EDB-FN Targeted Peptide–Drug Conjugates for Use against Prostate Cancer


 and
and 
Abstract
:1. Introduction
2. Results and Discussion
2.1. Chemistry
2.1.1. Synthesis of Targeting Ligand
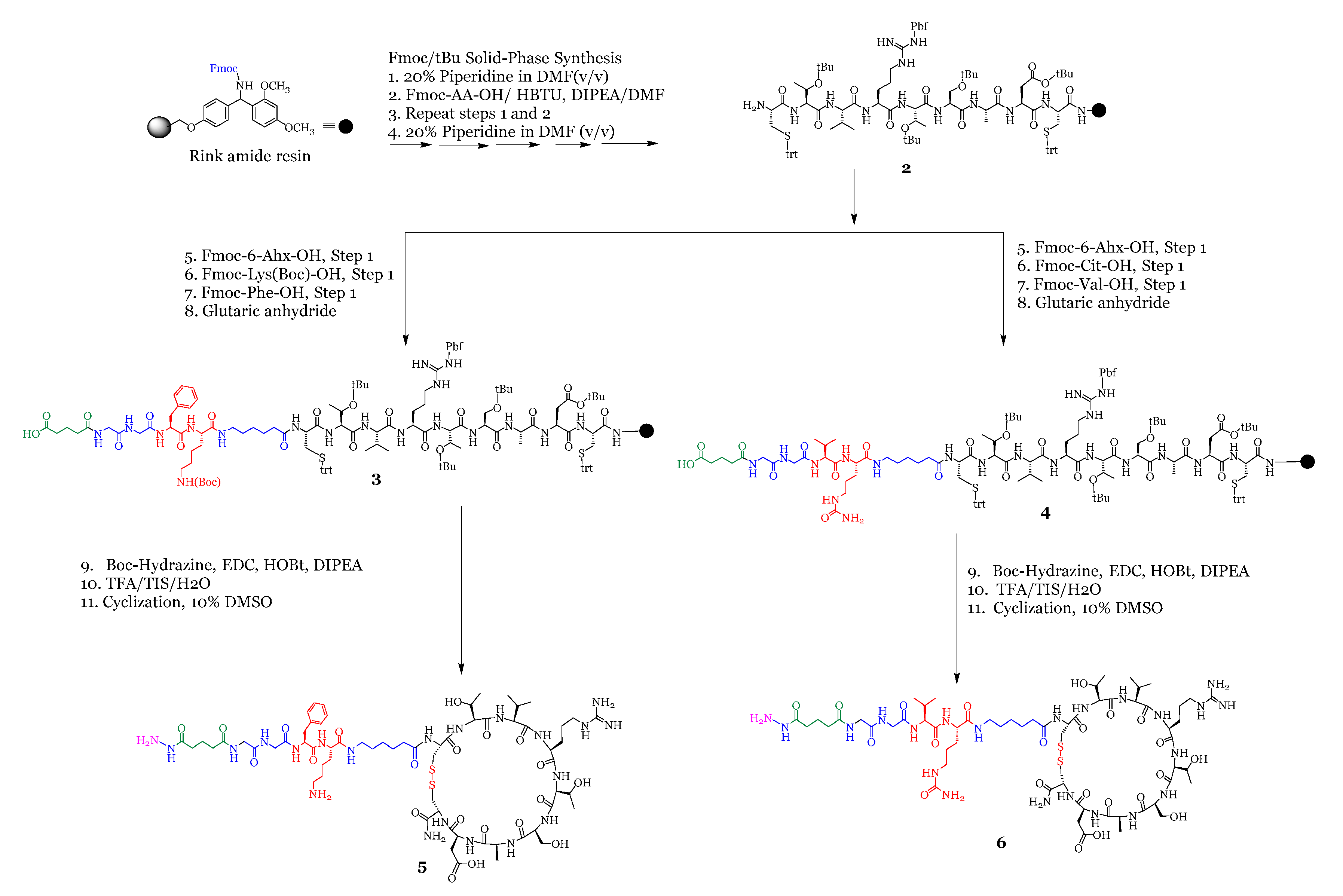
2.1.2. Synthesis of Peptide-Linker Conjugates
2.1.3. Conjugation of Anticancer Drugs with Peptide-Linker Conjugates
2.1.4. Conjugation of Anticancer Drugs with Peptides
2.1.5. Conjugation of 5(6)-Carboxyfluorescein (FAM) Motif with Peptides
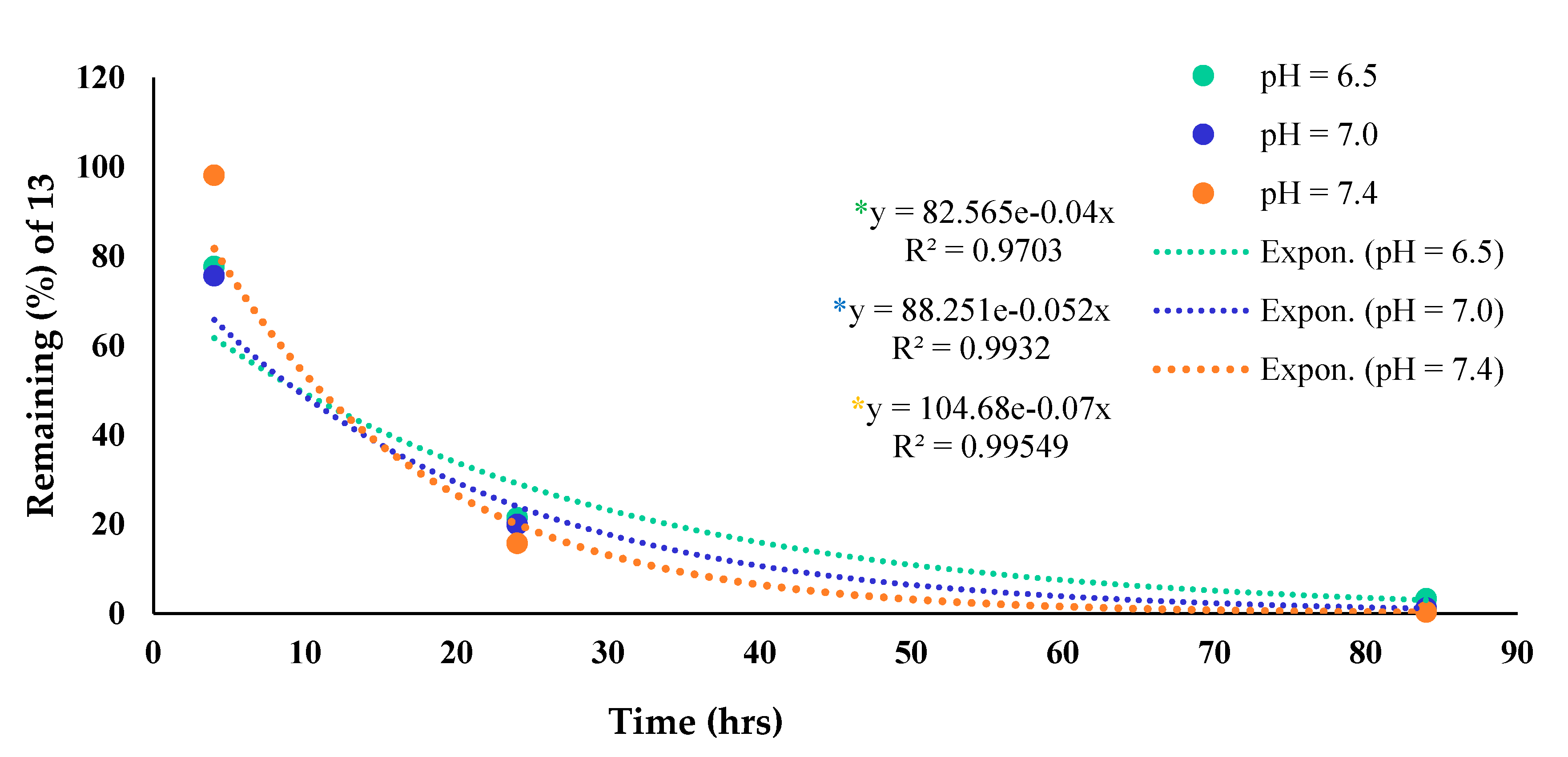
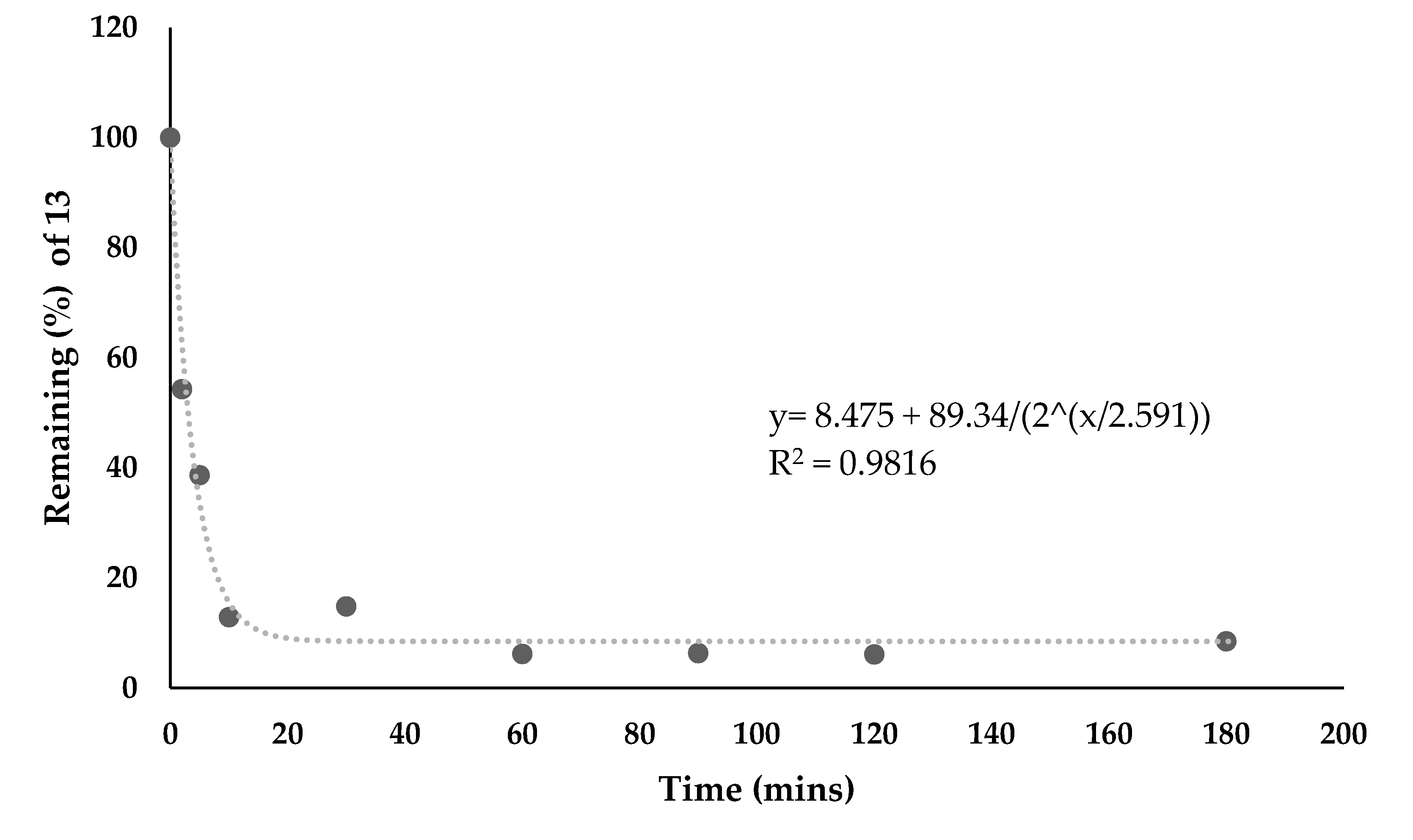
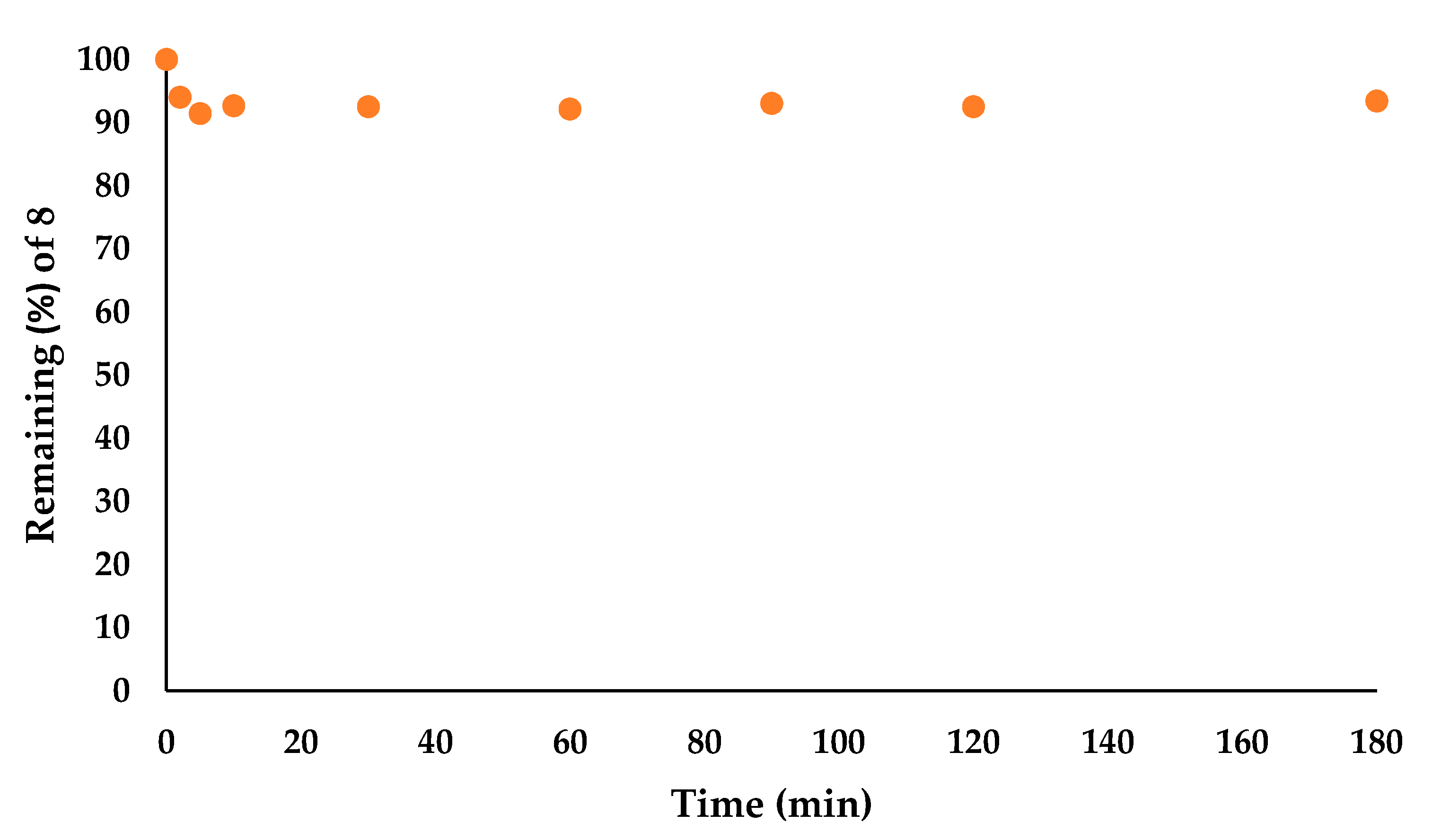
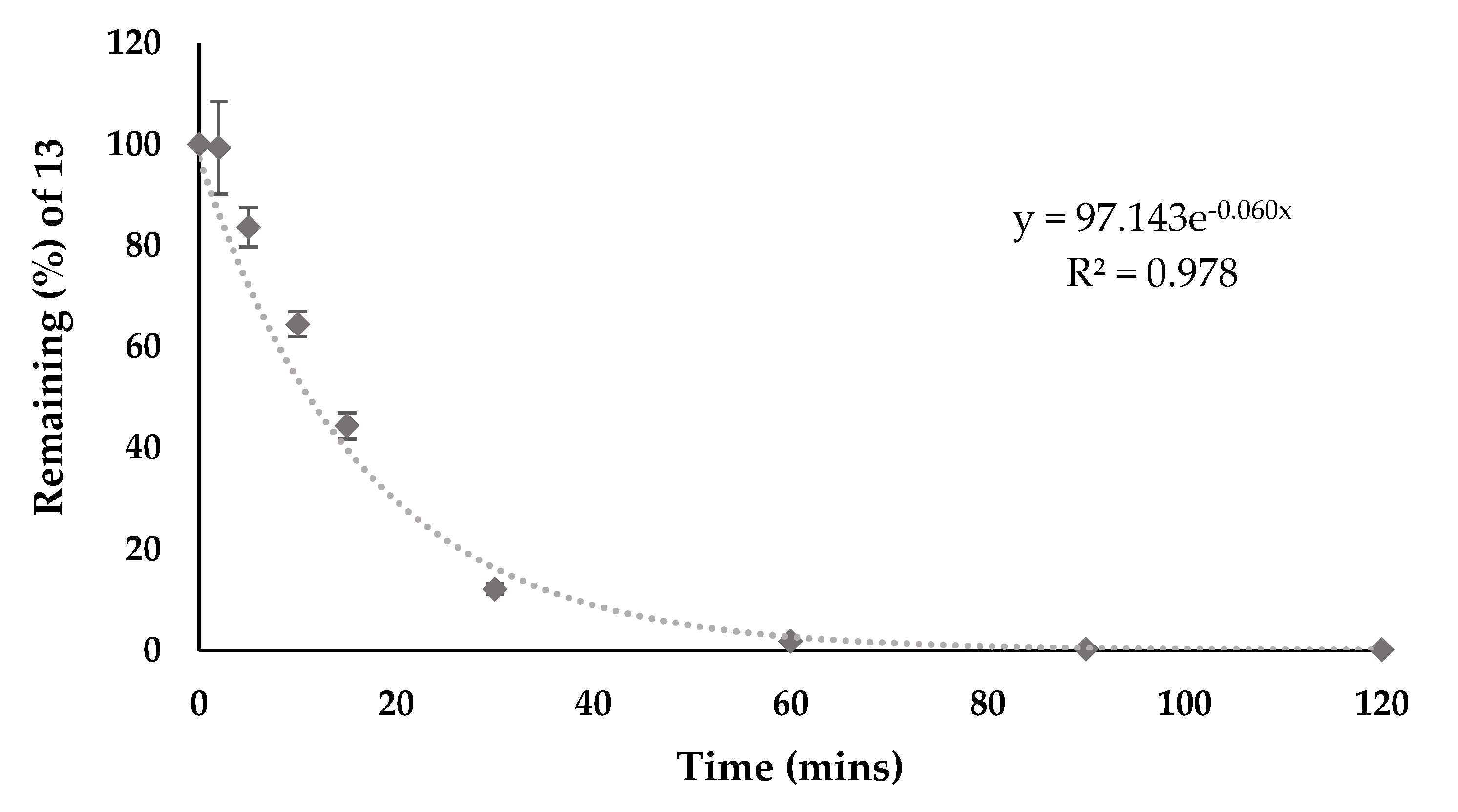
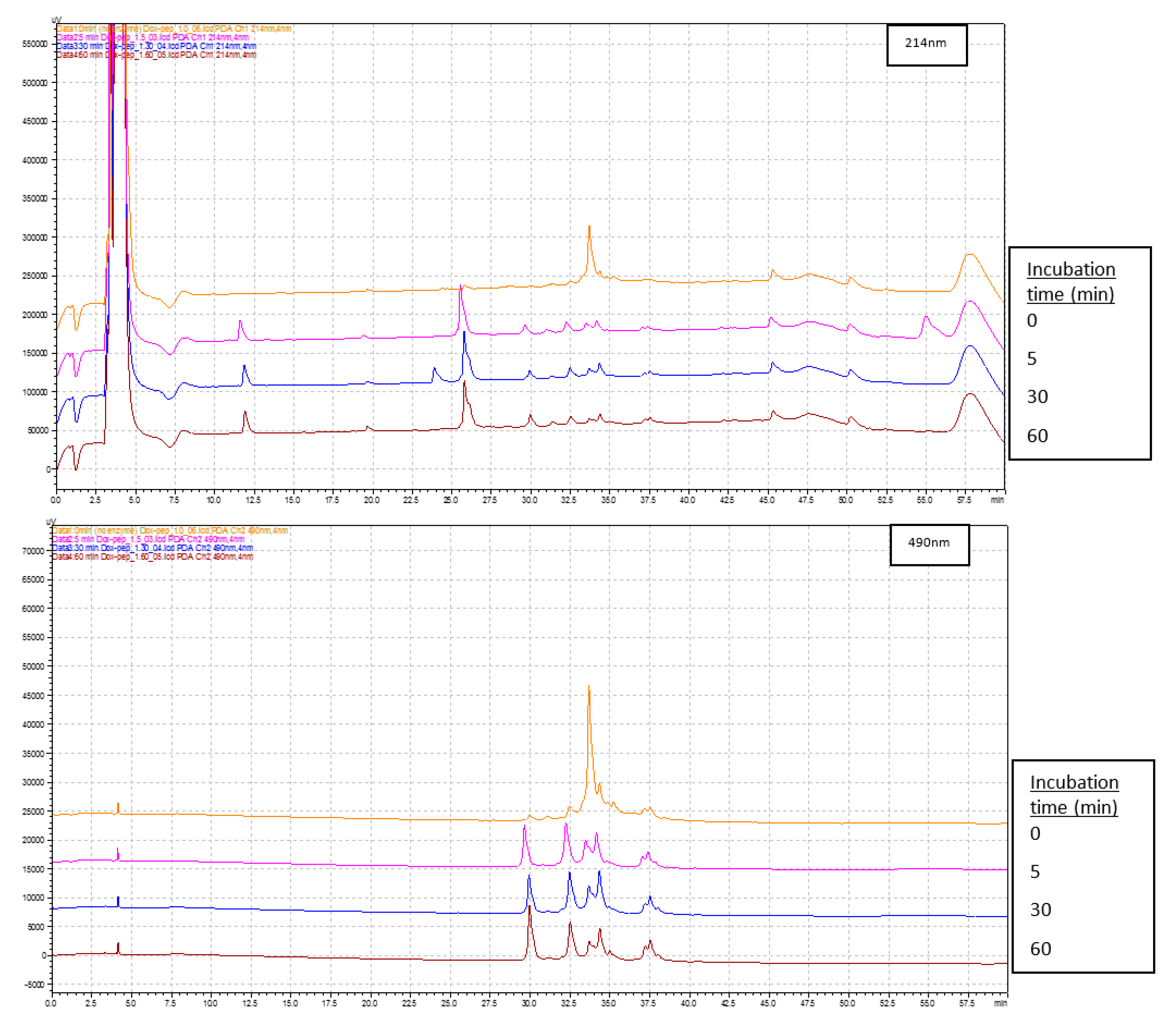
2.1.6. Stability and Hydrolysis
2.2. Cell-Based Assays
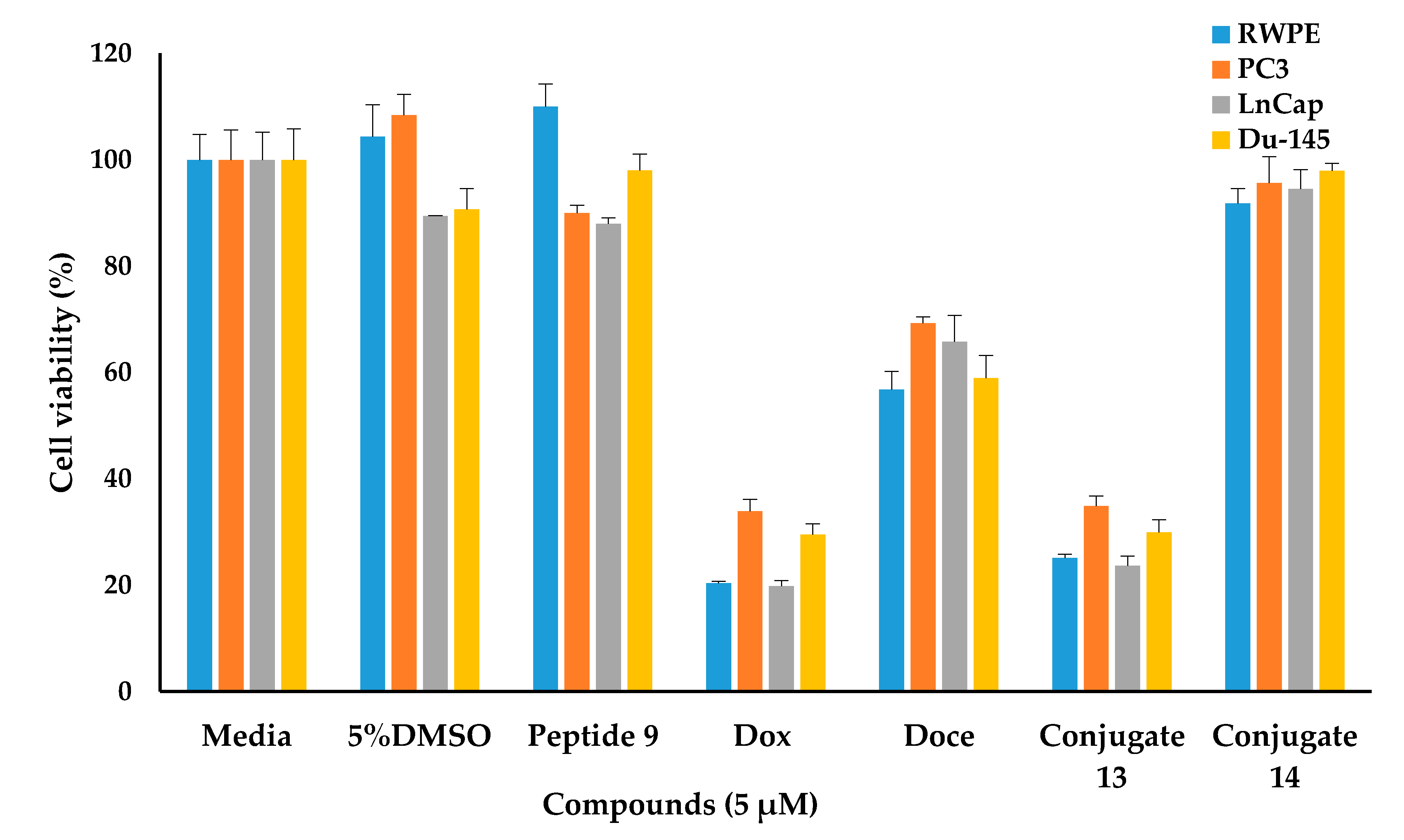
2.2.1. Peptide Cytotoxicity
2.2.2. Confocal Microscopy of the Fluorescently Tagged Compounds 17 and 18
3. Materials and Methods
3.1. Chemistry
3.1.1. Synthesis of Peptide 1 ([CTVRTSADC])
3.1.2. Synthesis of Peptide 8 ([KTVRTSADE])
3.1.3. Synthesis of Peptide 5 (Hydrazine-Glutarate-GG-FK-C6-[CTVRTSADC])
3.1.4. Synthesis of Peptide 6 (Hydrazine-Glutarate-GG-VCit-C6-[CTVRTSADC])
3.1.5. Synthesis of Peptides 19 (C-GFLG-C6-[CTVRTSADC]) and 9 (C-GFLG-C6-[KTVRTSADE])
3.1.6. Synthesis of Conjugate 12 (Dox-hydrazone-glutarate-GG-VCit-C6-[CTVRTSADC])
3.1.7. Synthesis of Conjugate 13 (Dox-s-s-CGFLG-C6-[KTVRTSADE])
3.1.8. Synthesis of Conjugate 14 (Doce-βA-thioether-CGFLG-C6-[KTVRTSADE])
3.1.9. Synthesis of FAM Conjugate Peptide 17 (FAM-GFLG-C6-[CTVRTSADC])
3.1.10. Synthesis of FAM Conjugate Peptide 18 (FAM-GFLG-C6-[KTVRTSADE])
3.2. Stability in PBS and Dithiothreitol (DTT)
3.3. Stability in Human Serum
3.4. Hydrolysis Studies with Cathepsin Enzyme
3.5. Cell Culture
3.5.1. Cell Viability Assays using MTS
3.5.2. Overexpression of EDB-FN using TGF-β
3.5.3. Confocal Microscopy
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References and Note
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- American Cancer Society Prostate Cancer. Available online: https://www.cancer.org/cancer/prostate-cancer.html (accessed on 16 February 2018).
- Gravis, G. Systemic treatment for metastatic prostate cancer. Asian J. Urol. 2019, 6, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Evison, B.J.; Sleebs, B.E.; Watson, K.G.; Phillips, D.R.; Cutts, S.M. Mitoxantrone, More than Just Another Topoisomerase II Poison. Med. Res. Rev. 2016, 36, 248–299. [Google Scholar] [CrossRef] [PubMed]
- Summers, N.; Vanderpuye-Orgle, J.; Reinhart, M.; Gallagher, M.; Sartor, O. Efficacy and safety of post-docetaxel therapies in metastatic castration-resistant prostate cancer: a systematic review of the literature. Curr. Med. Res. Opin. 2017, 33, 1995–2008. [Google Scholar] [CrossRef] [PubMed]
- Alavi, M.; Hamidi, M. Passive and active targeting in cancer therapy by liposomes and lipid nanoparticles. Drug Metab. Pers. Ther. 2019, 34. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, L.; You, X.; Xian, T.; Wu, J.; Pang, J. Nanoparticle Therapy for Prostate Cancer: Overview and Perspectives. Curr. Top. Med. Chem. 2019, 19, 57–73. [Google Scholar] [CrossRef] [PubMed]
- Nasrolahi Shirazi, A.; Tiwari, R.; Chhikara, B.S.; Mandal, D.; Parang, K. Design and biological evaluation of cell-penetrating peptide–doxorubicin conjugates as prodrugs. Mol. Pharm. 2013, 10, 488–499. [Google Scholar] [CrossRef]
- Schally, A.V.; Nagy, A. Cancer chemotherapy based on targeting of cytotoxic peptide conjugates to their receptors on tumors. Eur. J. Endocrinol. 1999, 141, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Arap, W.; Pasqualini, R.; Ruoslahti, E. Cancer treatment by targeted drug delivery to tumor vasculature in a mouse model. Science 1998, 279, 377–380. [Google Scholar] [CrossRef]
- Srinivasarao, M.; Low, P.S. Ligand-targeted drug delivery. Chem. Rev. 2017, 117, 12133–12164. [Google Scholar] [CrossRef]
- Åkerfelt, M.; Härmä, V.; Nees, M. Advanced Models for Target Validation & Drug Discovery in Prostate Cancer. In Prostate Cancer-From Bench to Bedside; IntechOpen: Rijeka, Croatia, 2011. [Google Scholar]
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 786. [Google Scholar] [CrossRef] [PubMed]
- Kumra, H.; Reinhardt, D.P. Fibronectin-targeted drug delivery in cancer. Adv. Drug Deliv. Rev. 2016, 97, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.A.; Guo, W.; Liao, M.-J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The Epithelial-Mesenchymal Transition Generates Cells with Properties of Stem Cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albrecht, M.; Renneberg, H.; Wennemuth, G.; Möschler, O.; Janssen, M.; Aumüller, G.; Konrad, L. Fibronectin in human prostatic cells in vivo and in vitro: expression, distribution, and pathological significance. Histochem. Cell Biol. 1999, 112, 51–61. [Google Scholar] [CrossRef]
- Kaspar, M.; Zardi, L.; Neri, D. Fibronectin as target for tumor therapy. Int. J. Cancer 2006, 118, 1331–1339. [Google Scholar] [CrossRef]
- Han, Z.; Zhou, Z.; Shi, X.; Wang, J.; Wu, X.; Sun, D.; Chen, Y.; Zhu, H.; Magi-Galluzzi, C.; Lu, Z.-R. EDB Fibronectin Specific Peptide for Prostate Cancer Targeting. Bioconjug. Chem. 2015, 26, 830–838. [Google Scholar] [CrossRef]
- Staudacher, A.H.; Brown, M.P. Antibody drug conjugates and bystander killing: is antigen-dependent internalisation required? Br. J. Cancer 2017, 117, 1736. [Google Scholar] [CrossRef]
- Dubowchik, G.M.; Firestone, R.A.; Padilla, L.; Willner, D.; Hofstead, S.J.; Mosure, K.; Knipe, J.O.; Lasch, S.J.; Trail, P.A. Cathepsin B-Labile Dipeptide Linkers for Lysosomal Release of Doxorubicin from Internalizing Immunoconjugates: Model Studies of Enzymatic Drug Release and Antigen-Specific In Vitro Anticancer Activity. Bioconjug. Chem. 2002, 13, 855–869. [Google Scholar] [CrossRef]
- Shao, L.-H.; Liu, S.-P.; Hou, J.-X.; Zhang, Y.-H.; Peng, C.-W.; Zhong, Y.-J.; Liu, X.; Liu, X.-L.; Hong, Y.-P.; Firestone, R.A.; et al. Cathepsin B cleavable novel prodrug Ac-Phe-Lys-PABC-ADM enhances efficacy at reduced toxicity in treating gastric cancer peritoneal carcinomatosis. Cancer 2012, 118, 2986–2996. [Google Scholar] [CrossRef]
- Zhong, Y.; Shao, L.; Li, Y. Cathepsin B-cleavable doxorubicin prodrugs for targeted cancer therapy. Int. J. Oncol. 2013, 42, 373–383. [Google Scholar] [CrossRef]
- Tsakalozou, E.; Eckman, A.M.; Bae, Y. Combination Effects of Docetaxel and Doxorubicin in Hormone-Refractory Prostate Cancer Cells. Biochem. Res. Int. 2012, 2012, 10. [Google Scholar] [CrossRef] [PubMed]
- Wohl, A.R.; Michel, A.R.; Kalscheuer, S.; Macosko, C.W.; Panyam, J.; Hoye, T.R. Silicate esters of paclitaxel and docetaxel: Synthesis, hydrophobicity, hydrolytic stability, cytotoxicity, and prodrug potential. J. Med. Chem. 2014, 57, 2368–2379. [Google Scholar] [CrossRef] [PubMed]
- Skwarczynski, M.; Hayashi, Y.; Kiso, Y. Paclitaxel prodrugs: toward smarter delivery of anticancer agents. J. Med. Chem. 2006, 49, 7253–7269. [Google Scholar] [CrossRef] [PubMed]
- Kingston, D.G.I. Taxol, a molecule for all seasons. Chem. Commun. 2001. [Google Scholar] [CrossRef]
- Inoue, T.; Cavanaugh, P.G.; Steck, P.A.; Brünner, N.; Nicolson, G.L. Differences in transferrin response and numbers of transferrin receptors in rat and human mammary carcinoma lines of different metastatic potentials. J. Cell. Physiol. 1993, 156, 212–217. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.; Yang, J.; Kopeckova, P.; Kopecek, J. Backbone Degradable Multiblock N-(2-Hydroxypropyl)methacrylamide Copolymer Conjugates via Reversible Addition−Fragmentation Chain Transfer Polymerization and Thiol−ene Coupling Reaction. Biomacromolecules 2011, 12, 247–252. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, D.; You, Z. In vitro and in vivo model systems used in prostate cancer research. J. Biol. Methods 2015. [Google Scholar] [CrossRef]
- Tai, S.; Sun, Y.; Squires, J.M.; Zhang, H.; Oh, W.K.; Liang, C.Z.; Huang, J. PC3 is a cell line characteristic of prostatic small cell carcinoma. Prostate 2011, 71, 1668–1679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bisoffi, M.; Klima, I.; Gresko, E.; Durfee, P.N.; Hines, W.C.; Griffith, J.K.; Studer, U.E.; Thalmann, G.N. Expression profiles of androgen independent bone metastatic prostate cancer cells indicate up-regulation of the putative serine-threonine kinase GS3955. J. Urol. 2004, 172, 1145–1150. [Google Scholar] [CrossRef]
- Han, Z.; Lu, Z.-R. Targeting fibronectin for cancer imaging and therapy. J. Mater. Chem. B 2017, 5, 639–654. [Google Scholar] [CrossRef]


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | Peptide Sequence | Chemical Formula | Exact Mass | Found (M/Z) |
|---|---|---|---|---|
| 1 | [CTVRTSADC] | C35H62N13O14S2 | 952.3980 | 952.4786, [M + H]+ |
| 5 | Hydrzb-Glutrtb-GG-FK-C6a-[CTVRTSADC] | C65H111N21O21S2 | 1585.7704 | 1585.5270, [M +H]+ |
| 6 | Hydrzb-Glutrtb-GG–VCita–C6a-[CTVRTSADC] | C61H109N22O22S2 | 1565.7527 | 1565.5920, [M + H]+ |
| 8 | [KTVRTSADE] | C40H71N14O15 | 987.5223 | 987.2699, [M + H]+ |
| 9 | CGFLG-C6a-[KTVRTSADE] | C68H113N20O21S | 1577.8110 | 1577.6815, [M]+ |
| 10 | Dox-s-s-Pyridine | C36H40N3O11S2 | 754.2104 | 754.4053, [M + H]+ |
| 11 | Doce-βA-SMCC | C58H72N3O18 | 1098.4811 | 1098.1675, [M + H]+ |
| 12 | Dox-hydrzc-Glutrtb–GG–VCita–C6a-[CTVRTSADC] | C88H136N23O32S2 | 2090.9003 | 2090.7130, [M + H]+ |
| 13 | Dox-s-s-CGFLG-C6a-[KTVRTSADE] | C99H150N22O32S2 | 2223.0227 | 2222.3398, [M +H]+ |
| 14 | Doce-βA-thioether-CGFLG-C6a-[KTVRTSADE] | C126H184N23O39S | 2675.2842 | 2675.8490, [M + H]+ |
| 15 | GFLG-C6a-[CTVRTSADC] | C60H100N18O19S2 | 1440.6615 | 1440.5944, [M + H]+ |
| 16 | GFLG-C6a-[KTVRTSADE] | C65H109N19O20 | 1475.8096 | 1475.8738, [M + H]+ |
| 17 | FAM-GFLG-C6a-[CTVRTSADC] | C81H111N18O25S2 | 1799.7249 | 1799.928, [M + H]+ |
| 18 | FAM-GFLG-C6a-[KTVRTSADE] | C86H118N19O26 | 1832.8495 | 1832.3740, [M + H]+ |
| 19 | CGFLG-C6a-[CTVRTSADC] | C63H106N19O20S3 | 1544.6863 | 1544.1794, [M + H]+ |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, S.E.; Shamloo, K.; Kristedja, T.A.; Darwish, S.; Bisoffi, M.; Parang, K.; Tiwari, R.K. EDB-FN Targeted Peptide–Drug Conjugates for Use against Prostate Cancer. Int. J. Mol. Sci. 2019, 20, 3291. https://doi.org/10.3390/ijms20133291
Park SE, Shamloo K, Kristedja TA, Darwish S, Bisoffi M, Parang K, Tiwari RK. EDB-FN Targeted Peptide–Drug Conjugates for Use against Prostate Cancer. International Journal of Molecular Sciences. 2019; 20(13):3291. https://doi.org/10.3390/ijms20133291
Chicago/Turabian StylePark, Shang Eun, Kiumars Shamloo, Timothy A. Kristedja, Shaban Darwish, Marco Bisoffi, Keykavous Parang, and Rakesh Kumar Tiwari. 2019. "EDB-FN Targeted Peptide–Drug Conjugates for Use against Prostate Cancer" International Journal of Molecular Sciences 20, no. 13: 3291. https://doi.org/10.3390/ijms20133291
APA StylePark, S. E., Shamloo, K., Kristedja, T. A., Darwish, S., Bisoffi, M., Parang, K., & Tiwari, R. K. (2019). EDB-FN Targeted Peptide–Drug Conjugates for Use against Prostate Cancer. International Journal of Molecular Sciences, 20(13), 3291. https://doi.org/10.3390/ijms20133291