosstalk and regulation of multiple mitophagy pathways. Biol. Chem. 2017, 399, 29–45. [Google Scholar] [CrossRef]
- Gautier, C.A.; Corti, O.; Brice, A. Mitochondrial dysfunctions in Parkinson’s disease. Rev. Neurol. 2014, 170, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Hattori, N. Genetic mutations and mitochondrial toxins shed new light on the pathogenesis of Parkinson’s disease. Parkinson’s Dis. 2011, 2011, 979231. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Wang, G. Mitochondrial dysfunction in Parkinson’s disease. Transl. Neurodegener. 2016, 5, 14. [Google Scholar] [CrossRef] [PubMed]
- Larsen, S.B.; Hanss, Z.; Kruger, R. The genetic architecture of mitochondrial dysfunction in Parkinson’s disease. Cell Tissue Res. 2018, 373, 21–37. [Google Scholar] [CrossRef] [PubMed]
- Giannoccaro, M.P.; La Morgia, C.; Rizzo, G.; Carelli, V. Mitochondrial DNA and primary mitochondrial dysfunction in Parkinson’s disease. Mov. Disord. Off. J. Mov. Disord. Soc. 2017, 32, 346–363. [Google Scholar] [CrossRef] [PubMed]
- Ricciardi, L.; Petrucci, S.; Guidubaldi, A.; Ialongo, T.; Serra, L.; Ferraris, A.; Spano, B.; Bozzali, M.; Valente, E.M.; Bentivoglio, A.R. Phenotypic variability of PINK1 expression: 12 Years’ clinical follow-up of two Italian families. Mov. Disord. Off. J. Mov. Disord. Soc. 2014, 29, 1561–1566. [Google Scholar] [CrossRef] [PubMed]
- Puschmann, A.; Fiesel, F.C.; Caulfield, T.R.; Hudec, R.; Ando, M.; Truban, D.; Hou, X.; Ogaki, K.; Heckman, M.G.; James, E.D.; et al. Heterozygous PINK1 p.G411S increases risk of Parkinson’s disease via a dominant-negative mechanism. Brain 2017, 140, 98–117. [Google Scholar] [CrossRef]
- Huttenlocher, J.; Stefansson, H.; Steinberg, S.; Helgadottir, H.T.; Sveinbjornsdottir, S.; Riess, O.; Bauer, P.; Stefansson, K. Heterozygote carriers for CNVs in PARK2 are at increased risk of Parkinson’s disease. Hum. Mol. Genet. 2015, 24, 5637–5643. [Google Scholar] [CrossRef]
- Meyer, J.N.; Leuthner, T.C.; Luz, A.L. Mitochondrial fusion, fission, and mitochondrial toxicity. Toxicology 2017, 391, 42–53. [Google Scholar] [CrossRef]
- Bagli, E.; Zikou, A.K.; Agnantis, N.; Kitsos, G. Mitochondrial Membrane Dynamics and Inherited Optic Neuropathies. In Vivo (Athens, Greece) 2017, 31, 511–525. [Google Scholar] [CrossRef]
- Lee, S.; Sterky, F.H.; Mourier, A.; Terzioglu, M.; Cullheim, S.; Olson, L.; Larsson, N.-G. Mitofusin 2 is necessary for striatal axonal projections of midbrain dopamine neurons. Human Mol. Genet. 2012, 21, 4827–4835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Dorn, G.W. PINK1-Phosphorylated Mitofusin 2 Is a Parkin Receptor for Culling Damaged Mitochondria. Science (New York, N.Y.) 2013, 340, 471–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Hunt, C.P.; Leung, M.C.; Bodhicharla, R.K.; McKeever, M.G.; Arrant, A.E.; Margillo, K.M.; Ryde, I.T.; Cyr, D.D.; Kosmaczewski, S.G.; Hammarlund, M.; et al. Exposure to mitochondrial genotoxins and dopaminergic neurodegeneration in Caenorhabditis elegans. PLoS ONE 2014, 9, e114459. [Google Scholar] [CrossRef] [PubMed]
- Nass, R.; Hall, D.H.; Miller, D.M., 3rd; Blakely, R.D. Neurotoxin-induced degeneration of dopamine neurons in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 2002, 99, 3264–3269. [Google Scholar] [CrossRef] [PubMed]
- Bess, A.S.; Crocker, T.L.; Ryde, I.T.; Meyer, J.N. Mitochondrial dynamics and autophagy aid in removal of persistent mitochondrial DNA damage in Caenorhabditis elegans. Nucleic Acids Res. 2012, 40, 7916–7931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luz, A.L.; Rooney, J.P.; Kubik, L.L.; Gonzalez, C.P.; Song, D.H.; Meyer, J.N. Mitochondrial Morphology and Fundamental Parameters of the Mitochondrial Respiratory Chain Are Altered in Caenorhabditis elegans Strains Deficient in Mitochondrial Dynamics and Homeostasis Processes. PLoS ONE 2015, 10, e0130940. [Google Scholar] [CrossRef] [PubMed]
- Luz, A.L.; Godebo, T.R.; Smith, L.L.; Leuthner, T.C.; Maurer, L.L.; Meyer, J.N. Deficiencies in mitochondrial dynamics sensitize Caenorhabditis elegans to arsenite and other mitochondrial toxicants by reducing mitochondrial adaptability. Toxicology 2017, 387, 81–94. [Google Scholar] [CrossRef]
- Leung, M.; Rooney, J.; Ryde, I.; Bernal, A.; Bess, A.; Crocker, T.; Ji, A.; Meyer, J. Effects of early life exposure to ultraviolet C radiation on mitochondrial DNA content, transcription, ATP production, and oxygen consumption in developing Caenorhabditis elegans. BMC Pharmacol. Toxicol. 2013, 14, 9. [Google Scholar] [CrossRef]
- Yang, C.C.; Chen, D.; Lee, S.S.; Walter, L. The dynamin-related protein DRP-1 and the insulin signaling pathway cooperate to modulate Caenorhabditis elegans longevity. Aging Cell 2011, 10, 724–728. [Google Scholar] [CrossRef] [Green Version]
- Civelek, M.; Mehrkens, J.F.; Carstens, N.M.; Fitzenberger, E.; Wenzel, U. Inhibition of mitophagy decreases survival of Caenorhabditis elegans by increasing protein aggregation. Mol. Cell. Biochem. 2019, 452, 123–131. [Google Scholar] [CrossRef]
- Stepkowski, T.M.; Meczynska-Wielgosz, S.; Kruszewski, M. mitoLUHMES: An Engineered Neuronal Cell Line for the Analysis of the Motility of Mitochondria. Cell. Mol. Neurobiol. 2017, 37, 1055–1066. [Google Scholar] [CrossRef]
- Gomez-Lazaro, M.; Bonekamp, N.A.; Galindo, M.F.; Jordan, J.; Schrader, M. 6-Hydroxydopamine (6-OHDA) induces Drp1-dependent mitochondrial fragmentation in SH-SY5Y cells. Free. Radic. Biol. Med. 2008, 44, 1960–1969. [Google Scholar] [CrossRef] [PubMed]
- Xi, Y.; Feng, D.; Tao, K.; Wang, R.; Shi, Y.; Qin, H.; Murphy, M.P.; Yang, Q.; Zhao, G. MitoQ protects dopaminergic neurons in a 6-OHDA induced PD model by enhancing Mfn2-dependent mitochondrial fusion via activation of PGC-1alpha. Biochim. et Biophys. Acta Mol. Basis Dis. 2018, 1864, 2859–2870. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Wen, M.; Lin, X.; Chen, Y.H.; Gou, Y.; Li, Y.; Zhang, Y.; Li, H.W.; Tang, L. Alpha Lipoamide Ameliorates Motor Deficits and Mitochondrial Dynamics in the Parkinson’s Disease Model Induced by 6-Hydroxydopamine. Neurotox. Res. 2018, 33, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Rappold, P.M.; Cui, M.; Grima, J.C.; Fan, R.Z.; de Mesy-Bentley, K.L.; Chen, L.; Zhuang, X.; Bowers, W.J.; Tieu, K. Drp1 inhibition attenuates neurotoxicity and dopamine release deficits in vivo. Nat. Commun 2014, 5, 5244. [Google Scholar] [CrossRef]
- Bido, S.; Soria, F.N.; Fan, R.Z.; Bezard, E.; Tieu, K. Mitochondrial division inhibitor-1 is neuroprotective in the A53T-alpha-synuclein rat model of Parkinson’s disease. Sci. Rep. 2017, 7, 7495. [Google Scholar] [CrossRef]
- Kim, H.; Perentis, R.J.; Caldwell, G.A.; Caldwell, K.A. Gene-by-environment interactions that disrupt mitochondrial homeostasis cause neurodegeneration in C. elegans Parkinson’s models. Cell Death Dis. 2018, 9, 555. [Google Scholar] [CrossRef]
- Pham, A.H.; Meng, S.; Chu, Q.N.; Chan, D.C. Loss of Mfn2 results in progressive, retrograde degeneration of dopaminergic neurons in the nigrostriatal circuit. Human Mol. Genet. 2012, 21, 4817–4826. [Google Scholar] [CrossRef]
- Galloway, C.A.; Lee, H.; Nejjar, S.; Jhun, B.S.; Yu, T.; Hsu, W.; Yoon, Y. Transgenic Control of Mitochondrial Fission Induces Mitochondrial Uncoupling and Relieves Diabetic Oxidative Stress. Diabetes 2012, 61, 2093–2104. [Google Scholar] [CrossRef] [Green Version]
- Cho, I.; Song, H.O.; Cho, J.H. Mitochondrial Uncoupling Attenuates Age-Dependent Neurodegeneration in C. elegans. Mol. Cells 2017, 40, 864–870. [Google Scholar] [CrossRef]
- Cooper, J.F.; Machiela, E.; Dues, D.J.; Spielbauer, K.K.; Senchuk, M.M.; Van Raamsdonk, J.M. Activation of the mitochondrial unfolded protein response promotes longevity and dopamine neuron survival in Parkinson’s disease models. Sci. Rep. 2017, 7, 16441. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, S.; Jin, X.; Furukawa, K.; Hamasaki, M.; Nezu, A.; Otera, H.; Saigusa, T.; Yoshimori, T.; Sakai, Y.; Mihara, K.; et al. Mitochondrial division occurs concurrently with autophagosome formation but independently of Drp1 during mitophagy. J. Cell Biol. 2016, 215, 649. [Google Scholar] [CrossRef] [PubMed]
- Goldman, S.M. Environmental toxins and Parkinson’s disease. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 141–164. [Google Scholar] [CrossRef] [PubMed]
- Pinto, M.; Nissanka, N.; Moraes, C.T. Lack of Parkin Anticipates the Phenotype and Affects Mitochondrial Morphology and mtDNA Levels in a Mouse Model of Parkinson’s Disease. J. Neurosci 2018, 38, 1042–1053. [Google Scholar] [CrossRef] [PubMed]
- Pickrell, A.M.; Huang, C.H.; Kennedy, S.R.; Ordureau, A.; Sideris, D.P.; Hoekstra, J.G.; Harper, J.W.; Youle, R.J. Endogenous Parkin Preserves Dopaminergic Substantia Nigral Neurons following Mitochondrial DNA Mutagenic Stress. Neuron 2015, 87, 371–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiavi, A.; Maglioni, S.; Palikaras, K.; Shaik, A.; Strappazzon, F.; Brinkmann, V.; Torgovnick, A.; Castelein, N.; De Henau, S.; Braeckman, B.P.; et al. Iron-Starvation-Induced Mitophagy Mediates Lifespan Extension upon Mitochondrial Stress in C. elegans. Curr. Biol. CB 2015, 25, 1810–1822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, J.F.; Spielbauer, K.K.; Senchuk, M.M.; Nadarajan, S.; Colaiacovo, M.P.; Van Raamsdonk, J.M. alpha-synuclein expression from a single copy transgene increases sensitivity to stress and accelerates neuronal loss in genetic models of Parkinson’s disease. Exp. Neurol. 2018, 310, 58–69. [Google Scholar] [CrossRef]
- Bornhorst, J.; Chakraborty, S.; Meyer, S.; Lohren, H.; Brinkhaus, S.G.; Knight, A.L.; Caldwell, K.A.; Caldwell, G.A.; Karst, U.; Schwerdtle, T.; et al. The effects of pdr1, djr1.1 and pink1 loss in manganese-induced toxicity and the role of alpha-synuclein in C. elegans. Met. Integr. Biometal Sci. 2014, 6, 476–490. [Google Scholar] [CrossRef]
- Martinez-Finley, E.J.; Chakraborty, S.; Slaughter, J.C.; Aschner, M. Early-life exposure to methylmercury in wildtype and pdr-1/parkin knockout C. elegans. Neurochem. Res. 2013, 38, 1543–1552. [Google Scholar] [CrossRef]
- Wu, S.; Lei, L.; Song, Y.; Liu, M.; Lu, S.; Lou, D.; Shi, Y.; Wang, Z.; He, D. Mutation of hop-1 and pink-1 attenuates vulnerability of neurotoxicity in C. elegans: The role of mitochondria-associated membrane proteins in Parkinsonism. Exp. Neurol. 2018, 309, 67–78. [Google Scholar] [CrossRef]
- Hamamichi, S.; Rivas, R.N.; Knight, A.L.; Cao, S.; Caldwell, K.A.; Caldwell, G.A. Hypothesis-based RNAi screening identifies neuroprotective genes in a Parkinson’s disease model. Proc. Natl. Acad. Sci. USA 2008, 105, 728–733. [Google Scholar] [CrossRef] [PubMed]
- Gautier, C.A.; Kitada, T.; Shen, J. Loss of PINK1 causes mitochondrial functional defects and increased sensitivity to oxidative stress. Proc. Natl. Acad. Sci. USA 2008, 105, 11364–11369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morais, V.A.; Haddad, D.; Craessaerts, K.; De Bock, P.-J.; Swerts, J.; Vilain, S.; Aerts, L.; Overbergh, L.; Grünewald, A.; Seibler, P.; et al. PINK1 Loss-of-Function Mutations Affect Mitochondrial Complex I Activity via NdufA10 Ubiquinone Uncoupling. Science (New York, N.Y.) 2014, 344, 203–207. [Google Scholar] [CrossRef] [PubMed]
- Morais, V.A.; Verstreken, P.; Roethig, A.; Smet, J.; Snellinx, A.; Vanbrabant, M.; Haddad, D.; Frezza, C.; Mandemakers, W.; Vogt-Weisenhorn, D.; et al. Parkinson’s disease mutations in PINK1 result in decreased Complex I activity and deficient synaptic function. EMBO Mol. Med. 2009, 1, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Dagda, R.K.; Pien, I.; Wang, R.; Zhu, J.; Wang, K.Z.Q.; Callio, J.; Banerjee, T.D.; Dagda, R.Y.; Chu, C.T. Beyond the mitochondrion: Cytosolic PINK1 remodels dendrites through Protein Kinase A. J. Neurochem. 2014, 128, 864–877. [Google Scholar] [CrossRef]
- Menges, S.; Minakaki, G.; Schaefer, P.M.; Meixner, H.; Prots, I.; Schlotzer-Schrehardt, U.; Friedland, K.; Winner, B.; Outeiro, T.F.; Winklhofer, K.F.; et al. Alpha-synuclein prevents the formation of spherical mitochondria and apoptosis under oxidative stress. Sci. Rep. 2017, 7, 42942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, P.L.; Dusenbery, D.B. Using the nematode Caenorhabditis elegans to predict mammalian acute lethality to metallic salts. Toxicol. Ind. Health 1988, 4, 469–478. [Google Scholar] [CrossRef]
- Lewis, J.A.; Fleming, J.T. Basic culture methods. Methods Cell Biol. 1995, 48, 3–29. [Google Scholar]
- Boyd, W.A.; Smith, M.V.; Kissling, G.E.; Rice, J.R.; Snyder, D.W.; Portier, C.J.; Freedman, J.H. Application of a Mathematical Model to Describe the Effects of Chlorpyrifos on Caenorhabditis elegans Development. PLoS ONE 2009, 4, e7024. [Google Scholar] [CrossRef]
- Sulston, J.E.; Horvitz, H.R. Post-embryonic cell lineages of the nematode, Caenorhabditis elegans. Dev. Biol. 1977, 56, 110–156. [Google Scholar] [CrossRef]
- Meyer, J.N.; Boyd, W.A.; Azzam, G.A.; Haugen, A.C.; Freedman, J.H.; Van Houten, B. Decline of nucleotide excision repair capacity in aging Caenorhabditis elegans. Genome Biol. 2007, 8, R70. [Google Scholar] [CrossRef] [PubMed]
- Meyer, J.N.; Lord, C.A.; Yang, X.Y.; Turner, E.A.; Badireddy, A.R.; Marinakos, S.M.; Chilkoti, A.; Wiesner, M.R.; Auffan, M. Intracellular uptake and associated toxicity of silver nanoparticles in Caenorhabditis elegans. Aquat. Toxicol. 2010, 100, 140–150. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Timepoint | Dose (J/m2) | fzo-1 vs. WT | drp-1 vs. WT |
|---|---|---|---|
| 48 h | 0 | 0.7991 | 0.6889 |
| 7.5 | N.D. 1 | 0.2973 | |
| 10 | N.D. | <0.001 * | |
| 96 h | 0 | 0.1127 | 0.1361 |
| 7.5 | N.D. | 0.0071 * | |
| 10 | N.D. | 0.3512 | |
| 9 d | 0 | 0.0237 * | 0.1157 |
| 7.5 | N.D. | 0.8769 | |
| 10 | N.D. | 0.0222 * |
| Timepoint | Dose (mM) | fzo-1 vs. WT | drp-1 vs. WT |
|---|---|---|---|
| 48 h | 0 | <0.0001 * | 0.1740 |
| 50 | <0.0001 * | 0.0018 * | |
| 100 | <0.0001 * | <0.0001 * | |
| 6 d | 0 | 0.4128 | 0.0053 * |
| 50 | <0.0001 * | 0.9166 | |
| 100 | <0.0001 * | 0.0040 * |
| Timepoint | Dose (J/m2) | pdr-1 vs. WT | pink-1 vs. WT |
|---|---|---|---|
| 24 h | 0 | 0.8599 | 0.7267 |
| 25 | 0.0438 * | 0.2373 | |
| 50 | 0.0119 * | 0.0005 * | |
| 75 | <0.0001 * | <0.0001 * | |
| 48 h | 0 | 0.0517 | 0.1928 |
| 25 | 0.7275 | 0.4445 | |
| 50 | 0.1419 | 0.5351 | |
| 75 | 0.3884 | 0.3332 | |
| 72 h | 0 | 0.5833 | 0.7540 |
| 25 | 0.0124 * | 0.9593 | |
| 50 | 0.0002 * | 0.9467 | |
| 75 | 0.0001 * | <0.0001 * |
| Timepoint | Dose (mM) | pdr-1 vs. WT | pink-1 vs. WT |
|---|---|---|---|
| 24 h | 0 | <0.0001 * | 0.6336 |
| 33 | <0.0001 * | 0.3738 | |
| 100 | <0.0001 * | <0.0001 * | |
| 48 h | 0 | 0.0030 * | 0.1894 |
| 33 | <0.0001 * | 0.0108 * | |
| 100 | <0.0001 * | <0.0001 * | |
| 72 h | 0 | 0.4511 | 0.2050 |
| 33 | 0.0680 | <0.0001 * | |
| 100 | <0.0001 * | <0.0001 * |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hartman, J.H.; Gonzalez-Hunt, C.; Hall, S.M.; Ryde, I.T.; Caldwell, K.A.; Caldwell, G.A.; Meyer, J.N. Genetic Defects in Mitochondrial Dynamics in Caenorhabditis elegans Impact Ultraviolet C Radiation- and 6-hydroxydopamine-Induced Neurodegeneration. Int. J. Mol. Sci. 2019, 20, 3202. https://doi.org/10.3390/ijms20133202
Hartman JH, Gonzalez-Hunt C, Hall SM, Ryde IT, Caldwell KA, Caldwell GA, Meyer JN. Genetic Defects in Mitochondrial Dynamics in Caenorhabditis elegans Impact Ultraviolet C Radiation- and 6-hydroxydopamine-Induced Neurodegeneration. International Journal of Molecular Sciences. 2019; 20(13):3202. https://doi.org/10.3390/ijms20133202
Chicago/Turabian StyleHartman, Jessica H., Claudia Gonzalez-Hunt, Samantha M. Hall, Ian T. Ryde, Kim A. Caldwell, Guy A. Caldwell, and Joel N. Meyer. 2019. "Genetic Defects in Mitochondrial Dynamics in Caenorhabditis elegans Impact Ultraviolet C Radiation- and 6-hydroxydopamine-Induced Neurodegeneration" International Journal of Molecular Sciences 20, no. 13: 3202. https://doi.org/10.3390/ijms20133202
APA StyleHartman, J. H., Gonzalez-Hunt, C., Hall, S. M., Ryde, I. T., Caldwell, K. A., Caldwell, G. A., & Meyer, J. N. (2019). Genetic Defects in Mitochondrial Dynamics in Caenorhabditis elegans Impact Ultraviolet C Radiation- and 6-hydroxydopamine-Induced Neurodegeneration. International Journal of Molecular Sciences, 20(13), 3202. https://doi.org/10.3390/ijms20133202