Dynamics and Mechanisms in the Recruitment and Transference of Histone Chaperone CIA/ASF1


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. There Exists Three Metastable Conformational States for DBD(CCG1)
2.2. Exploring the Free Energy Landscape
2.2.1. The High Energy Barrier between the Closed and Half-Open Bound States of DBD(CCG1) Enables CIA/ASF1 to Stably Bind to the Half-Open State during the Recruitment
2.2.2. The Moderate Energy Barrier between the Half-open State and the Open State Facilitates the Change of the Interacting Partner for CIA/ASF1
2.3. Asymmetric Binding of CIA/ASF1 Results in the Open State
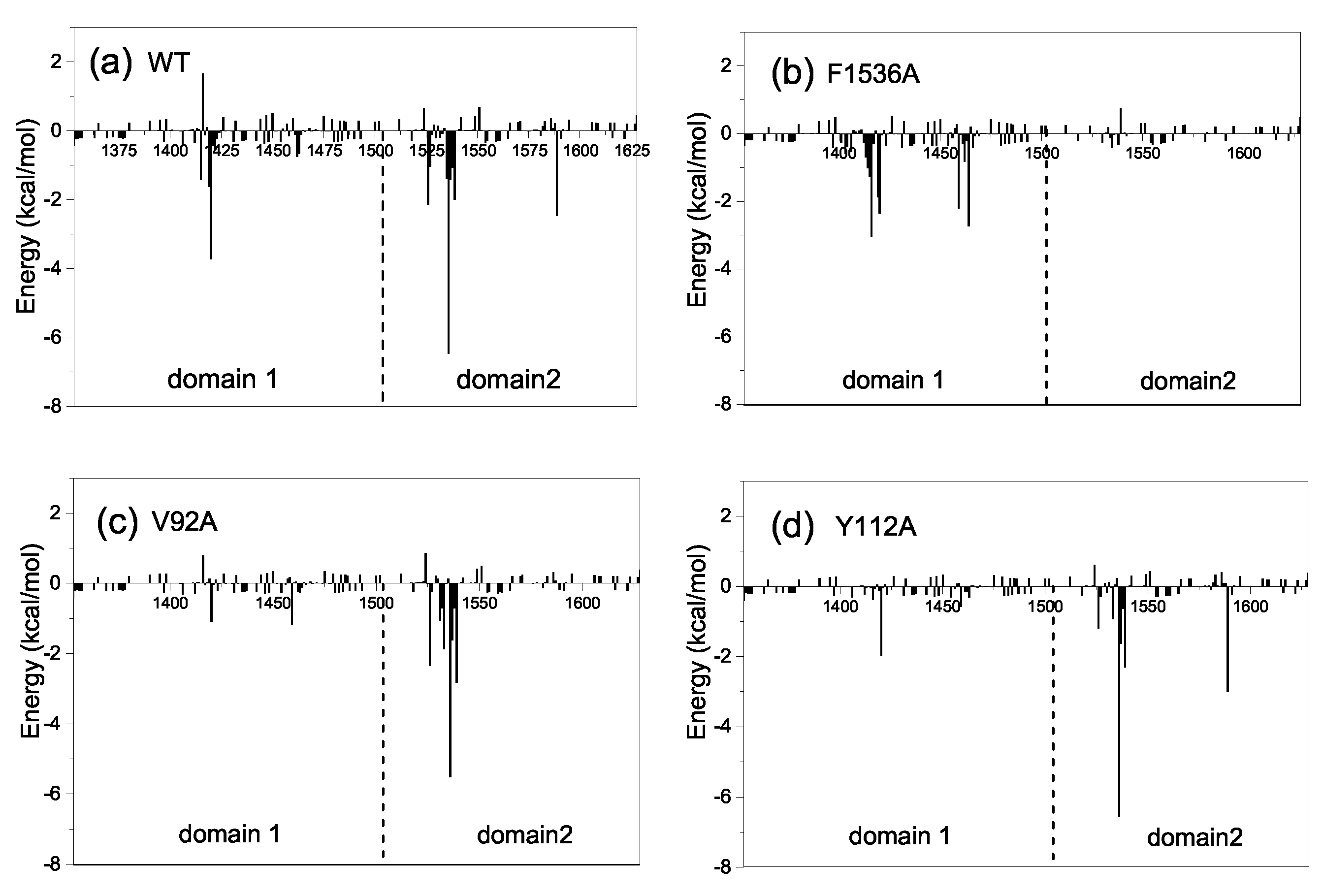
2.4. Mechanism of the Effect of Key Residue Mutations on the Change of Binding State
3. Discussion
4. Materials and Methods
4.1. Protein Systems Preparation
4.2. Molecular Dynamics Simulation Protocol
4.3. MD Trajectory Analysis
4.4. Free Energy Calculations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Waksman, G. Proteomics and Protein-Protein Interactions: Biology, Chemistry, Bioinformatics, and Drug Design; Springer: Berlin, Germany, 2005. [Google Scholar]
- Miki, Y.; Iwabuchi, E.; Ono, K.; Sasano, H.; Ito, K. Exploring Protein-Protein Interaction in the Study of Hormone-Dependent Cancers. Int. J. Mol. Sci. 2018, 19, 3173. [Google Scholar] [CrossRef] [PubMed]
- Sorgen, P.L.; Trease, A.J.; Spagnol, G.; Delmar, M.; Nielsen, M.S. Protein-Protein Interactions with Connexin 43: Regulation and Function. Int. J. Mol. Sci. 2018, 19, 1428. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.-L.; Harris, J.L.; Khanna, K.K.; Hong, J.-H. A Comprehensive Review on Current Advances in Peptide Drug Development and Design. Int. J. Mol. Sci. 2019, 20, 2383. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.-Y. Search strategies and evaluation in protein-protein docking: principles, advances and challenges. Drug Discov. Today 2014, 19, 1081–1096. [Google Scholar] [CrossRef]
- Huang, S.Y.; Zou, X. Advances and challenges in protein-ligand docking. Int. J. Mol. Sci. 2010, 11, 3016–3034. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Jin, B.; Li, H.; Huang, S.-Y. HPEPDOCK: A web server for blind peptide-protein docking based on a hierarchical algorithm. Nucleic Acids Res. 2018, 46, W443–W450. [Google Scholar] [CrossRef]
- Wen, Z.; He, J.; Tao, H.; Huang, S.-Y. PepBDB: A comprehensive structural database of biological peptide-protein interactions. Bioinformatics 2019, 35, 175–177. [Google Scholar] [CrossRef]
- Davey, C.A.; Sargent, D.F.; Luger, K.; Maeder, A.W.; Richmond, T.J. Solvent mediated interactions in the structure of the nucleosome core particle at 1.9 angstrom resolution. J. Mol. Biol. 2002, 319, 1097–1113. [Google Scholar] [CrossRef]
- Luger, K.; Mader, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal structure of the nucleosome core particle at 2.8 angstrom resolution. Nature 1997, 389, 251–260. [Google Scholar] [CrossRef]
- Olins, A.L.; Olins, D.E. E Spheroid chromatin units (v bodies). Science 1974, 183, 330–332. [Google Scholar] [CrossRef]
- Kornberg, R.D.; Thomas, J.O. Chromatin structure; oligomers of the histones. Science 1974, 184, 865–868. [Google Scholar] [CrossRef] [PubMed]
- Arents, G.; Moudrianakis, E.N. Topography of the histone octamer surface: Repeating structural motifs utilized in the docking of nucleosomal DNA. Proc. Natl. Acad. Sci. USA 1993, 90, 10489–10493. [Google Scholar] [CrossRef] [PubMed]
- Simpson, R.T. Structure of the chromatosome, a chromatin particle containing 160 base pairs of DNA and all the histones. Biochemistry 1978, 17, 5524–5531. [Google Scholar] [CrossRef] [PubMed]
- Bálint, M.; Horváth, I.; Mészáros, N.; Hetényi, C. Towards Unraveling the Histone Code by Fragment Blind Docking. Int. J. Mol. Sci. 2019, 20, 422. [Google Scholar] [CrossRef] [PubMed]
- Von Knethen, A.; Brüne, B. Histone Deacetylation Inhibitors as Therapy Concept in Sepsis. Int. J. Mol. Sci. 2019, 20, 346. [Google Scholar] [CrossRef] [PubMed]
- English, C.M.; Adkins, M.W.; Carson, J.J.; Churchill, M.E.A.; Tyler, J.K. Structural basis for the histone chaperone activity of Asf1. Cell 2006, 127, 495–508. [Google Scholar] [CrossRef]
- Huang, T.H.; Fowler, F.; Chen, C.C.; Shen, Z.J.; Sleckman, B.; Tyler, J.K. The Histone Chaperones ASF1 and CAF-1 Promote MMS22L-TONSL-Mediated Rad51 Loading onto ssDNA during Homologous Recombination in Human Cells. Mol. Cell 2018, 69, 879–892. [Google Scholar] [CrossRef]
- Okuwaki, M.; Kato, K.; Shimahara, H.; Tate, S.; Nagata, K. Assembly and disassembly of nucleosome core particles containing histone variants by human nucleosome assembly protein I. Mol. Cell. Biol. 2005, 25, 10639–10651. [Google Scholar] [CrossRef]
- Hatanaka, Y.; Inoue, K.; Oikawa, M.; Kamimura, S.; Ogonuki, N.; Kodama, E.N.; Ohkawa, Y.; Tsukada, Y.; Ogura, A. Histone chaperone CAF-1 mediates repressive histone modifications to protect preimplantation mouse embryos from endogenous retrotransposons. Proc. Natl. Acad. Sci. USA 2015, 112, 14641–14646. [Google Scholar] [CrossRef] [Green Version]
- Das, C.; Roy, S.; Namjoshi, S.; Malarkey, C.S.; Jones, D.N.; Kutateladze, T.G.; Churchill, M.E.; Tyler, J.K. Binding of the histone chaperone ASF1 to the CBP bromodomain promotes histone acetylation. Proc. Natl. Acad. Sci. USA 2014, 111, E1072–E1081. [Google Scholar] [CrossRef] [Green Version]
- Hammond, C.M.; Sundaramoorthy, R.; Larance, M.; Lamond, A.; Stevens, M.A.; El-Mkami, H.; Norman, D.G.; Owen-Hughes, T. The histone chaperone Vps75 forms multiple oligomeric assemblies capable of mediating exchange between histone H3-H4 tetramers and Asf1-H3-H4 complexes. Nucleic Acids Res. 2016, 44, 6157–6172. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.H.; Roemer, S.C.; Port, A.M.; Churchill, M.E. CAF-1-induced oligomerization of histones H3/H4 and mutually exclusive interactions with Asf1 guide H3/H4 transitions among histone chaperones and DNA. Nucleic Acids Res. 2012, 40, 11229–11239. [Google Scholar] [CrossRef] [PubMed]
- Ricketts, M.D.; Frederick, B.; Hoff, H.; Tang, Y.; Schultz, D.C.; Singh Rai, T.; Grazia, V.M.; Adams, P.D.; Marmorstein, R. Ubinuclein-1 confers histone H3.3-specific-binding by the HIRA histone chaperone complex. Nat. Commun. 2015, 6, 7711. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Deng, Z.; Vladimirova, O.; Wiedmer, A.; Lu, F.; Lieberman, P.M.; Patel, D.J. Structural basis underlying viral hijacking of a histone chaperone complex. Nat. Commun. 2016, 7, 12707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgess, R.J.; Zhang, Z. Histone chaperones in nucleosome assembly and human disease. Nat. Struct. Mol. Biol. 2013, 20, 14–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammond, C.M.; Strmme, C.B.; Huang, H.; Patel, D.J.; Groth, A. Histone chaperone networks shaping chromatin function. Nat. Rev. Mol. Cell. Biol. 2017, 18, 141–158. [Google Scholar] [CrossRef] [Green Version]
- Valieva, M.E.; Armeev, G.A.; Kudryashova, K.S.; Gerasimova, N.S.; Shaytan, A.K.; Kulaeva, O.I.; McCullough, L.L.; Formosa, T.; Georgiev, P.G.; Kirpichnikov, M.P.; et al. Large-scale ATP-independent nucleosome unfolding by a histone chaperone. Nat. Struct. Mol. Biol. 2016, 23, 1111–1116. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Oh, S.; Jeong, K.; Jo, H.; Choi, Y.; Seo, H.D.; Kim, M.; Choe, J.; Kwon, C.S.; Lee, D. Dot1 regulates nucleosome dynamics by its inherent histone chaperone activity in yeast. Nat. Commun. 2018, 9, 240. [Google Scholar] [CrossRef] [Green Version]
- Klimovskaia, I.M.; Young, C.; Stromme, C.B.; Menard, P.; Jasencakova, Z.; Mejlvang, J.; Ask, K.; Ploug, M.; Nielsen, M.L.; Jensen, O.N.; et al. Tousled-like kinases phosphorylate Asf1 to promote histone supply during DNA replication. Nat. Commun. 2014, 5, 13. [Google Scholar] [CrossRef]
- Fan, H.F.; Liu, Z.N.; Chow, S.Y.; Lu, Y.H.; Li, H. Histone chaperone-mediated nucleosome assembly process. PLoS ONE 2015, 10, e0115007. [Google Scholar] [CrossRef]
- Grover, P.; Asa, J.S.; Campos, E.I. H3-H4 Histone Chaperone Pathways. Annu. Rev. Genet. 2018, 52, 109–130. [Google Scholar] [CrossRef] [PubMed]
- De, K.L.; Corpet, A.; Haber, J.E.; Almouzni, G. Histone chaperones: An escort network regulating histone traffic. Nat. Struct. Mol. Biol. 2007, 14, 997–1007. [Google Scholar]
- Chimura, T.; Kuzuhara, T.; Horikoshi, M. Identification and characterization of CIA/ASF1 as an interactor of bromodomains associated with TFIID. Proc. Natl. Acad. Sci. USA 2002, 99, 9334–9339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishikawa, K.; Ohsumi, T.; Tada, S.; Natsume, R.; Kundu, L.R.; Nozaki, N.; Senda, T.; Enomoto, T.; Horikoshi, M.; Seki, M. Roles of histone chaperone CIA/Asf1 in nascent DNA elongation during nucleosome replication. Genes Cells 2011, 16, 1050–1062. [Google Scholar] [CrossRef] [PubMed]
- Mousson, F.; Lautrette, A.; Thuret, J.Y.; Agez, M.; Courbeyrette, R.; Amigues, B.; Becker, E.; Neumann, J.M.; Guerois, R.; Mann, C.; et al. Structural basis for the interaction of Asf1 with histone H3 and its functional implications. Proc. Natl. Acad. Sci. USA 2005, 102, 5975–5980. [Google Scholar] [CrossRef] [Green Version]
- O’Sullivan, R.J.; Arnoult, N.; Lackner, D.H.; Oganesian, L.; Haggblom, C.; Corpet, A.; Almouzni, G.; Karlseder, J. Rapid induction of alternative lengthening of telomeres by depletion of the histone chaperone ASF1. Nat. Struct. Mol. Biol. 2014, 21, 167–174. [Google Scholar] [CrossRef]
- Grigsby, I.F.; Rutledge, E.M.; Morton, C.A.; Finger, F.P. Functional redundancy of two C. elegans homologs of the histone chaperone Asf1 in germline DNA replication. Dev. Biol. 2009, 329, 64–79. [Google Scholar] [CrossRef] [Green Version]
- Schulz, L.L.; Tyler, J.K. The histone chaperone ASF1 localizes to active DNA replication forks to mediate efficient DNA replication. FASEB J. 2006, 20, 488–490. [Google Scholar] [CrossRef]
- Richet, N.; Liu, D.; Legrand, P.; Velours, C.; Corpet, A.; Gaubert, A.; Bakail, M.; Moal-Raisin, G.; Guerois, R.; Compper, C.; et al. Structural insight into how the human helicase subunit MCM2 may act as a histone chaperone together with ASF1 at the replication fork. Nucleic Acids Res. 2015, 43, 1905–1917. [Google Scholar] [CrossRef] [Green Version]
- Lercher, L.; Danilenko, N.; Kirkpatrick, J.; Carlomagno, T. Structural characterization of the Asf1-Rtt109 interaction and its role in histone acetylation. Nucleic Acids Res. 2018, 46, 2279–2289. [Google Scholar] [CrossRef]
- Akai, Y.; Adachi, N.; Hayashi, Y.; Eitoku, M.; Sano, N.; Natsume, R.; Kudo, N.; Tanokura, M.; Senda, T.; Horikoshi, M. Structure of the histone chaperone CIA/ASF1-double bromodomain complex linking histone modifications and site-specific histone eviction. Proc. Natl. Acad. Sci. USA 2010, 107, 8153–8158. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, R.H.; Ladurner, A.G.; King, D.S.; Tjian, R. Structure and function of a human TAFII250 double bromodomain module. Science 2000, 288, 1422–1425. [Google Scholar] [CrossRef] [PubMed]
- Brand, M.; Measures, A.R.; Wilson, B.G.; Cortopassi, W.A.; Alexander, R.; Höss, M.; Hewings, D.S.; Rooney, T.P.; Paton, R.S.; Conway, S.J. Small molecule inhibitors of bromodomain-acetyl-lysine interactions. ACS Chem. Biol. 2015, 10, 22–39. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Knapp, S. The bromodomain interaction module. FEBS Lett. 2012, 586, 2692–2704. [Google Scholar] [CrossRef] [PubMed]
- Filippakopoulos, P.; Picaud, S.; Mangos, M.; Keates, T.; Lambert, J.P.; Barsyte-Lovejoy, D.; Felletar, I.; Volkmer, R.; Muller, S.; Pawson, T.; et al. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell 2012, 149, 214–231. [Google Scholar] [CrossRef] [PubMed]
- Owen, D.J.; Ornaghi, P.; Yang, J.C.; Lowe, N.; Evans, P.R.; Ballario, P.; Neuhaus, D.; Filetici, P.; Travers, A.A. The structural basis for the recognition of acetylated histone H4 by the bromodomain of histone acetyltransferase gcn5p. EMBO J. 2000, 19, 6141–6149. [Google Scholar] [CrossRef] [PubMed]
- Clément, C.; Almouzni, G. MCM2 binding to histones H3-H4 and ASF1 supports a tetramer-to-dimer model for histone inheritance at the replication fork. Nat. Struct. Mol. Biol. 2015, 22, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, M.; Yang, N.; Xu, R.M. Structure of the quaternary complex of histone H3-H4 heterodimer with chaperone ASF1 and the replicative helicase subunit MCM2. Protein Cell 2015, 6, 693–697. [Google Scholar] [CrossRef]
- Natsume, R.; Eitoku, M.; Akai, Y.; Sano, N.; Horikoshi, M.; Senda, T. Structure and function of the histone chaperone CIA/ASF1 complexed with histones H3 and H4. Nature 2007, 446, 338–341. [Google Scholar] [CrossRef]
- Fenwick, R.B.; Esteban-Martín, S.; Salvatella, X. Understanding biomolecular motion, recognition, and allostery by use of conformational ensembles. Eur. Biophys. J. 2011, 40, 1339–1355. [Google Scholar] [CrossRef] [Green Version]
- Gaines, C.S.; York, D.M. Ribozyme Catalysis with a Twist: Active State of the Twister Ribozyme in Solution Predicted from Molecular Simulation. J. Am. Chem. Soc. 2016, 138, 3058–3065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiao, Q.; Bowman, G.R.; Huang, X. Dynamics of an intrinsically disordered protein reveal metastable conformations that potentially seed aggregation. J. Am. Chem. Soc. 2013, 135, 16092–16101. [Google Scholar] [CrossRef] [PubMed]
- Heo, L.; Feig, M. Experimental accuracy in protein structure refinement via molecular dynamics simulations. Proc. Natl. Acad. Sci. USA 2013, 115, 13276–13281. [Google Scholar] [CrossRef] [PubMed]
- Sali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef] [PubMed]
- Schrodinger, LLC. The PyMOL Molecular Graphics System, Version 1.8; Schrodinger, LLC: New York, NY, USA, 2015. [Google Scholar]
- Gordon, J.C.; Myers, J.B.; Folta, T.; Shoja, V.; Heath, L.S.; Onufriev, A. H++: A server for estimating pKas and adding missing hydrogens to macromolecules. Nucleic Acids Res. 2005, 33, W368–W371. [Google Scholar] [CrossRef]
- Anandakrishnan, R.; Aguilar, B.; Onufriev, A.V. H++ 3.0: Automating pK prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulations. Nucleic Acids Res. 2012, 40, W537–W541. [Google Scholar] [CrossRef]
- Myers, J.; Grothaus, G.; Narayanan, S.; Onufriev, A. A simple clustering algorithm can be accurate enough for use in calculations of pKs in macromolecules. Proteins 2006, 64, 928–938. [Google Scholar] [CrossRef]
- Case, D.A.; Berryman, J.T.; Betz, R.M.; Cai, Q.; Cerutti, D.S.; Cheatham, T.E., III; Darden, T.A.; Duke, R.E.; Goetz, A.W.; Gusarow, S.; et al. AMBER 14; University of California, San Francisco: San Francisco, CA, USA, 2014. [Google Scholar]
- William, L.; Jorgensen, J.C.; Jeffry, D.M.; Roger, W.I.; Michael, L.K. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An Nlog(N) method for Ewald sums in large systems. J. Chem. Phys. 1933, 98, 10089–10092. [Google Scholar] [CrossRef]
- Ryckaert, J.P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Tina, K.G.; Bhadra, R.; Srinivasan, N. PIC: Protein Interactions Calculator. Nucleic Acids Res. 2007, 35, W473–W476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alhadeff, R.; Vorobyov, I.; Yoon, H.W.; Warshel, A. Exploring the free-energy landscape of GPCR activation. Proc. Natl. Acad. Sci. USA 2018, 115, 10327–10332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czub, J.; Grubmüller, H. Rotation triggers nucleotide-independent conformational transition of the empty β subunit of F-ATPase. J. Am. Chem. Soc. 2014, 136, 6960–6968. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; McCammon, J.A. Graded activation and free energy landscapes of a muscarinic G-protein-coupled receptor. Proc. Natl. Acad. Sci. USA 2016, 113, 12162–12167. [Google Scholar] [CrossRef] [PubMed]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W.; et al. Calculating structures and free energies of complex molecules: Combining molecular mechanics and continuum models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef] [PubMed]
- Homeyer, N.; Gohlke, H. Free Energy Calculations by the Molecular Mechanics Poisson-Boltzmann Surface Area Method. Mol. Inform. 2012, 31, 114–122. [Google Scholar] [CrossRef] [PubMed]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Tao, H.; Huang, S.-Y. Dynamics and Mechanisms in the Recruitment and Transference of Histone Chaperone CIA/ASF1. Int. J. Mol. Sci. 2019, 20, 3325. https://doi.org/10.3390/ijms20133325
Zhang Y, Tao H, Huang S-Y. Dynamics and Mechanisms in the Recruitment and Transference of Histone Chaperone CIA/ASF1. International Journal of Molecular Sciences. 2019; 20(13):3325. https://doi.org/10.3390/ijms20133325
Chicago/Turabian StyleZhang, Yanjun, Huanyu Tao, and Sheng-You Huang. 2019. "Dynamics and Mechanisms in the Recruitment and Transference of Histone Chaperone CIA/ASF1" International Journal of Molecular Sciences 20, no. 13: 3325. https://doi.org/10.3390/ijms20133325
APA StyleZhang, Y., Tao, H., & Huang, S. -Y. (2019). Dynamics and Mechanisms in the Recruitment and Transference of Histone Chaperone CIA/ASF1. International Journal of Molecular Sciences, 20(13), 3325. https://doi.org/10.3390/ijms20133325