Regulation of Gene Expression under Hypoxic Conditions



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
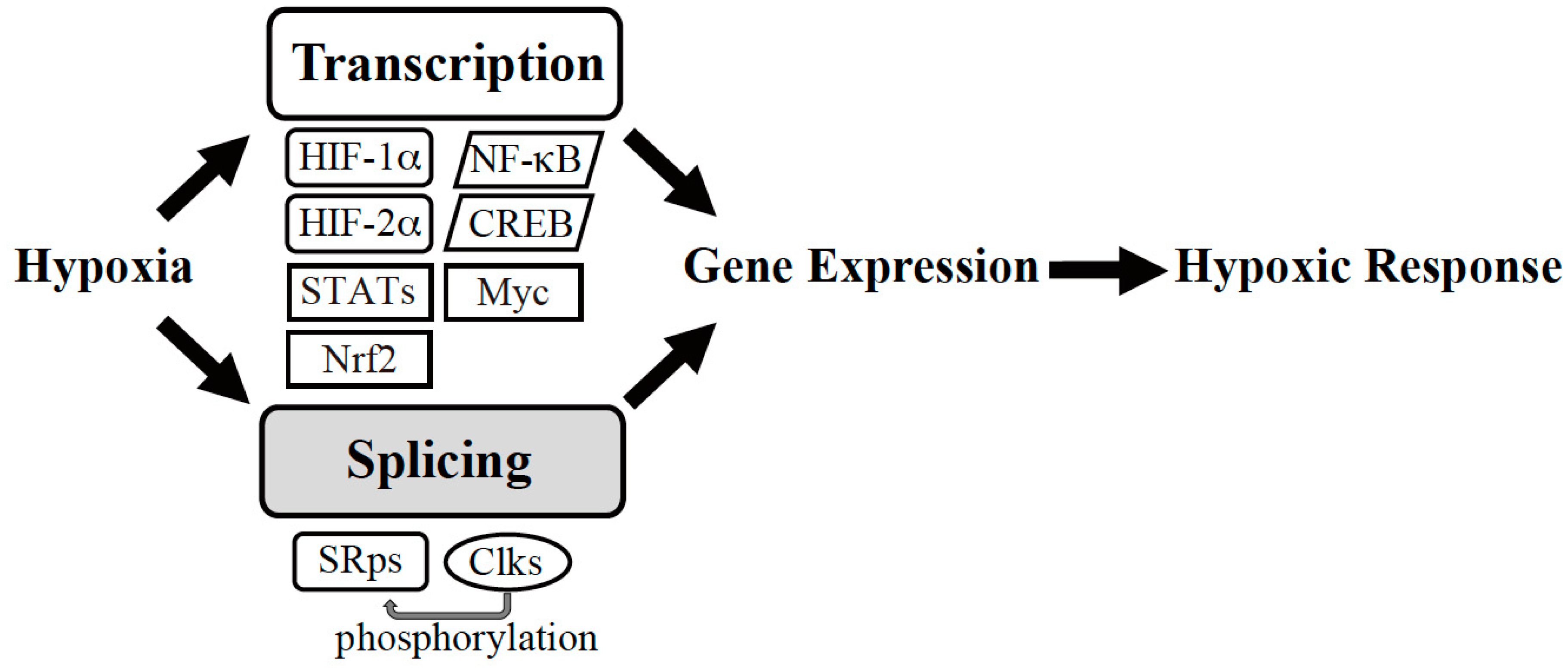
:1. Introduction
2. Transcriptional Regulation under Hypoxia
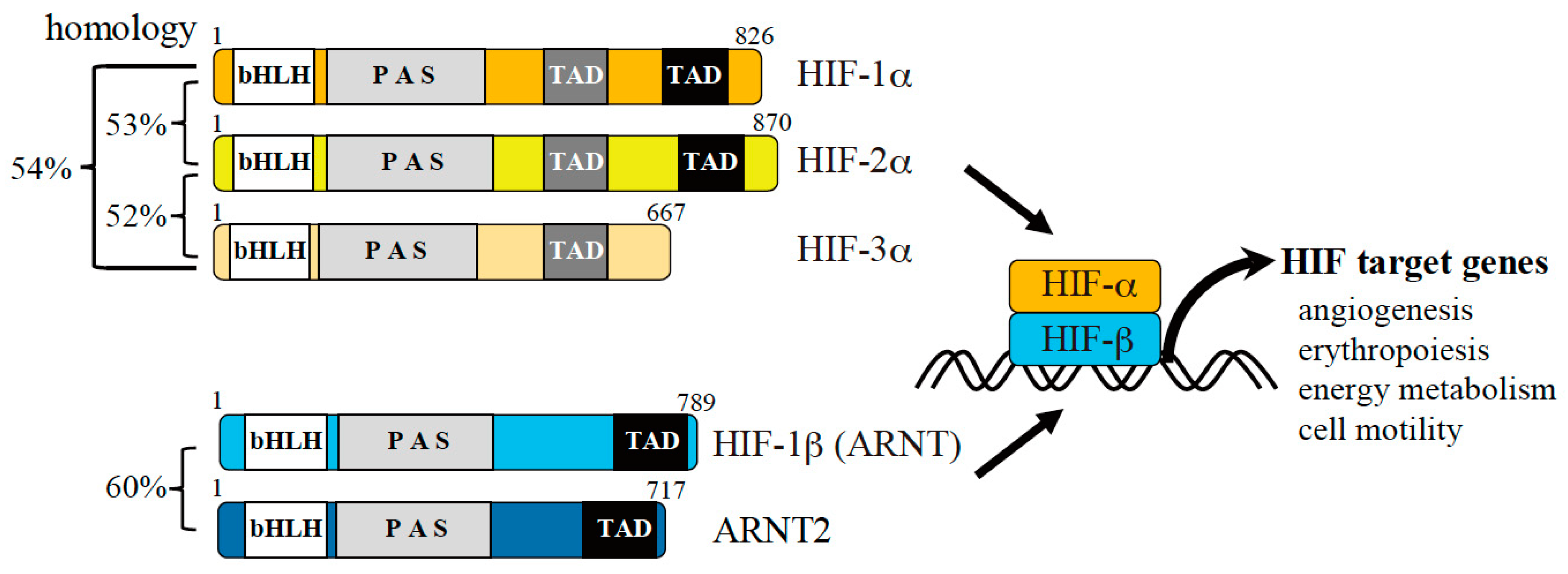
2.1. Hypoxia-Inducible Factor (HIF)
2.2. NF-κB
2.3. CREB
2.4. Roles of other Transcription Factors in Hypoxia
3. Post-Transcriptional Regulation under Hypoxia
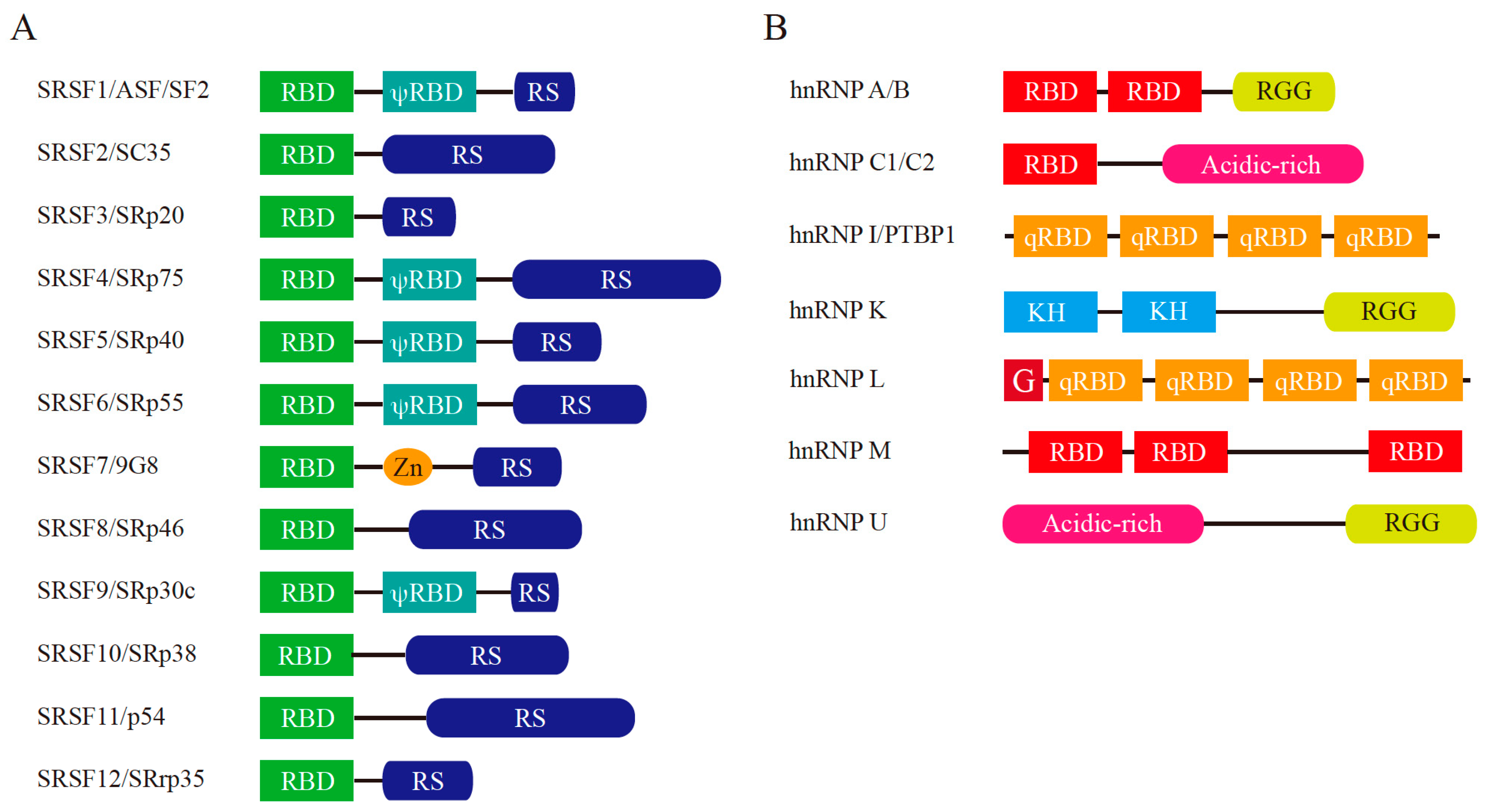
3.1. Pre-mRNA Splicing and Splicing Regulators
3.2. Pre-mRNA Splicing in the Hypoxic Response
3.3. Splicing Regulators in the Hypoxic Response
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Simon, M.C.; Keith, B. The role of oxygen availability in embryonic development and stem cell function. Nat. Rev. Mol. Cell Biol. 2008, 9, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Pugh, C.W.; Ratcliffe, P.J. New horizons in hypoxia signaling pathways. Exp. Cell Res. 2017, 356, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 2003, 3, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Prabhakar, N.R.; Semenza, G.L. Oxygen Sensing and Homeostasis. Physiology 2015, 30, 340–348. [Google Scholar] [CrossRef] [PubMed]
- Johansson, C.; Tumber, A.; Che, K.; Cain, P.; Nowak, R.; Gileadi, C.; Oppermann, U. The roles of Jumonji-type oxygenases in human disease. Epigenomics 2014, 6, 89–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arsham, A.M.; Howell, J.J.; Simon, M.C. A novel hypoxia-inducible factor-independent hypoxic response regulating mammalian target of rapamycin and its targets. J. Biol. Chem. 2003, 278, 29655–29660. [Google Scholar] [CrossRef]
- Brugarolas, J.; Lei, K.; Hurley, R.L.; Manning, B.D.; Reiling, J.H.; Hafen, E.; Witters, L.A.; Ellisen, L.W.; Kaelin, W.G.J. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev. 2004, 18, 2893–2904. [Google Scholar] [CrossRef] [Green Version]
- Gingras, A.C.; Raught, B.; Sonenberg, N. Regulation of translation initiation by FRAP/mTOR. Genes Dev. 2001, 15, 807–826. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.L.; Semenza, G.L. Purification and characterization of hypoxia-inducible factor 1. J. Biol. Chem. 1995, 270, 1230–1237. [Google Scholar] [CrossRef]
- Samanta, D.; Prabhakar, N.R.; Semenza, G.L. Systems biology of oxygen homeostasis. Wiley Interdiscip. Rev. Syst. Biol. Med. 2017, 9, e1382. [Google Scholar] [CrossRef]
- Koyasu, S.; Kobayashi, M.; Goto, Y.; Hiraoka, M.; Harada, H. Regulatory mechanisms of hypoxia-inducible factor 1 activity: Two decades of knowledge. Cancer Sci. 2018, 109, 560–571. [Google Scholar] [CrossRef] [PubMed]
- Kamura, T.; Sato, S.; Iwai, K.; Czyzyk–Krzeska, M.; Conaway, R.C.; Conaway, J.W. Activation of HIF1alpha ubiquitination by a reconstituted von Hippel–Lindau (VHL) tumor suppressor complex. Proc. Natl. Acad. Sci. USA 2000, 97, 10430–10435. [Google Scholar] [CrossRef] [PubMed]
- Ohh, M.; Park, C.W.; Ivan, M.; Hoffman, M.A.; Kim, T.Y.; Huang, L.E.; Pavletich, N.; Chau, V.; Kaelin, W.G. Ubiquitination of hypoxia-inducible factor requires direct binding to the beta-domain of the von Hippel–Lindau protein. Nat. Cell Biol. 2000, 2, 423–427. [Google Scholar] [CrossRef] [PubMed]
- Masson, N.; Willam, C.; Maxwell, P.H.; Pugh, C.W.; Ratcliffe, P.J. Independent function of two destruction domains in hypoxia-inducible factor-alpha chains activated by prolyl hydroxylation. EMBO J. 2001, 20, 5197–5206. [Google Scholar] [CrossRef] [PubMed]
- Tanimoto, K.; Makino, Y.; Pereira, T.; Poellinger, L. Mechanism of regulation of the hypoxia-inducible factor-1 alpha by the von Hippel–Lindau tumor suppressor protein. EMBO J. 2000, 19, 4298–4309. [Google Scholar] [CrossRef] [PubMed]
- Ivan, M.; Kondo, K.; Yang, H.; Kim, W.; Valiando, J.; Ohh, M.; Salic, A.; Asara, J.M.; Lane, W.S.; Kaelin, W.G.J. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: Implications for O2 sensing. Science 2001, 292, 464–468. [Google Scholar] [CrossRef] [PubMed]
- Jaakkola, P.; Mole, D.R.; Tian, Y.M.; Wilson, M.I.; Gielbert, J.; Gaskell, S.J.; Kriegsheim, A.; Hebestreit, H.F.; Mukherji, M.; Schofield, C.J.; et al. Targeting of HIF–alpha to the von Hippel–Lindau ubiquitylation complex by O2–regulated prolyl hydroxylation. Science 2001, 292, 468–472. [Google Scholar] [CrossRef]
- Epstein, A.C.; Gleadle, J.M.; McNeill, L.A.; Hewitson, K.S.; O’Rourke, J.; Mole, D.R.; Mukherji, M.; Metzen, E.; Wilson, M.I.; Dhanda, A.; et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell 2001, 107, 43–54. [Google Scholar] [CrossRef]
- Kaelin, W.G.J.; Ratcliffe, P.J. Oxygen sensing by metazoans: The central role of the HIF hydroxylase pathway. Mol. Cell 2008, 30, 393–402. [Google Scholar] [CrossRef]
- Maxwell, P.H.; Wiesener, M.S.; Chang, G.W.; Clifford, S.C.; Vaux, E.C.; Cockman, M.E.; Wykoff, C.C.; Pugh, C.W.; Maher, E.R.; Ratcliffe, P.J. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999, 399, 271–275. [Google Scholar] [CrossRef]
- Kamura, T.; Koepp, D.M.; Conrad, M.N.; Skowyra, D.; Moreland, R.J.; Iliopoulos, O.; Lane, W.S.; Kaelin, W.G.J.; Elledge, S.J.; Conaway, R.C.; et al. Rbx1, a component of the VHL tumor suppressor complex and SCF ubiquitin ligase. Science 1999, 284, 657–661. [Google Scholar] [CrossRef] [PubMed]
- Gordeuk, V.R.; Miasnikova, G.Y.; Sergueeva, A.I.; Niu, X.; Nouraie, M.; Okhotin, D.J.; Polyakova, L.A.; Ammosova, T.; Nekhai, S.; Ganz, T.; et al. Chuvash polycythemia VHLR200W mutation is associated with down–regulation of hepcidin expression. Blood 2011, 118, 5278–5282. [Google Scholar] [CrossRef] [PubMed]
- Lonser, R.R.; Glenn, G.M.; Walther, M.; Chew, E.Y.; Libutti, S.K.; Linehan, W.M.; Oldfield, E.H. von Hippel–Lindau disease. Lancet 2003, 361, 2059–2067. [Google Scholar] [CrossRef]
- Lando, D.; Peet, D.J.; Gorman, J.J.; Whelan, D.A.; Whitelaw, M.L.; Bruick, R.K. FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev. 2002, 16, 1466–1471. [Google Scholar] [CrossRef] [Green Version]
- Iyer, N.V.; Kotch, L.E.; Agani, F.; Leung, S.W.; Laughner, E.; Wenger, R.H.; Gassmann, M.; Gearhart, J.D.; Lawler, A.M.; Yu, A.Y.; et al. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 alpha. Genes Dev. 1998, 12, 149–162. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Zhang, L.; Drysdale, L.; Fong, G.H. The transcription factor EPAS-1/hypoxia-inducible factor 2alpha plays an important role in vascular remodeling. Proc. Natl. Acad. Sci. USA 2000, 97, 8386–8391. [Google Scholar] [CrossRef] [PubMed]
- Ryan, H.E.; Lo, J.; Johnson, R.S. HIF-1 alpha is required for solid tumor formation and embryonic vascularization. EMBO J. 1998, 17, 3005–3015. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Hammer, R.E.; Matsumoto, A.M.; Russell, D.W.; McKnight, S.L. The hypoxia-responsive transcription factor EPAS1 is essential for catecholamine homeostasis and protection against heart failure during embryonic development. Genes Dev. 1998, 12, 3320–3324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palazon, A.; Goldrath, A.W.; Nizet, V.; Johnson, R.S. HIF transcription factors, inflammation, and immunity. Immunity 2014, 41, 518–528. [Google Scholar] [CrossRef]
- Gilmore, T.D. Introduction to NF-kappaB: Players, pathways, perspectives. Oncogene 2006, 25, 6680–6684. [Google Scholar] [CrossRef]
- Taniguchi, K.; Karin, M. NF-kappaB, inflammation, immunity and cancer: Coming of age. Nat. Rev. Immunol. 2018, 18, 309–324. [Google Scholar] [CrossRef] [PubMed]
- Belaiba, R.S.; Bonello, S.; Zahringer, C.; Schmidt, S.; Hess, J.; Kietzmann, T.; Gorlach, A. Hypoxia up-regulates hypoxia-inducible factor-1alpha transcription by involving phosphatidylinositol 3-kinase and nuclear factor kappaB in pulmonary artery smooth muscle cells. Mol. Biol. Cell 2007, 18, 4691–4697. [Google Scholar] [CrossRef] [PubMed]
- Rius, J.; Guma, M.; Schachtrup, C.; Akassoglou, K.; Zinkernagel, A.S.; Nizet, V.; Johnson, R.S.; Haddad, G.G.; Karin, M. NF-kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha. Nature 2008, 453, 807–811. [Google Scholar] [CrossRef] [PubMed]
- Frede, S.; Stockmann, C.; Freitag, P.; Fandrey, J. Bacterial lipopolysaccharide induces HIF-1 activation in human monocytes via p44/42 MAPK and NF-kappaB. Biochem. J. 2006, 396, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Walmsley, S.R.; Print, C.; Farahi, N.; Peyssonnaux, C.; Johnson, R.S.; Cramer, T.; Sobolewski, A.; Condliffe, A.M.; Cowburn, A.S.; Johnson, N.; et al. Hypoxia-induced neutrophil survival is mediated by HIF-1alpha-dependent NF-kappaB activity. J. Exp. Med. 2005, 201, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, K. Camp-response element-binding protein (CREB) and NF-kappaB transcription factors are activated during prolonged hypoxia and cooperatively regulate the induction of matrix metalloproteinase MMP1. J. Biol. Chem. 2013, 288, 22584–22595. [Google Scholar] [CrossRef] [PubMed]
- Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix metalloproteinases: Regulators of the tumor microenvironment. Cell 2010, 141, 52–67. [Google Scholar] [CrossRef] [PubMed]
- Cummins, E.P.; Berra, E.; Comerford, K.M.; Ginouves, A.; Fitzgerald, K.T.; Seeballuck, F.; Godson, C.; Nielsen, J.E.; Moynagh, P.; Pouyssegur, J.; et al. Prolyl hydroxylase-1 negatively regulates IkappaB kinase-beta, giving insight into hypoxia-induced NFkappaB activity. Proc. Natl. Acad. Sci. USA 2006, 103, 18154–18159. [Google Scholar] [CrossRef]
- Fitzpatrick, S.F.; Fabian, Z.; Schaible, B.; Lenihan, C.R.; Schwarzl, T.; Rodriguez, J.; Zheng, X.; Li, Z.; Tambuwala, M.M.; Higgins, D.G.; et al. Prolyl hydroxylase-1 regulates hepatocyte apoptosis in an NF-kappaB-dependent manner. Biochem. Biophys. Res. Commun. 2016, 474, 579–586. [Google Scholar] [CrossRef]
- Liu, J.; Tao, X.; Zhang, J.; Wang, P.; Sha, M.; Ma, Y.; Geng, X.; Feng, L.; Shen, Y.; Yu, Y.; et al. Small ubiquitin-related modifier 1 is involved in hepatocellular carcinoma progression via mediating p65 nuclear translocation. Oncotarget 2016, 7, 22206–22218. [Google Scholar] [CrossRef] [Green Version]
- Chan, D.A.; Kawahara, T.L.; Sutphin, P.D.; Chang, H.Y.; Chi, J.T.; Giaccia, A.J. Tumor vasculature is regulated by PHD2-mediated angiogenesis and bone marrow-derived cell recruitment. Cancer Cell 2009, 15, 527–538. [Google Scholar] [CrossRef] [PubMed]
- Patel, H.; Zaghloul, N.; Lin, K.; Liu, S.F.; Miller, E.J.; Ahmed, M. Hypoxia-induced activation of specific members of the NF-kB family and its relevance to pulmonary vascular remodeling. Int. J. Biochem. Cell Biol. 2017, 92, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Murata, K.; Fang, C.; Terao, C.; Giannopoulou, E.G.; Lee, Y.J.; Lee, M.J.; Mun, S.H.; Bae, S.; Qiao, Y.; Yuan, R.; et al. Hypoxia-Sensitive COMMD1 Integrates Signaling and Cellular Metabolism in Human Macrophages and Suppresses Osteoclastogenesis. Immunity 2017, 47, 66–79.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dukel, M.; Streitfeld, W.S.; Tang, T.C.; Backman, L.R.; Ai, L.; May, W.S.; Brown, K.D. The Breast Cancer Tumor Suppressor TRIM29 Is Expressed via ATM-dependent Signaling in Response to Hypoxia. J. Biol. Chem. 2016, 291, 21541–21552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, P.; Shin, S.H.; Chun, Y.S.; Shin, H.W.; Shin, Y.J.; Lee, Y.; Kim, D.; Nam, D.H.; Park, J.W. Astrocyte-derived CCL20 reinforces HIF-1-mediated hypoxic responses in glioblastoma by stimulating the CCR6-NF-kappaB signaling pathway. Oncogene 2018, 37, 3070–3087. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Xu, W.; Wang, Z.; Qi, X.; Wang, Y.; Ni, Y.; Shen, H.; Hu, Q.; Han, W. Crosstalk between the HIF-1 and Toll-like receptor/nuclear factor–kappaB pathways in the oral squamous cell carcinoma microenvironment. Oncotarget 2016, 7, 37773–37789. [Google Scholar] [PubMed]
- Mayr, B.; Montminy, M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nature reviews. Mol. Cell. Biol. 2001, 2, 599–609. [Google Scholar]
- Johannessen, M.; Delghandi, M.P.; Moens, U. What turns CREB on? Cell. Signal. 2004, 16, 1211–1227. [Google Scholar] [CrossRef]
- Comerford, K.M.; Leonard, M.O.; Karhausen, J.; Carey, R.; Colgan, S.P.; Taylor, C.T. Small ubiquitin–related modifier-1 modification mediates resolution of CREB-dependent responses to hypoxia. Proc. Natl. Acad. Sci. USA 2003, 100, 986–991. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, S.M.; Leonard, M.O.; Kieran, N.; Comerford, K.M.; Cummins, E.; Pouliot, M.; Lee, S.B.; Taylor, C.T. Hypoxia induces epithelial amphiregulin gene expression in a CREB-dependent manner. American journal of physiology. Cell Physiol. 2006, 290, C592–C600. [Google Scholar] [CrossRef]
- Kikuchi, D.; Tanimoto, K.; Nakayama, K. CREB is activated by ER stress and modulates the unfolded protein response by regulating the expression of IRE1alpha and PERK. Biochem. Biophys. Res. Commun. 2016, 469, 243–250. [Google Scholar] [CrossRef] [PubMed]
- De Jesus, D.S.; DeVallance, E.; Li, Y.; Falabella, M.; Guimaraes, D.; Shiva, S.; Kaufman, B.A.; Gladwin, M.T.; Pagano, P.J. Nox1/Ref-1-mediated activation of CREB promotes Gremlin1-driven endothelial cell proliferation and migration. Redox Biol. 2019, 22, 101138. [Google Scholar] [CrossRef] [PubMed]
- Ramamoorthy, P.; Xu, G.; Shi, H. Expression of Hypoxia Inducible Factor 1alpha Is Protein Kinase A-dependent in Primary Cortical Astrocytes Exposed to Severe Hypoxia. Neurochem. Res. 2019, 44, 258–268. [Google Scholar] [CrossRef] [PubMed]
- Luo, Q.Q.; Qian, Z.M.; Zhou, Y.F.; Zhang, M.W.; Wang, D.; Zhu, L.; Ke, Y. Expression of Iron Regulatory Protein 1 Is Regulated not only by HIF-1 but also pCREB under Hypoxia. Int. J. Biol. Sci. 2016, 12, 1191–1202. [Google Scholar] [CrossRef] [PubMed]
- Steven, A.; Leisz, S.; Sychra, K.; Hiebl, B.; Wickenhauser, C.; Mougiakakos, D.; Kiessling, R.; Denkert, C.; Seliger, B. Hypoxia-mediated alterations and their role in the HER-2/neuregulated CREB status and localization. Oncotarget 2016, 7, 52061–52084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sekine, H.; Okazaki, K.; Ota, N.; Shima, H.; Katoh, Y.; Suzuki, N.; Igarashi, K.; Ito, M.; Motohashi, H.; Yamamoto, M. The Mediator Subunit MED16 Transduces NRF2-Activating Signals into Antioxidant Gene Expression. Mol. Cell. Biol. 2016, 36, 407–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, X.; Wang, H.; Zhu, J.; Zhu, L.; Pan, H.; Li, W.; Zhou, Y.; Cong, Z.; Yan, F.; Chen, S. Knockdown of Nrf2 suppresses glioblastoma angiogenesis by inhibiting hypoxia-induced activation of HIF-1alpha. International journal of cancer. J. Int. Cancer 2014, 135, 574–584. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.H.; Hur, E.G.; Kang, S.J.; Kim, J.A.; Thapa, D.; Lee, Y.M.; Ku, S.K.; Jung, Y.; Kwak, M.K. NRF2 blockade suppresses colon tumor angiogenesis by inhibiting hypoxia-induced activation of HIF-1alpha. Cancer Res. 2011, 71, 2260–2275. [Google Scholar] [CrossRef] [PubMed]
- Ji, W.; Wang, L.; He, S.; Yan, L.; Li, T.; Wang, J.; Kong, A.T.; Yu, S.; Zhang, Y. Effects of acute hypoxia exposure with different durations on activation of Nrf2-ARE pathway in mouse skeletal muscle. PLoS ONE 2018, 13, e0208474. [Google Scholar] [CrossRef]
- Fan, J.; Lv, H.; Li, J.; Che, Y.; Xu, B.; Tao, Z.; Jiang, W. Roles of Nrf2/HO-1 and HIF-1alpha/VEGF in lung tissue injury and repair following cerebral ischemia/reperfusion injury. J. Cell Physiol. 2019, 234, 7695–7707. [Google Scholar] [CrossRef]
- Syu, J.P.; Chi, J.T.; Kung, H.N. Nrf2 is the key to chemotherapy resistance in MCF7 breast cancer cells under hypoxia. Oncotarget 2016, 7, 14659–14672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levy, D.E.; Darnell, J.E.J. Stats: Transcriptional control and biological impact. Nature reviews. Mol. Cell. Biol. 2002, 3, 651–662. [Google Scholar]
- Bromberg, J.F.; Wrzeszczynska, M.H.; Devgan, G.; Zhao, Y.; Pestell, R.G.; Albanese, C.; Darnell, J.E.J. Stat3 as an oncogene. Cell 1999, 98, 295–303. [Google Scholar] [CrossRef]
- McCann, G.A.; Naidu, S.; Rath, K.S.; Bid, H.K.; Tierney, B.J.; Suarez, A.; Varadharaj, S.; Zhang, J.; Hideg, K.; Houghton, P.; et al. Targeting constitutively-activated STAT3 in hypoxic ovarian cancer, using a novel STAT3 inhibitor. Oncoscience 2014, 1, 216–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.Y.; Joung, Y.H.; Lim, E.J.; Park, J.H.; Ye, S.K.; Park, T.; Zhang, Z.; Park, D.K.; Lee, K.J.; Yang, Y.M. Phosphorylation and activation of STAT proteins by hypoxia in breast cancer cells. Breast 2006, 15, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Damrauer, J.S.; Bailey, S.T.; Hadzic, T.; Jeong, Y.; Clark, K.; Fan, C.; Murphy, L.; Lee, C.Y.; Troester, M.A.; et al. Erythropoietin promotes breast tumorigenesis through tumor–initiating cell self–renewal. J. Clin. Investig. 2014, 124, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Nagaraju, G.P.; Park, W.; Wen, J.; Mahaseth, H.; Landry, J.; Farris, A.B.; Willingham, F.; Sullivan, P.S.; Proia, D.A.; El–Hariry, I.; et al. Antiangiogenic effects of ganetespib in colorectal cancer mediated through inhibition of HIF-1alpha and STAT–3. Angiogenesis 2013, 16, 903–917. [Google Scholar] [CrossRef]
- Yamasaki, M.; Nomura, T.; Sato, F.; Mimata, H. Chronic hypoxia induces androgen-independent and invasive behavior in LNCaP human prostate cancer cells. Urol. Oncol. Semin. Orig. Investig. 2013, 31, 1124–1131. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; McCormick, J.; Connolly, M.; Balogh, E.; Veale, D.J.; Fearon, U. Hypoxia and STAT3 signalling interactions regulate pro–inflammatory pathways in rheumatoid arthritis. Ann. Rheum. Dis. 2015, 74, 1275–1283. [Google Scholar] [CrossRef] [PubMed]
- Bai, L.; Yu, Z.; Qian, G.; Qian, P.; Jiang, J.; Wang, G.; Bai, C. SOCS3 was induced by hypoxia and suppressed STAT3 phosphorylation in pulmonary arterial smooth muscle cells. Respir. Physiol. Neurobiol. 2006, 152, 83–91. [Google Scholar] [CrossRef]
- Baudino, T.A.; McKay, C.; Pendeville–Samain, H.; Nilsson, J.A.; Maclean, K.H.; White, E.L.; Davis, A.C.; Ihle, J.N.; Cleveland, J.L. c-Myc is essential for vasculogenesis and angiogenesis during development and tumor progression. Genes Dev. 2002, 16, 2530–2543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osthus, R.C.; Shim, H.; Kim, S.; Li, Q.; Reddy, R.; Mukherjee, M.; Xu, Y.; Wonsey, D.; Lee, L.A.; Dang, C.V. Deregulation of glucose transporter 1 and glycolytic gene expression by c-Myc. J. Biol. Chem. 2000, 275, 21797–21800. [Google Scholar] [CrossRef] [PubMed]
- Koshiji, M.; Kageyama, Y.; Pete, E.A.; Horikawa, I.; Barrett, J.C.; Huang, L.E. HIF-1alpha induces cell cycle arrest by functionally counteracting Myc. EMBO J. 2004, 23, 1949–1956. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.J.; Qiu, B.; Nakazawa, M.S.; Qing, G.; Simon, M.C. MYC degradation under low O2 tension promotes survival by evading hypoxia-induced cell death. Mol. Cell. Biol. 2013, 33, 3494–3504. [Google Scholar] [CrossRef] [PubMed]
- Gordan, J.D.; Bertout, J.A.; Hu, C.J.; Diehl, J.A.; Simon, M.C. HIF–2alpha promotes hypoxic cell proliferation by enhancing c–myc transcriptional activity. Cancer Cell 2007, 11, 335–347. [Google Scholar] [CrossRef] [PubMed]
- Mongiardi, M.P.; Savino, M.; Falchetti, M.L.; Illi, B.; Bozzo, F.; Valle, C.; Helmer-Citterich, M.; Ferre, F.; Nasi, S.; Levi, A. c-MYC inhibition impairs hypoxia response in glioblastoma multiforme. Oncotarget 2016, 7, 33257–33271. [Google Scholar] [CrossRef]
- Xiang, S.; Gu, H.; Jin, L.; Thorne, R.F.; Zhang, X.D.; Wu, M. LncRNA IDH1-AS1 links the functions of c-Myc and HIF1alpha via IDH1 to regulate the Warburg effect. Proc. Natl. Acad. Sci. USA 2018, 115, E1465–E1474. [Google Scholar] [CrossRef]
- Wahl, M.C.; Will, C.L.; Luhrmann, R. The spliceosome: Design principles of a dynamic RNP machine. Cell 2009, 136, 701–718. [Google Scholar] [CrossRef]
- Cartegni, L.; Chew, S.L.; Krainer, A.R. Listening to silence and understanding nonsense: Exonic mutations that affect splicing. Nat. Rev. Genet. 2002, 3, 285–298. [Google Scholar] [CrossRef]
- Fu, X.D.; Ares, M.J. Context-dependent control of alternative splicing by RNA-binding proteins. Nat. Rev. Genet. 2014, 15, 689–701. [Google Scholar] [CrossRef]
- Kataoka, N. Modulation of aberrant splicing in human RNA diseases by chemical compounds. Hum. Genet. 2017, 136, 1237–1245. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Rio, D.C. Mechanisms and Regulation of Alternative Pre-mRNA Splicing. Annu. Rev. Biochem. 2015, 84, 291–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howard, J.M.; Sanford, J.R. The RNAissance family: SR proteins as multifaceted regulators of gene expression. Wiley Interdiscip. Rev. RNA 2015, 6, 93–110. [Google Scholar] [CrossRef] [PubMed]
- Manley, J.L.; Krainer, A.R. A rational nomenclature for serine/arginine-rich protein splicing factors (SR proteins). Genes Dev. 2010, 24, 1073–1074. [Google Scholar] [CrossRef] [PubMed]
- De Conti, L.; Baralle, M.; Buratti, E. Exon and intron definition in pre-mRNA splicing. Wiley Interdiscip. Rev. RNA 2013, 4, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Geuens, T.; Bouhy, D.; Timmerman, V. The hnRNP family: Insights into their role in health and disease. Hum. Genet. 2016, 135, 851–867. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Mayeda, A.; Krainer, A.R. Exon identity established through differential antagonism between exonic splicing silencer-bound hnRNP A1 and enhancer-bound SR proteins. Mol. Cell 2001, 8, 1351–1361. [Google Scholar] [CrossRef]
- David, C.J.; Manley, J.L. Alternative pre-mRNA splicing regulation in cancer: Pathways and programs unhinged. Genes Dev. 2010, 24, 2343–2364. [Google Scholar] [CrossRef] [PubMed]
- Kanopka, A. Cell survival: Interplay between hypoxia and pre-mRNA splicing. Exp. Cell Res. 2017, 356, 187–191. [Google Scholar] [CrossRef]
- Bates, D.O.; Cui, T.G.; Doughty, J.M.; Winkler, M.; Sugiono, M.; Shields, J.D.; Peat, D.; Gillatt, D.; Harper, S.J. VEGF165b, an inhibitory splice variant of vascular endothelial growth factor, is down-regulated in renal cell carcinoma. Cancer Res. 2002, 62, 4123–4131. [Google Scholar]
- Harper, S.J.; Bates, D.O. VEGF-A splicing: The key to anti-angiogenic therapeutics? Nat. Rev. Cancer 2008, 8, 880–887. [Google Scholar] [CrossRef] [PubMed]
- Woolard, J.; Wang, W.Y.; Bevan, H.S.; Qiu, Y.; Morbidelli, L.; Pritchard-Jones, R.O.; Cui, T.G.; Sugiono, M.; Waine, E.; Perrin, R.; et al. VEGF165b, an inhibitory vascular endothelial growth factor splice variant: Mechanism of action, in vivo effect on angiogenesis and endogenous protein expression. Cancer Res. 2004, 64, 7822–7835. [Google Scholar] [CrossRef] [PubMed]
- Pritchard-Jones, R.O.; Dunn, D.B.; Qiu, Y.; Varey, A.H.; Orlando, A.; Rigby, H.; Harper, S.J.; Bates, D.O. Expression of VEGF(xxx)b, the inhibitory isoforms of VEGF, in malignant melanoma. Br. J. Cancer 2007, 97, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Makino, Y.; Cao, R.; Svensson, K.; Bertilsson, G.; Asman, M.; Tanaka, H.; Cao, Y.; Berkenstam, A.; Poellinger, L. Inhibitory PAS domain protein is a negative regulator of hypoxia-inducible gene expression. Nature 2001, 414, 550–554. [Google Scholar] [CrossRef] [PubMed]
- Makino, Y.; Kanopka, A.; Wilson, W.J.; Tanaka, H.; Poellinger, L. Inhibitory PAS domain protein (IPAS) is a hypoxia-inducible splicing variant of the hypoxia-inducible factor-3alpha locus. J. Biol. Chem. 2002, 277, 32405–32408. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Ohneda, O.; Nagano, M.; Iemitsu, M.; Makino, Y.; Tanaka, H.; Miyauchi, T.; Goto, K.; Ohneda, K.; Fujii-Kuriyama, Y.; et al. Abnormal heart development and lung remodeling in mice lacking the hypoxia-inducible factor-related basic helix-loop-helix PAS protein NEPAS. Mol. Cell. Biol. 2008, 28, 1285–1297. [Google Scholar] [CrossRef] [PubMed]
- Dales, J.P.; Beaufils, N.; Silvy, M.; Picard, C.; Pauly, V.; Pradel, V.; Formisano-Treziny, C.; Bonnier, P.; Giusiano, S.; Charpin, C.; et al. Hypoxia inducible factor 1alpha gene (HIF-1alpha) splice variants: Potential prognostic biomarkers in breast cancer. BMC Med. 2010, 8, 44. [Google Scholar] [CrossRef] [PubMed]
- Depping, R.; Hagele, S.; Wagner, K.F.; Wiesner, R.J.; Camenisch, G.; Wenger, R.H.; Katschinski, D.M. A dominant-negative isoform of hypoxia-inducible factor-1 alpha specifically expressed in human testis. Biol. Reprod. 2004, 71, 331–339. [Google Scholar] [CrossRef]
- Pasanen, A.; Heikkila, M.; Rautavuoma, K.; Hirsila, M.; Kivirikko, K.I.; Myllyharju, J. Hypoxia-inducible factor (HIF)-3alpha is subject to extensive alternative splicing in human tissues and cancer cells and is regulated by HIF-1 but not HIF-2. Int. J. Biochem. Cell Biol. 2010, 42, 1189–1200. [Google Scholar] [CrossRef]
- Heikkila, M.; Pasanen, A.; Kivirikko, K.I.; Myllyharju, J. Roles of the human hypoxia-inducible factor (HIF)-3alpha variants in the hypoxia response. Cell Mol. Life Sci. 2011, 68, 3885–3901. [Google Scholar] [CrossRef]
- Maynard, M.A.; Evans, A.J.; Hosomi, T.; Hara, S.; Jewett, M.A.; Ohh, M. Human HIF-3alpha4 is a dominant-negative regulator of HIF-1 and is down-regulated in renal cell carcinoma. FASEB J. 2005, 19, 1396–1406. [Google Scholar] [CrossRef] [PubMed]
- Hang, X.; Li, P.; Li, Z.; Qu, W.; Yu, Y.; Li, H.; Shen, Z.; Zheng, H.; Gao, Y.; Wu, Y.; et al. Transcription and splicing regulation in human umbilical vein endothelial cells under hypoxic stress conditions by exon array. BMC Genom. 2009, 10, 126. [Google Scholar] [CrossRef] [PubMed]
- Sena, J.A.; Wang, L.; Heasley, L.E.; Hu, C.J. Hypoxia regulates alternative splicing of HIF and non-HIF target genes. Mol. Cancer Res. 2014, 12, 1233–1243. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Li, J.; Ho, J.C.; Chia, G.S.; Kato, H.; Jha, S.; Yang, H.; Poellinger, L.; Lee, K.L. Hypoxia is a Key Driver of Alternative Splicing in Human Breast Cancer Cells. Sci. Rep. 2017, 7, 4108. [Google Scholar] [CrossRef] [PubMed]
- Jakubauskiene, E.; Vilys, L.; Makino, Y.; Poellinger, L.; Kanopka, A. Increased Serine-Arginine (SR) Protein Phosphorylation Changes Pre-mRNA Splicing in Hypoxia. J. Biol. Chem. 2015, 290, 18079–18089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naro, C.; Sette, C. Phosphorylation-mediated regulation of alternative splicing in cancer. Int. J. Cell Biol. 2013, 2013, 151839. [Google Scholar] [CrossRef] [PubMed]
- Bowler, E.; Porazinski, S.; Uzor, S.; Thibault, P.; Durand, M.; Lapointe, E.; Rouschop, K.M.A.; Hancock, J.; Wilson, I.; Ladomery, M. Hypoxia leads to significant changes in alternative splicing and elevated expression of CLK splice factor kinases in PC3 prostate cancer cells. BMC Cancer 2018, 18, 355. [Google Scholar] [CrossRef] [PubMed]
- Peciuliene, I.; Vilys, L.; Jakubauskiene, E.; Zaliauskiene, L.; Kanopka, A. Hypoxia alters splicing of the cancer associated Fas gene. Exp. Cell Res. 2019, 380, 29–35. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakayama, K.; Kataoka, N. Regulation of Gene Expression under Hypoxic Conditions. Int. J. Mol. Sci. 2019, 20, 3278. https://doi.org/10.3390/ijms20133278
Nakayama K, Kataoka N. Regulation of Gene Expression under Hypoxic Conditions. International Journal of Molecular Sciences. 2019; 20(13):3278. https://doi.org/10.3390/ijms20133278
Chicago/Turabian StyleNakayama, Koh, and Naoyuki Kataoka. 2019. "Regulation of Gene Expression under Hypoxic Conditions" International Journal of Molecular Sciences 20, no. 13: 3278. https://doi.org/10.3390/ijms20133278
APA StyleNakayama, K., & Kataoka, N. (2019). Regulation of Gene Expression under Hypoxic Conditions. International Journal of Molecular Sciences, 20(13), 3278. https://doi.org/10.3390/ijms20133278