DMSO Reductase Family: Phylogenetics and Applications of Extremophiles

 , ,
, ,  and
and
Abstract
:1. Introduction
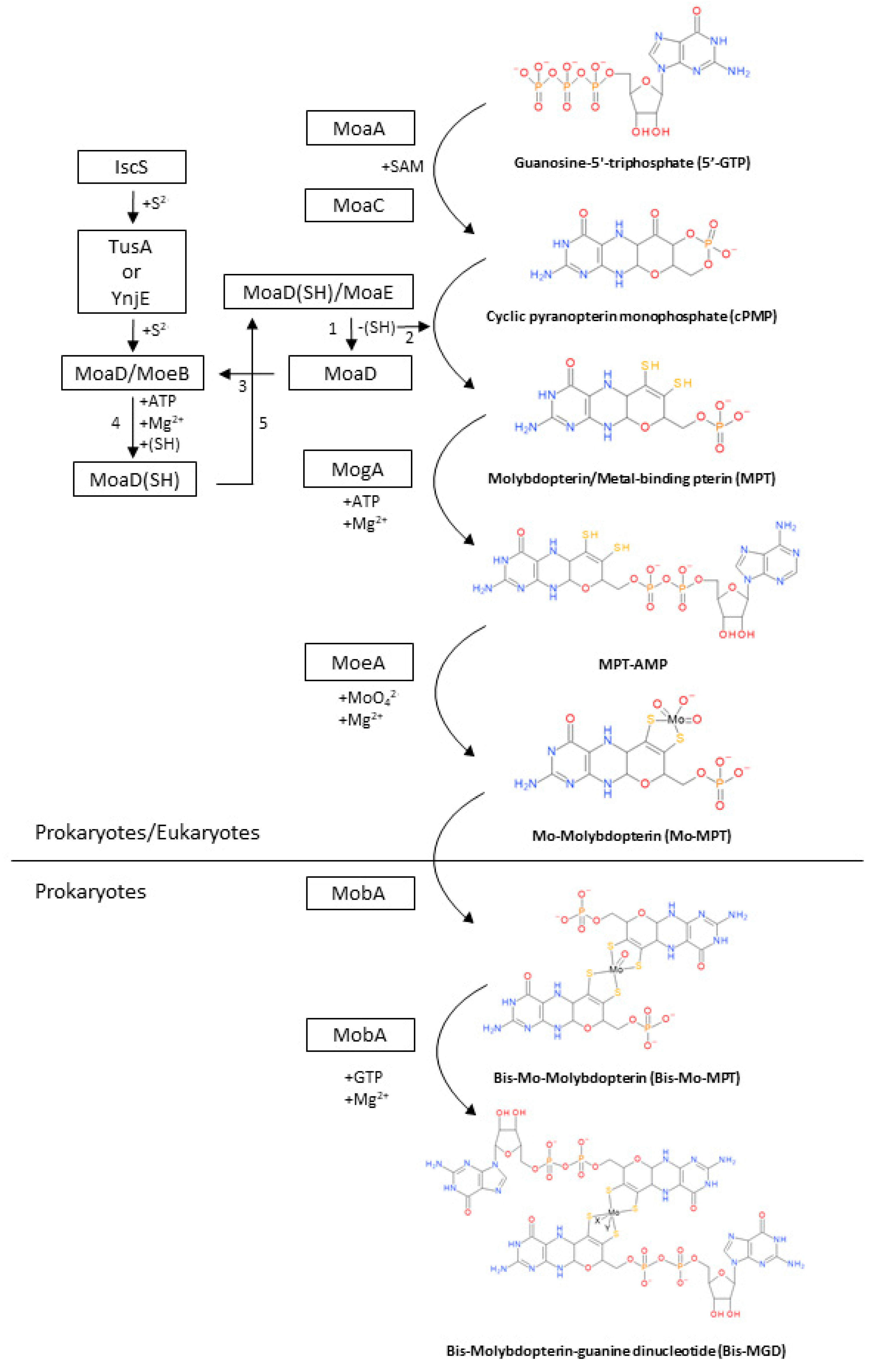
2. Biosynthesis of the Bis-MGD Cofactor and Maturation of Molybdoenzymes
2.1. Synthesis of cPMP from 5′-GTP
2.2. Conversion of cPMP into MPT
2.3. Metal Insertion in MPT to Synthetize the Mo-MPT
2.4. Formation of Bis-MGD
2.5. Maturation of DMSO Reductases
3. Families of Enzymes Containing Molybdenum or Tungsten
3.1. Sulfite Oxidase Family
3.2. Xanthine Oxidase Family
3.3. DMSO Reductase Family
3.4. Tungsten-Enzyme Family
4. Connections between DMSO Reductases and N-Cycle
4.1. Nitrate Reduction
4.2. (Per)Chlorate Reduction
4.3. Arsenate Reduction
4.4. Polysulfide Reduction
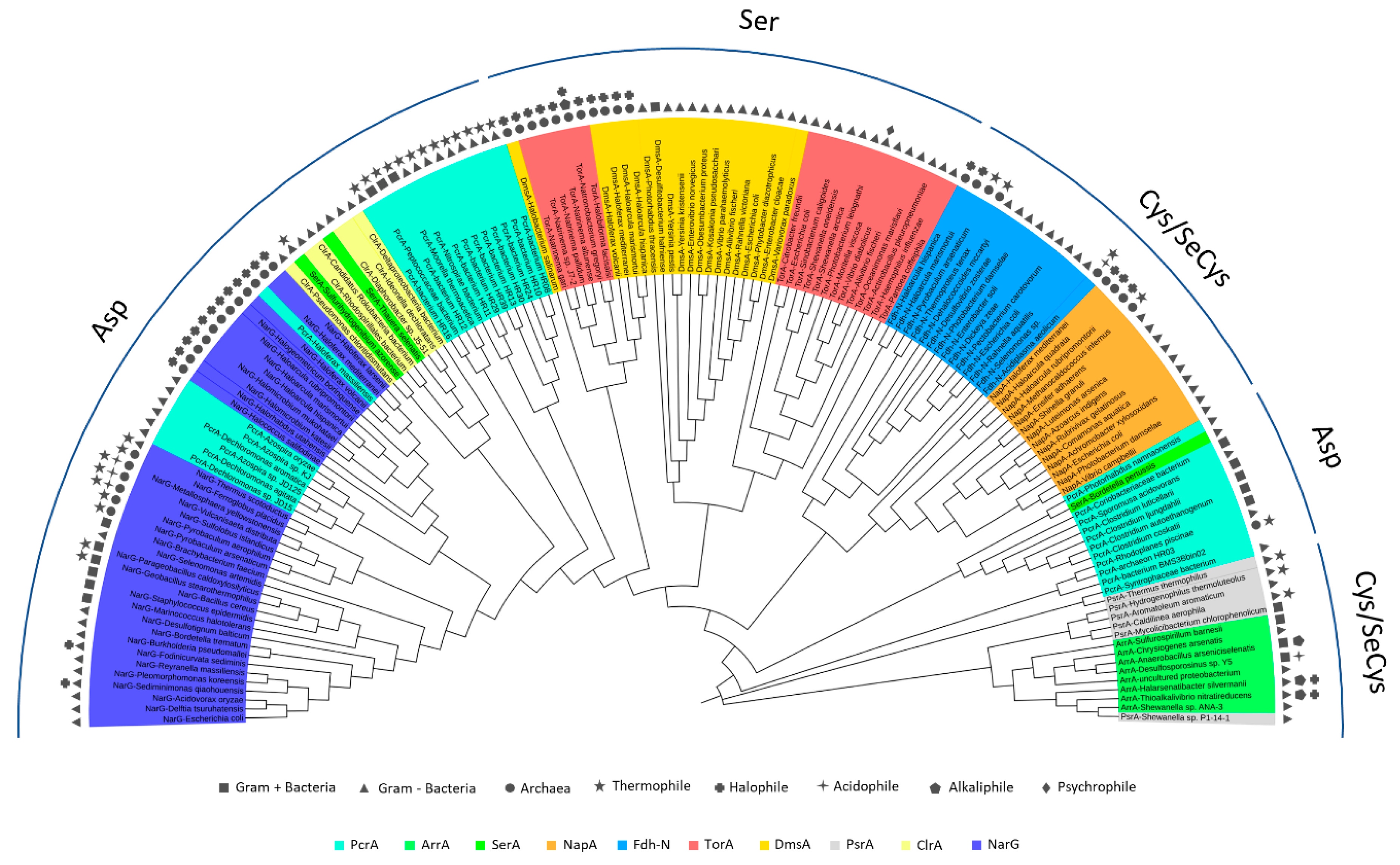
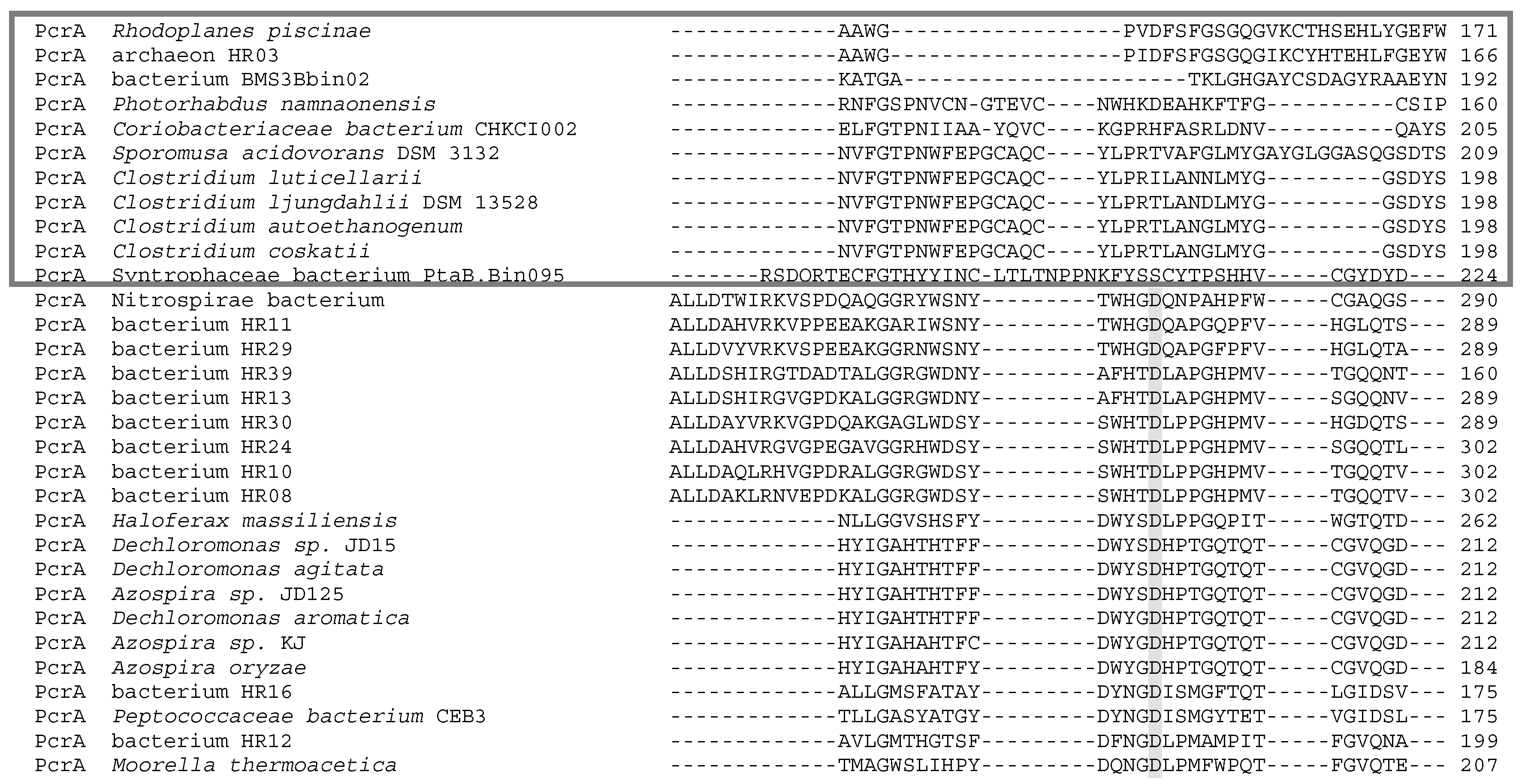
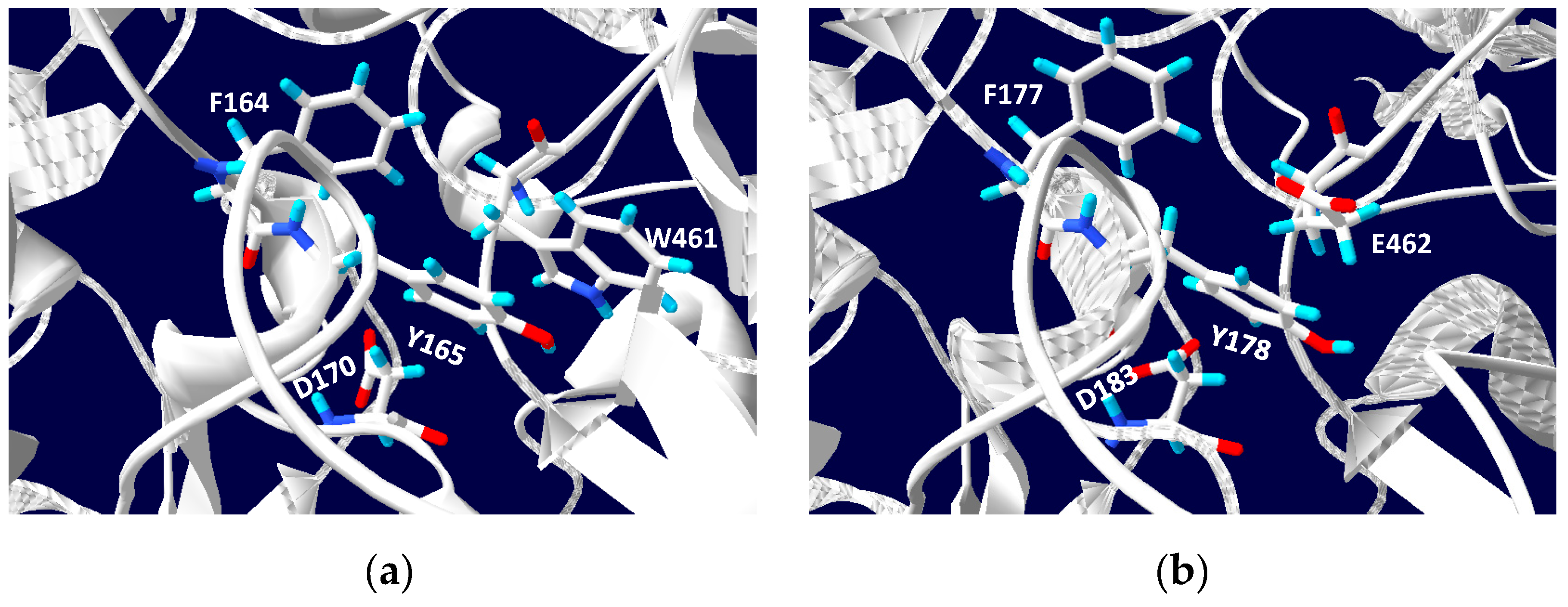
5. Phylogenetics of DMSO Reductases: An Updated Overview
6. DMSO Reductases: Bioremediation with Extremophiles
6.1. (Per)chlorates and Nitrates
6.2. Arsenates and Sulphates
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| FeMoCo | iron-molybdenum Cofactor |
| MoCo | molybdenum pterin-based Cofactor |
| cPMP | cyclic pyranopterin monophosphate |
| 5′-GTP | guanosine triphosphate |
| MPT | molybdopterin / metal-Binding Pterin |
| FeVCo | iron-vanadium cofactor |
| WCo | tungsten cofactor |
| DMSO | dimethyl sulfoxide |
| mARC | mitochondrial amidoxime reducing components |
| Bis-MGD | bis-molybdopterin-guanine dinucleotide |
| 3′,8-cH2GTP | 3′,8-cyclo-7,8-dihydro-guanosine 5′-triphosphate |
| Bis-Mo-MPT | bis-mo-molybdopterin |
| TMAO | trimethylamine N-oxide |
| NapA | periplasmic nitrate reductase |
| NarG | respiratory nitrate reductase |
| Nas | assimilatory nitrate reductase |
| PsrA | polysulfide reductases |
| ArrA | respiratory arsenate reductases |
| Fdh-N | formate dehydrogenases N |
| PcrA | perchlorate reductases |
| SerA | respiratory selenate reductases |
| ClrA | chlorate reductases |
| DmsA | DMSO reductases |
| TorA | TMAO reductases |
References
- Mendel, R.R. Metabolism of molybdenum. In Metallomics and the Cell; Banci, L., Sigel, H., Sigel, A., Sigel, R., Eds.; Springer: Dordrecht, The Netherlands, 2013; Volume 12, pp. 503–528. [Google Scholar]
- Dos Santos, P.C.; Dean, D.R.; Hu, Y.; Ribbe, M.W. Formation and insertion of the nitrogenase iron—Molybdenum cofactor. Chem. Rev. 2004, 104, 1159–1174. [Google Scholar] [CrossRef] [PubMed]
- Leimkühler, S.; Mendel, R.R. Molybdenum cofactor biosynthesis. In Molybdenum and Tungsten Enzymes: Biochemistry; Hille, R., Schulzke, C., Kirk, M.L., Eds.; Royal Society of Chemistry: Cambridge, UK, 2017; pp. 100–116. [Google Scholar]
- Hille, R. Molybdenum and tungsten in biology. Trends Biochem. Sci. 2002, 27, 360–367. [Google Scholar] [CrossRef]
- Bursakov, S.A.; Gavel, O.Y.; Di Rocco, G.; Lampreia, J.; Calvete, J.; Pereira, A.S.; Moura, J.J.G.; Moura, I. Antagonists Mo and Cu in a heterometallic cluster present on a novel protein (orange protein) isolated from Desulfovibrio gigas. J. Inorg. Biochem. 2004, 98, 833–840. [Google Scholar] [CrossRef] [PubMed]
- Maiti, B.K.; Almeida, R.M.; Maia, L.B.; Moura, I.; Moura, J.J. Insights into the molybdenum/copper heterometallic cluster assembly in the orange protein: Probing intermolecular interactions with an artificial metal-binding ATCUN tag. Inorg. Chem. 2017, 56, 8900–8911. [Google Scholar] [CrossRef] [PubMed]
- Nason, A.; Lee, K.Y.; Pan, S.S.; Ketchum, P.A.; Lamberti, A.; DeVries, J. Invitro formation of assimilatory reduced nicotinamide adenine dinucleotide phosphate: Nitrate reductase from a Neurospora mutant and a component of molybdenum-enzymes. Proc. Natl. Acad. Sci. USA 1971, 68, 3242–3246. [Google Scholar] [CrossRef] [PubMed]
- Dassault Systèmes BIOVIA BIOVIA Draw 2019. Available online: https://www.3dsbiovia.com/products/collaborative-science/biovia-draw/ (accessed on 12 June 2019).
- Grimaldi, S.; Schoepp-Cothenet, B.; Ceccaldi, P.; Guigliarelli, B.; Magalon, A. The prokaryotic Mo/W-bisPGD enzymes family: A catalytic workhorse in bioenergetic. Biochim. Biophys. Acta Bioenergy 2013, 1827, 1048–1085. [Google Scholar] [CrossRef] [Green Version]
- Jiménez-Vicente, E.; Hernandez, J.A.; Echavarri-Erasun, C.; Rubio, L.M. Biosynthesis of the iron-molybdenum cofactor of nitrogenase. In Biological Nitrogen Fixation; de Bruijn, F., Ed.; John Wiley & Sons, Inc.: New York, NY, USA, 2015; pp. 75–86. [Google Scholar]
- Maia, L.B.; Moura, I.; Moura, J.J.G. Molybdenum and tungsten-containing enzymes: An overview. In Molybdenum and Tungsten Enzymes: Biochemistry; Hille, R., Schulzke, C., Kirk, M., Eds.; Royal Society of Chemistry: Cambridge, UK, 2017; pp. 1–80. [Google Scholar]
- Holm, R.H.; Solomon, E.I.; Majumdar, A.; Tenderholt, A. Comparative molecular chemistry of molybdenum and tungsten and its relation to hydroxylase and oxotransferase enzymes. Coord. Chem. Rev. 2011, 255, 993–1015. [Google Scholar] [CrossRef]
- Schoepp-Cothenet, B.; Van Lis, R.; Philippot, P.; Magalon, A.; Russell, M.J.; Nitschke, W. The ineluctable requirement for the trans-iron elements molybdenum and/or tungsten in the origin of life. Sci. Rep. 2012, 2, 263. [Google Scholar] [CrossRef]
- Kishida, K.; Sohrin, Y.; Okamura, K.; Ishibashi, J. Tungsten enriched in submarine hydrothermal fluids. Earth Planet. Sci. Lett. 2004, 222, 819–827. [Google Scholar] [CrossRef]
- Roy, R.; Adams, M.W.W. Tungsten-dependent aldehyde oxidoreductase: A new family of enzymes containing the pterin cofactor. Met. Ions Biol. Syst. 2002, 39, 673–697. [Google Scholar]
- Pang, H.; Yokoyama, K. Lessons from the studies of a C-C bond forming radical SAM enzyme in molybdenum cofactor biosynthesis. In Radical SAM Enzymes; Bandarian, V., Ed.; Academic Press: Cambridge, UK, 2018; pp. 485–522. [Google Scholar]
- Schwarz, G.; Mendel, R.R.; Ribbe, M.W. Molybdenum cofactors, enzymes and pathways. Nature 2009, 460, 839–847. [Google Scholar] [CrossRef] [PubMed]
- Hover, B.M.; Lilla, E.A.; Yokoyama, K. Mechanistic investigation of cPMP synthase in molybdenum cofactor biosynthesis using an uncleavable substrate analogue. Biochemistry 2015, 54, 7229–7236. [Google Scholar] [CrossRef] [PubMed]
- Hover, B.M.; Tonthat, N.K.; Schumacher, M.A.; Yokoyama, K. Mechanism of pyranopterin ring formation in molybdenum cofactor biosynthesis. Proc. Natl. Acad. Sci. USA 2015, 112, 6347–6352. [Google Scholar] [CrossRef] [Green Version]
- Wuebbens, M.M.; Rajagopalan, K.V. Mechanistic and mutational studies of Escherichia coli molybdopterin synthase clarify the final step of molybdopterin biosynthesis. J. Biol. Chem. 2003, 278, 14523–14532. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Urban, A.; Mihara, H.; Leimkühler, S.; Kurihara, T.; Esaki, N. IscS functions as a primary sulfur-donating enzyme by interacting specifically with MoeBand MoaD in the biosynthesis of molybdopterin in Escherichia coli. J. Biol. Chem. 2010, 285, 2302–2308. [Google Scholar] [CrossRef]
- Bühning, M.; Valleriani, A.; Leimkühler, S. The role of SufS is restricted to Fe-S cluster biosynthesis in Escherichia coli. Biochemistry 2017, 56, 1987–2000. [Google Scholar] [CrossRef] [PubMed]
- Dahl, J.; Radon, C.; Bühning, M.; Nimtz, M.; Leichert, L.; Denis, Y.; Jourlin-Castelli, C.; Iobbi-Nivol, C.; Méjean, V.; Leimkühler, S. The sulfur carrier protein TusA has a pleiotropic role in Escherichia coli that also affects molybdenum cofactor biosynthesis. J. Biol. Chem. 2013, 228, 5426–5442. [Google Scholar] [CrossRef] [PubMed]
- Dahl, J.U.; Urban, A.; Bolte, A.; Sriyabhaya, P.; Donahue, J.L.; Nimtz, M.; Larson, T.J.; Leimkühler, S. The identification of a novel protein involved in molybdenum cofactor biosynthesis in Escherichia coli. J. Biol. Chem. 2011, 286, 35801–35812. [Google Scholar] [CrossRef]
- Nichols, J.D.; Rajagopalan, K.V. In vitro molybdenum ligation to molybdopterin using purified components. J. Biol. Chem. 2005, 280, 7817–7822. [Google Scholar] [CrossRef]
- Iobbi-Nivol, C.; Leimkühler, S. Molybdenum enzymes, their maturation and molybdenum cofactor biosynthesis in Escherichia coli. Biochim. Biophys. Acta Bioenergy 2013, 1829, 1086–1101. [Google Scholar] [CrossRef] [PubMed]
- McLuskey, K.; Harrison, J.A.; Schüttelkopf, A.W.; Boxer, D.H.; Hunter, W.N. Insight into the role of Escherichia coli MobB in molybdenum cofactor biosynthesis based on the high resolution crystal structure. J. Biol. Chem. 2003, 278, 23706–23713. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, C.E.M.; Sargent, F.; Buchanan, G.; Palmer, T.; Lawson, D.M. Crystal structure of the molybdenum cofactor biosynthesis protein MobA from Escherichia coli at near-atomic resolution. Structure 2000, 8, 1115–1125. [Google Scholar] [CrossRef]
- Lake, M.W.; Temple, C.A.; Rajagopalan, K.V.; Schindelin, H. The crystal structure of the Escherichia coli MobA protein provides insight into molybdopterin guanine dinucleotide biosynthesis. J. Biol. Chem. 2000, 275, 40211–40217. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Seduk, F.; Iobbi-Nivol, C.; Leimkühler, S. Molybdopterin dinucleotide biosynthesis in Escherichia coli: Identification of amino acid residues of molybdopterin dinucleotide transferases that determine specificity for binding of guanine or cytosine nucleotides. J. Biol. Chem. 2011, 286, 1400–1408. [Google Scholar] [CrossRef] [PubMed]
- Reschke, S.; Sigfridsson, K.G.V.; Kaufmann, P.; Leidel, N.; Horn, S.; Gast, K.; Schulzke, C.; Haumann, M.; Leimkühler, S. Identification of a bis-molybdopterin intermediate in molybdenum cofactor biosynthesis in Escherichia coli. J. Biol. Chem. 2013, 288, 29736–29745. [Google Scholar] [CrossRef] [PubMed]
- Jack, R.L.; Buchanan, G.; Dubini, A.; Hatzixanthis, K.; Palmer, T.; Sargent, F. Coordinating assembly and export of complex bacterial proteins. EMBO J. 2004, 23, 3962–3972. [Google Scholar] [CrossRef] [PubMed]
- Hatzixanthis, K.; Clarke, T.A.; Oubrie, A.; Richardson, D.J.; Turner, R.J.; Sargent, F. Signal peptide-chaperone interactions on the twin-arginine protein transport pathway. Proc. Natl. Acad. Sci. USA 2005, 102, 8460–8465. [Google Scholar] [CrossRef]
- Leimkühler, S.; Iobbi-Nivol, C. Bacterial molybdoenzymes: Old enzymes for new purposes. FEMS Microbiol. Rev. 2015, 40, 1–18. [Google Scholar] [CrossRef]
- Müller, J.A.; DasSarma, S. Genomic analysis of anaerobic respiration in the archaeon Halobacterium sp. strain NRC-1: Dimethyl sulfoxide and trimethylamine N-oxide as terminal electron acceptors. J. Bacteriol. 2005, 187, 1659–1667. [Google Scholar] [CrossRef]
- Yamamura, S.; Amachi, S. Microbiology of inorganic arsenic: From metabolism to bioremediation. J. Biosci. Bioeng. 2014, 118, 1–9. [Google Scholar] [CrossRef]
- Pinchbeck, B.J.; Soriano-Laguna, M.J.; Sullivan, M.J.; Luque-Almagro, V.M.; Rowley, G.; Ferguson, S.J.; Roldán, M.D.; Richardson, D.J.; Gates, A.J. A dual functional redox enzyme maturation protein for respiratory and assimilatory nitrate reductases in bacteria. Mol. Microbiol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Raymond, J.; Siefert, J.L.; Staples, C.R.; Blankenship, R.E. The natural history of nitrogen fixation. Mol. Biol. Evol. 2004, 21, 541–554. [Google Scholar] [CrossRef] [PubMed]
- Hille, R.; Hall, J.; Basu, P. The mononuclear molybdenum enzymes. Chem. Rev. 2014, 114, 3963–4038. [Google Scholar] [CrossRef] [PubMed]
- Havemeyer, A.; Bittner, F.; Wollers, S.; Mendel, R.; Kunze, T.; Clement, B. Identification of the missing component in the mitochondrial benzamidoxime prodrug-converting system as a novel molybdenum enzyme. J. Biol. Chem. 2006, 281, 34796–34802. [Google Scholar] [CrossRef] [PubMed]
- Gladyshev, V.N.; Zhang, Y. Abundance, ubiquity and evolution of molybdoenzymes. In RSC Metallobiology; Hille, R., Schulzke, C., Kirk, M., Eds.; Royal Society of Chemistry: Cambridge, UK, 2017; pp. 81–99. [Google Scholar]
- Wahl, B.; Reichmann, D.; Niks, D.; Krompholz, N.; Havemeyer, A.; Clement, B.; Messerschmidt, T.; Rothkegel, M.; Biester, H.; Hille, R.; et al. Biochemical and spectroscopic characterization of the human mitochondrial amidoxime reducing components hmARC-1 and hmARC-2 suggests the existence of a new molybdenum enzyme family in eukaryotes. J. Biol. Chem. 2010, 285, 37847–37859. [Google Scholar] [CrossRef] [PubMed]
- Kappler, K.; Schwarzb, G. The sulfite oxidase family of molybdenum enzymes. In Molybdenum and Tungsten Enzymes: Biochemistry; Hille, R., Schulzke, C., Kirk, M., Eds.; Royal Society of Chemistry: Cambridge, UK, 2017; pp. 240–273. [Google Scholar]
- Nishinoa, T.; Okamotoa, K.; Leimkühler, S. Enzymes of the xanthine oxidase family. In Molybdenum and Tungsten Enzymes: Biochemistry; Hille, R., Schulzke, C., Kirk, M., Eds.; Royal Society of Chemistry: Cambridge, UK, 2017; pp. 192–239. [Google Scholar]
- Meyer, O.; Dobbek, H.; Huber, R.; Gremer, L.; Kiefersauer, R. Catalysis at a dinuclear [CuSMo(O)OH] cluster in a CO dehydrogenase resolved at 1.1-A resolution. Proc. Natl. Acad. Sci. USA 2002, 99, 15971–15976. [Google Scholar]
- Zhang, Y.; Rump, S.; Gladyshev, V.N. Comparative genomics and evolution of molybdenum utilization. Coord. Chem. Rev. 2011, 255, 1206–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McEwan, A.G.; Ridge, J.P.; McDevitt, C.A.; Hugenholtz, P. The DMSO reductase family of microbial molybdenum enzymes; molecular properties and role in the dissimilatory reduction of toxic elements. Geomicrobiol. J. 2002, 19, 3–21. [Google Scholar] [CrossRef]
- Chan, M.; Mukund, S.; Kletzin, A.; Adams, M.W.; Rees, D.C. Structure of a hyperthermophilic tungstopterin enzyme, aldehyde ferredoxin oxidoreductase. Science 1995, 267, 1463–1469. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Espinosa, R.M.; Dridge, E.J.; Bonete, M.J.; Butt, J.N.; Butler, C.S.; Sargent, F.; Richardson, D.J. Look on the positive side! The orientation, identification and bioenergetics of “archaeal” membrane-bound nitrate reductases. FEMS Microbiol. Lett. 2007, 276, 129–139. [Google Scholar] [CrossRef]
- Martínez-Espinosa, R.M.; Richardson, D.J.; Bonete, M.J. Characterisation of chlorate reduction in the haloarchaeon Haloferax mediterranei. Biochim. Biophys. Acta Gen. Subj. 2015, 1850, 587–594. [Google Scholar] [CrossRef] [PubMed]
- Richardson, D.J.; Berks, B.C.; Russell, D.A.; Spiro, S.; Taylor, C.J. Functional, biochemical and genetic diversity of prokaryotic nitrate reductases. Cell. Mol. Life Sci. 2001, 58, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Gates, A.J.; Butler, C.S.; Richardson, D.J.; Butt, J.N. Electrocatalytic reduction of nitrate and selenate by NapAB. Biochem. Soc. Trans. 2011, 39, 236–242. [Google Scholar] [CrossRef] [PubMed]
- Sparacino-Watkins, C.; Stolz, J.F.; Basu, P. Nitrate and periplasmic nitrate reductases. Chem. Soc. Rev. 2014, 43, 676–706. [Google Scholar] [CrossRef] [PubMed]
- Cabello, P.; Roldán, M.D.; Moreno-Vivián, C. Nitrate reduction and the nitrogen cycle in archaea. Microbiology 2004, 150, 3527–3546. [Google Scholar] [CrossRef] [PubMed]
- Lledó, B.; Martı́nez-Espinosa, R.M.; Marhuenda-Egea, F.C.; Bonete, M.J. Respiratory nitrate reductase from haloarchaeon Haloferax mediterranei: Biochemical and genetic analysis. Biochim. Biophys. Acta Gen. Subj. 2004, 1674, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Yoshimatsu, K.; Iwasaki, T.; Fujiwara, T. Sequence and electron paramagnetic resonance analyses of nitrate reductase NarGH from a denitrifying halophilic euryarchaeote Haloarcula marismortui. FEBS Lett. 2002, 516, 145–150. [Google Scholar] [CrossRef]
- Alvarez, L.; Bricio, C.; Blesa, A.; Hidalgo, A.; Berenguer, J. Transferable denitrification capability of Thermus thermophilus. Appl. Environ. Microbiol. 2014, 80, 19–28. [Google Scholar] [CrossRef]
- Cava, F.; Laptenko, O.; Borukhov, S.; Chahlafi, Z.; Blas-Galindo, E.; Gómez-Puertas, P.; Berenguer, J. Control of the respiratory metabolism of Thermus thermophilus by the nitrate respiration conjugative element NCE. Mol. Microbiol. 2007, 64, 630–646. [Google Scholar] [CrossRef]
- Alvarez, L.; Bricio, C.; José Gómez, M.; Berenguer, J. Lateral transfer of the denitrification pathway genes among Thermus thermophilus strains. Appl. Environ. Microbiol. 2011, 77, 1352–1358. [Google Scholar] [CrossRef]
- Coates, J.D.; Achenbach, L.A. Microbial perchlorate reduction: Rocket-fuelled metabolism. Nat. Rev. Microbiol. 2004, 2, 569–580. [Google Scholar] [CrossRef] [PubMed]
- Thrash, J.C.; Pollock, J.; Torok, T.; Coates, J.D. Description of the novel perchlorate-reducing bacteria Dechlorobacter hydrogenophilus gen. nov., sp. nov. and Propionivibrio militaris, sp. nov. Appl. Microbiol. Biotechnol. 2010, 86, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Kengen, S.W.; Rikken, G.B.; Hagen, W.R.; van Ginkel, C.G.; Stams, A.J. Purification and characterization of (per)chlorate reductase from the chlorate-respiring strain GR-1. J. Bacteriol. 1999, 181, 6706–6711. [Google Scholar] [PubMed]
- Wolterink, A.F.W.M.; Schiltz, E.; Hagedoorn, P.-L.; Hagen, W.R.; Kengen, S.W.M.; Stams, A.J.M. Characterization of the chlorate reductase from Pseudomonas chloritidismutans. J. Bacteriol. 2003, 185, 3210–3213. [Google Scholar] [CrossRef] [PubMed]
- Okeke, B.C.; Frankenberger, W.T. Molecular analysis of a perchlorate reductase from a perchlorate-respiring bacterium Perc1ace. Microbiol. Res. 2003, 158, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, T.; Rova, M.; Smedja Bäcklund, A. Microbial metabolism of oxochlorates: A bioenergetic perspective. Biochim. Biophys. Acta Bioenergy 2013, 1827, 189–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bardiya, N.; Bae, J.-H. Dissimilatory perchlorate reduction: A review. Microbiol. Res. 2011, 166, 237–254. [Google Scholar] [CrossRef]
- Yoshimatsu, K.; Sakurai, T.; Fujiwara, T. Purification and characterization of dissimilatory nitrate reductase from a denitrifying halophilic archaeon, Haloarcula marismortui. FEBS Lett. 2000, 470, 216–220. [Google Scholar] [CrossRef]
- Afshar, S.; Johnson, E.; de Vries, S.; Schroder, I. Properties of a thermostable nitrate reductase from the hyperthermophilic archaeon Pyrobaculum aerophilum. J. Bacteriol. 2001, 183, 5491–5495. [Google Scholar] [CrossRef]
- Liebensteiner, M.G.; Pinkse, M.W.H.; Nijsse, B.; Verhaert, P.D.E.M.; Tsesmetzis, N.; Stams, A.J.M.; Lomans, B.P. Perchlorate and chlorate reduction by the crenarchaeon Aeropyrum pernix and two thermophilic Firmicutes. Environ. Microbiol. Rep. 2015, 7, 936–945. [Google Scholar] [CrossRef]
- Liebensteiner, M.G.; Pinkse, M.W.H.; Schaap, P.J.; Stams, A.J.M.; Lomans, B.P. Archaeal (per)chlorate reduction at high temperature: An interplay of biotic and abiotic reactions. Science (80-. ) 2013, 340, 85–87. [Google Scholar] [CrossRef] [PubMed]
- van Lis, R.; Nitschke, W.; Duval, S.; Schoepp-Cothenet, B. Arsenics as bioenergetic substrates. Biochim. Biophys. Acta Bioenergy 2013, 1827, 176–188. [Google Scholar] [CrossRef] [PubMed]
- Del Giudice, I.; Limauro, D.; Pedone, E.; Bartolucci, S.; Fiorentino, G. A novel arsenate reductase from the bacterium Thermus thermophilus HB27: Its role in arsenic detoxification. Biochim. Biophys. Acta Proteins Proteom. 2013, 1834, 2071–2079. [Google Scholar] [CrossRef] [PubMed]
- Ordoñez, O.F.; Rasuk, M.C.; Soria, M.N.; Contreras, M.; Farías, M.E. Haloarchaea from the Andean Puna: Biological role in the energy metabolism of arsenic. Microb. Ecol. 2018, 76, 695–705. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, R.; Rosen, B.P. Arsenate reductases in prokaryotes and eukaryotes. Environ. Health Perspect. 2002, 110, 745–748. [Google Scholar] [CrossRef] [PubMed]
- Stolz, J.F.; Oremland, R.S. Bacterial respiration of arsenic and selenium. FEMS Microbiol. Rev. 1999, 23, 615–627. [Google Scholar] [CrossRef]
- Pandey, S.; Shrivastava, A.K.; Singh, V.K.; Rai, R.; Singh, P.K.; Rai, S.; Rai, L.C. A new arsenate reductase involved in arsenic detoxification in Anabaena sp. PCC7120. Funct. Integr. Genom. 2013, 13, 43–55. [Google Scholar] [CrossRef]
- Takahashi, Y.; Suto, K.; Inoue, C. Polysulfide reduction by Clostridium relatives isolated from sulfate-reducing enrichment cultures. J. Biosci. Bioeng. 2010, 109, 372–380. [Google Scholar] [CrossRef]
- Braatsch, S.; Krafft, T.; Simon, J.; Gross, R.; Klimmek, O.; Kröger, A. PsrR, a member of the AraC family of transcriptional regulators, is required for the synthesis of Wolinella succinogenes polysulfide reductase. Arch. Microbiol. 2002, 178, 202–207. [Google Scholar] [CrossRef]
- Sugio, T.; Oda, K.; Matsumoto, K.; Takai, M.; Wakasa, S.; Kamimura, K. Purification and characterization of sulfur reductase from a moderately thermophilic bacterial strain, TI-1, that oxidizes iron. Biosci. Biotechnol. Biochem. 1998, 62, 705–709. [Google Scholar] [CrossRef]
- Laska, S.; Kletzin, A. Improved purification of the membrane-bound hydrogenase-sulfur-reductase complex from thermophilic archaea using epsilon-aminocaproic acid-containing chromatography buffers. J. Chromatogr. B Biomed. Sci. Appl. 2000, 737, 151–160. [Google Scholar] [CrossRef]
- Laska, S.; Lottspeich, F.; Kletzin, A. Membrane-bound hydrogenase and sulfur reductase of the hyperthermophilic and acidophilic archaeon Acidianus ambivalens. Microbiology 2003, 149, 2357–2371. [Google Scholar] [CrossRef] [PubMed]
- Findlay, A.J. Microbial impact on polysulfide dynamics in the environment. FEMS Microbiol. Lett. 2016, 363. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Soding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2014, 7, 539. [Google Scholar] [CrossRef] [PubMed]
- Soding, J. Protein homology detection by HMM-HMM comparison. Bioinformatics 2005, 21, 951–960. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019. [Google Scholar] [CrossRef] [PubMed]
- Duval, S.; Ducluzeau, A.-L.; Nitschke, W.; Schoepp-Cothenet, B. Enzyme phylogenies as markers for the oxidation state of the environment: The case of respiratory arsenate reductase and related enzymes. BMC Evol. Biol. 2008, 8, 206. [Google Scholar] [CrossRef] [PubMed]
- Afkar, E.; Lisak, J.; Saltikov, C.; Basu, P.; Oremland, R.S.; Stolz, J.F. The respiratory arsenate reductase from Bacillus selenitireducens strain MLS10. FEMS Microbiol. Lett. 2003, 226, 107–112. [Google Scholar] [CrossRef]
- Rothery, R.A.; Workun, G.J.; Weiner, J.H. The prokaryotic complex iron-sulfur molybdoenzyme family. Biochim. Biophys. Acta Biomembr. 2008, 1778, 1897–1929. [Google Scholar] [CrossRef] [PubMed]
- Hettmann, T.; Siddiqui, R.A.; von Langen, J.; Frey, C.; Romão, M.J.; Diekmann, S. Mutagenesis study on the role of a lysine residue highly conserved in formate dehydrogenases and periplasmic nitrate reductases. Biochem. Biophys. Res. Commun. 2003, 310, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Zeamari, K.; Gerbaud, G.; Grosse, S.; Fourmond, V.; Chaspoul, F.; Biaso, F.; Arnoux, P.; Sabaty, M.; Pignol, D.; Guigliarelli, B.; et al. Tuning the redox properties of a [4Fe-4S] center to modulate the activity of Mo-bisPGD periplasmic nitrate reductase. Biochim. Biophys. Acta Bioenergy 2019, 1860, 402–413. [Google Scholar] [CrossRef] [PubMed]
- McCrindle, S.L.; Kappler, U.; McEwan, A.G. Microbial dimethylsulfoxide and trimethylamine-N-oxide respiration. Adv. Microb. Physiol. 2005, 50, 147–201. [Google Scholar] [PubMed]
- Zhang, L.; Nelson, J.; Rajagopalan, K.; George, G. Structure of the molybdenum site of Escherichia coli trimethylamine N-oxide reductase. Inorg. Chem. 2007, 47, 1074–1078. [Google Scholar] [CrossRef] [PubMed]
- Kappler, U.; Schäfer, H. Transformations of dimethylsulfide. In The Metal-Driven Biogeochemistry of Gaseous Compounds in the Environment; Kroneck, P.M., Sosa Torres, M.E., Eds.; Springer: Dordrecht, The Netherlands, 2014; pp. 279–313. [Google Scholar]
- Schröder, I.; Rech, S.; Krafft, T.; Macy, J.M. Purification and characterization of the selenate reductase from Thauera selenatis. J. Biol. Chem. 1997, 272, 23765–23768. [Google Scholar] [CrossRef] [PubMed]
- Bender, K.S.; Shang, C.; Chakraborty, R.; Belchik, S.M.; Coates, J.D.; Achenbach, L.A. Identification, characterization, and classification of genes encoding perchlorate reductase. J. Bacteriol. 2005, 187, 5090–5096. [Google Scholar] [CrossRef] [PubMed]
- Clark, I.C.; Melnyk, R.A.; Engelbrektson, A.; Coates, J.D. Structure and evolution of chlorate reduction composite transposons. MBio 2013, 4, e00379-13. [Google Scholar] [CrossRef] [PubMed]
- Ize, B.; Coulthurst, S.J.; Hatzixanthis, K.; Caldelari, I.; Buchanan, G.; Barclay, E.C.; Richardson, D.J.; Palmer, T.; Sargent, F. Remnant signal peptides on non-exported enzymes: Implications for the evolution of prokaryotic respiratory chains. Microbiology 2009, 155, 3992–4004. [Google Scholar] [CrossRef] [PubMed]
- Lucker, S.; Wagner, M.; Maixner, F.; Pelletier, E.; Koch, H.; Vacherie, B.; Rattei, T.; Damste, J.S.S.; Spieck, E.; Le Paslier, D.; et al. A Nitrospira metagenome illuminates the physiology and evolution of globally important nitrite-oxidizing bacteria. Proc. Natl. Acad. Sci. USA 2010, 107, 13479–13484. [Google Scholar] [CrossRef]
- Melnyk, R.A.; Coates, J.D. The perchlorate reduction genomic island: Mechanisms and pathways of evolution by horizontal gene rransfer. BMC Genom. 2015, 16, 862. [Google Scholar] [CrossRef]
- Youngblut, M.D.; Wang, O.; Barnum, T.P.; Coates, J.D. (Per)chlorate in biology on Earth and beyond. Annu. Rev. Microbiol. 2016, 70, 435–457. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, Y. I-TASSER server: New development for protein structure and function predictions. Nucleic Acids Res. 2015, 43, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Freddolino, P.L.; Zhang, Y. COFACTOR: Improved protein function prediction by combining structure, sequence and protein—Protein interaction information. Nucleic Acids Res. 2017, 45, W291–W299. [Google Scholar] [CrossRef] [PubMed]
- Krzmarzick, M.J.; Taylor, D.K.; Fu, X.; McCutchan, A.L. Diversity and niche of archaea in bioremediation. Archaea 2018, 2018, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Wang, O.; Coates, J.D. Biotechnological applications of microbial (per)chlorate reduction. Microorganisms 2017, 5, 76. [Google Scholar] [CrossRef] [PubMed]
- Stepanov, V.G.; Xiao, Y.; Tran, Q.; Rojas, M.; Willson, R.C.; Fofanov, Y.; Fox, G.E.; Roberts, D.J. The presence of nitrate dramatically changed the predominant microbial community in perchlorate degrading cultures under saline conditions. BMC Microbiol. 2014, 14, 225. [Google Scholar] [CrossRef]
- Fox, S.; Bruner, T.; Oren, Y.; Gilron, J.; Ronen, Z. Concurrent microbial reduction of high concentrations of nitrate and perchlorate in an ion exchange membrane bioreactor. Biotechnol. Bioeng. 2016, 113, 1881–1891. [Google Scholar] [CrossRef]
- Oren, A.; Elevi Bardavid, R.; Mana, L. Perchlorate and halophilic prokaryotes: Implications for possible halophilic life on Mars. Extremophiles 2014, 18, 75–80. [Google Scholar] [CrossRef]
- Torregrosa-Crespo, J.; Pire, C.; Martínez-Espinosa, R.M.; Bergaust, L. Denitrifying haloarchaea within the genus Haloferax display divergent respiratory phenotypes, with implications for their release of nitrogenous gases. Environ. Microbiol. 2019, 21, 427–436. [Google Scholar] [CrossRef]
- Gal, H.; Ronen, Z.; Weisbrod, N.; Dahan, O.; Nativ, R. Perchlorate biodegradation in contaminated soils and the deep unsaturated zone. Soil Biol. Biochem. 2008, 40, 1751–1757. [Google Scholar] [CrossRef]
- Ricardo, A.R.; Carvalho, G.; Velizarov, S.; Crespo, J.G.; Reis, M.A.M. Kinetics of nitrate and perchlorate removal and biofilm stratification in an ion exchange membrane bioreactor. Water Res. 2012, 46, 4556–4568. [Google Scholar] [CrossRef]
- Jebelli, M.A.; Maleki, A.; Amoozegar, M.A.; Kalantar, E.; Shahmoradi, B.; Gharibi, F. Isolation and identification of indigenous prokaryotic bacteria from arsenic-contaminated water resources and their impact on arsenic transformation. Ecotoxicol. Environ. Saf. 2017, 140, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Serrano, J.; Leiva, E.; Serrano, J.; Leiva, E. Removal of arsenic using acid/metal-tolerant sulfate reducing bacteria: A new approach for bioremediation of high-arsenic acid mine waters. Water 2017, 9, 994. [Google Scholar] [CrossRef]
- Kumari, N.; Jagadevan, S. Genetic identification of arsenate reductase and arsenite oxidase in redox transformations carried out by arsenic metabolising prokaryotes—A comprehensive review. Chemosphere 2016, 163, 400–412. [Google Scholar] [CrossRef] [PubMed]
- Rosen, B.P. Biochemistry of arsenic detoxification. FEBS Lett. 2002, 529, 86–92. [Google Scholar] [CrossRef] [Green Version]
- Mitra, A.; Chatterjee, S.; Gupta, D.K. Potential role of microbes in bioremediation of arsenic. In Arsenic Contamination in the Environment; Gupta, D.K., Chatterjee, S., Eds.; Springer: Cham, Vietnam, 2017; pp. 195–213. [Google Scholar]
- Huber, R.; Sacher, M.; Vollmann, A.; Huber, H.; Rose, D. Respiration of arsenate and aelenate by hyperthermophilic archaea. Syst. Appl. Microbiol. 2000, 23, 305–314. [Google Scholar] [CrossRef]
- Zhou, E.-M.; Xian, W.-D.; Mefferd, C.C.; Thomas, S.C.; Adegboruwa, A.L.; Williams, N.; Murugapiran, S.K.; Dodsworth, J.A.; Ganji, R.; Li, M.-M.; et al. Thermus sediminis sp. nov., a thiosulfate-oxidizing and arsenate-reducing organism isolated from Little Hot Creek in the Long Valley Caldera, California. Extremophiles 2018, 22, 983–991. [Google Scholar] [CrossRef] [PubMed]
- Gibson, G.R. Physiology and ecology of the sulphate-reducing bacteria. J. Appl. Bacteriol. 1990, 69, 769–797. [Google Scholar] [CrossRef] [PubMed]
- Utgikar, V.P.; Harmon, S.M.; Chaudhary, N.; Tabak, H.H.; Govind, R.; Haines, J.R. Inhibition of sulfate-reducing bacteria by metal sulfide formation in bioremediation of acid mine drainage. Environ. Toxicol. 2002, 17, 40–48. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
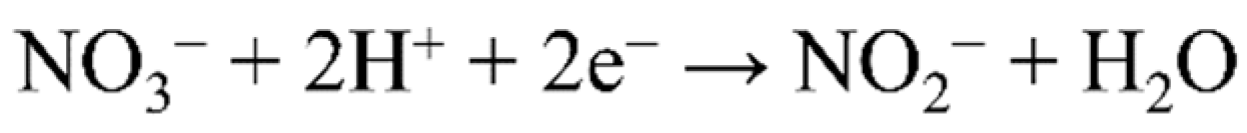{kind=link}
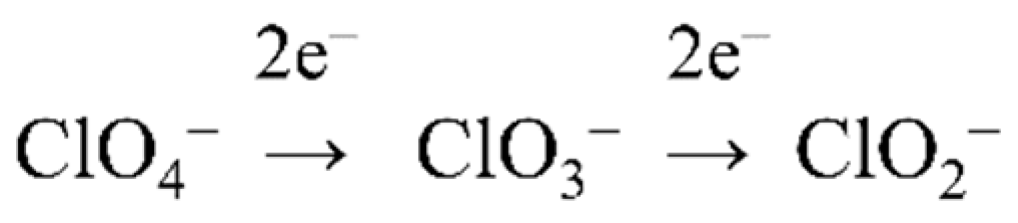{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family | Cofactor Structure | Ligands for Coordination | Example of Enzymes |
|---|---|---|---|
| Sulfite oxidase family |  | Cysteine | Sulfite oxidases Eukaryotic assimilatory nitrate reductases Sulfite dehydrogenase |
| Xanthine oxidase family |  | X: Sulfur, Selenium, Oxygen and S-Cu-S(Cys) | Xanthine oxidases Aldehyde oxidases 4-hydroxybenzoyl-CoA reductases Nicotine dehydrogenase |
| DMSO reductase family |  | X: Sulfur, Selenium, Oxygen, Y: Aspartate, Serine, Cysteine, and Selenocysteine | DMSO reductases Arsenate reductases Respiratory nitrate reductases Assimilatory nitrate reductases (Per)Chlorate reductases Polysulfide reductases |
| Tungstoenzymes family |  | X: Sulfur, Selenium, Oxygen, Y: Aspartate, Serine, Cysteine, and Selenocysteine | Aldehyde oxidoreductases Ferredoxin oxidoreductases Formate dehydrogenses Glyceraldehyde-3-phosphate oxidorreductase |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miralles-Robledillo, J.M.; Torregrosa-Crespo, J.; Martínez-Espinosa, R.M.; Pire, C. DMSO Reductase Family: Phylogenetics and Applications of Extremophiles. Int. J. Mol. Sci. 2019, 20, 3349. https://doi.org/10.3390/ijms20133349
Miralles-Robledillo JM, Torregrosa-Crespo J, Martínez-Espinosa RM, Pire C. DMSO Reductase Family: Phylogenetics and Applications of Extremophiles. International Journal of Molecular Sciences. 2019; 20(13):3349. https://doi.org/10.3390/ijms20133349
Chicago/Turabian StyleMiralles-Robledillo, Jose María, Javier Torregrosa-Crespo, Rosa María Martínez-Espinosa, and Carmen Pire. 2019. "DMSO Reductase Family: Phylogenetics and Applications of Extremophiles" International Journal of Molecular Sciences 20, no. 13: 3349. https://doi.org/10.3390/ijms20133349
APA StyleMiralles-Robledillo, J. M., Torregrosa-Crespo, J., Martínez-Espinosa, R. M., & Pire, C. (2019). DMSO Reductase Family: Phylogenetics and Applications of Extremophiles. International Journal of Molecular Sciences, 20(13), 3349. https://doi.org/10.3390/ijms20133349