Short Tandem Repeat Expansions and RNA-Mediated Pathogenesis in Myotonic Dystrophy


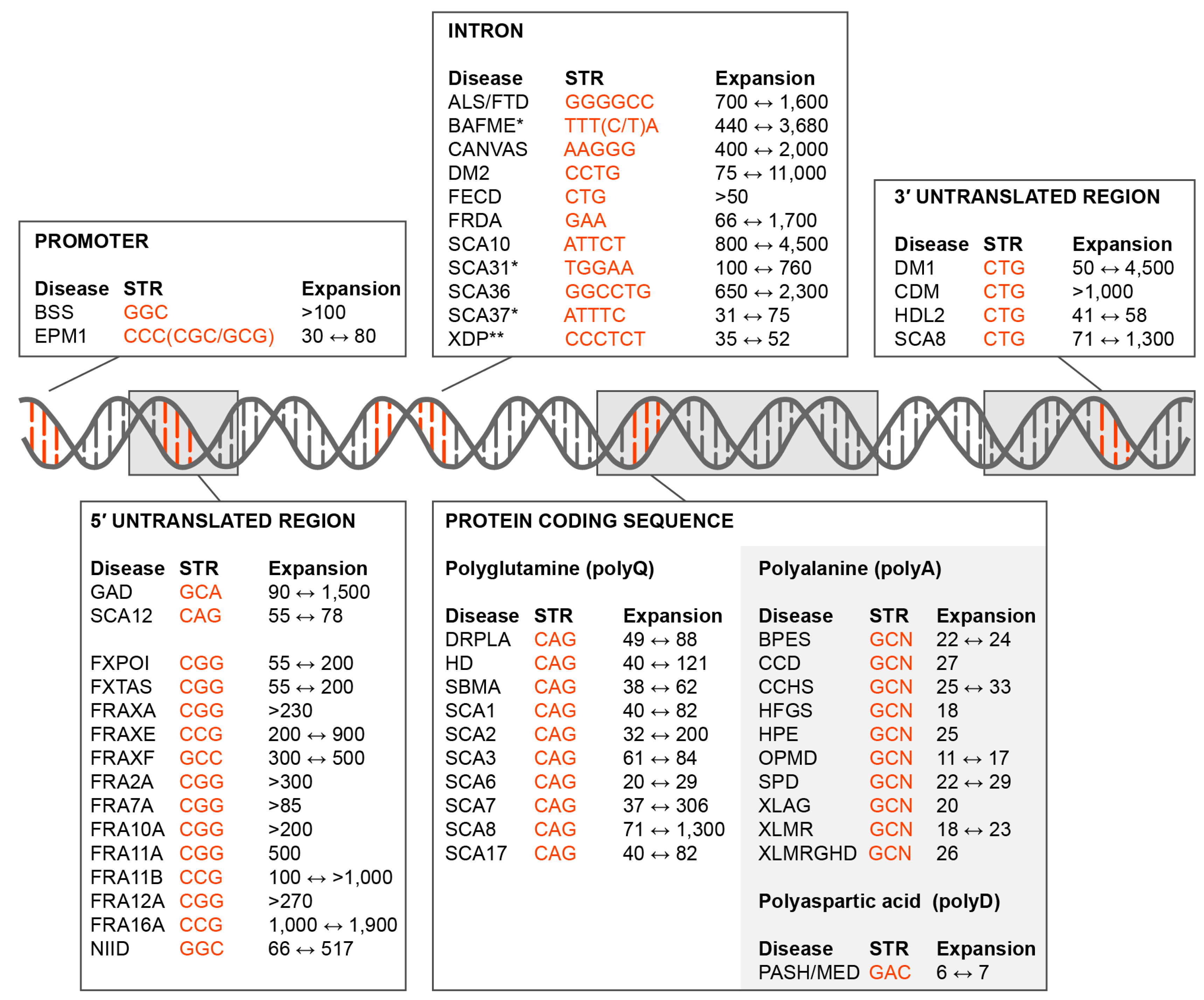{kind=link}
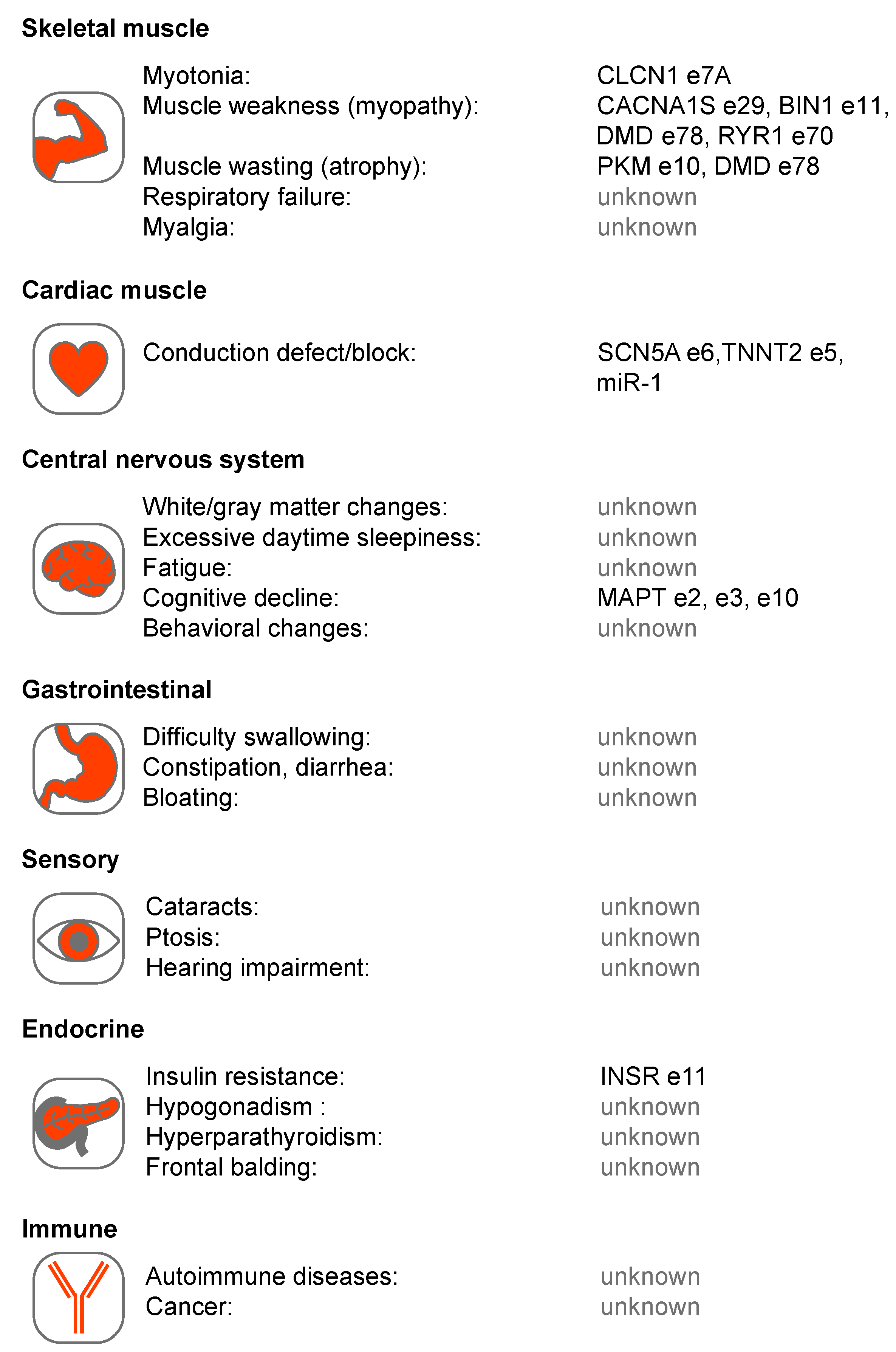{kind=link}
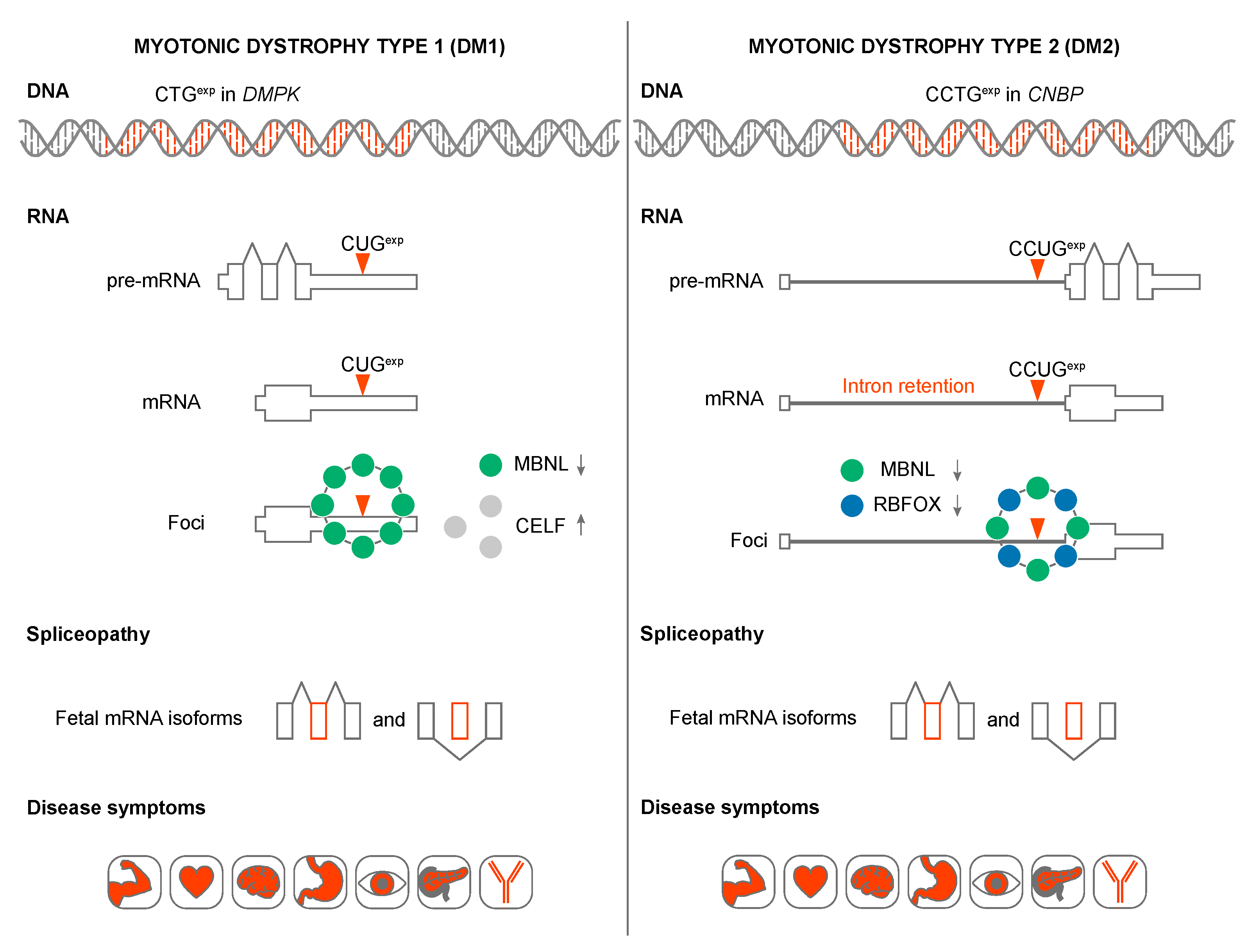{kind=link}
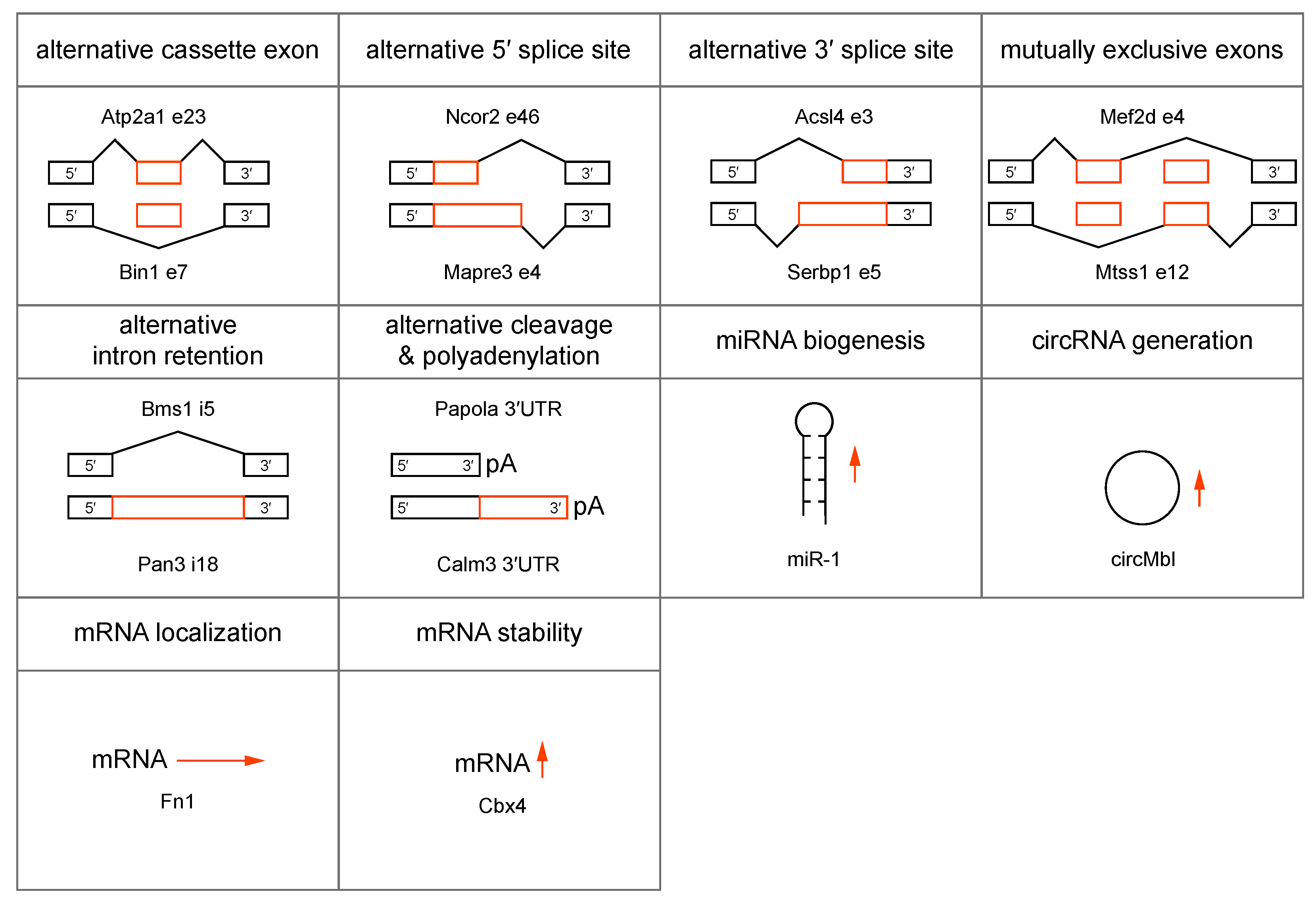{kind=link}
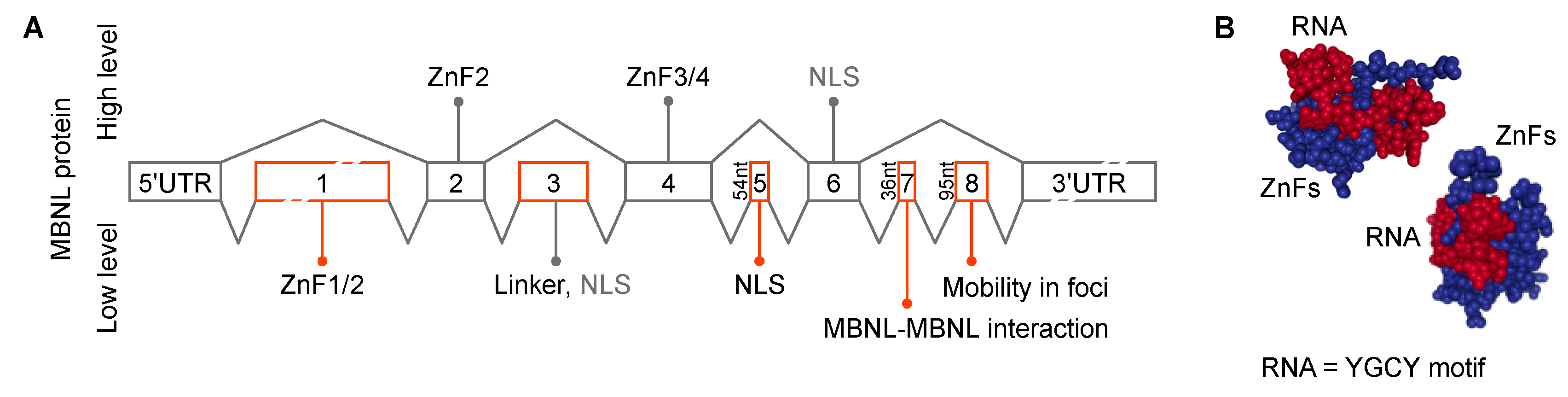{kind=link}
{kind=link}
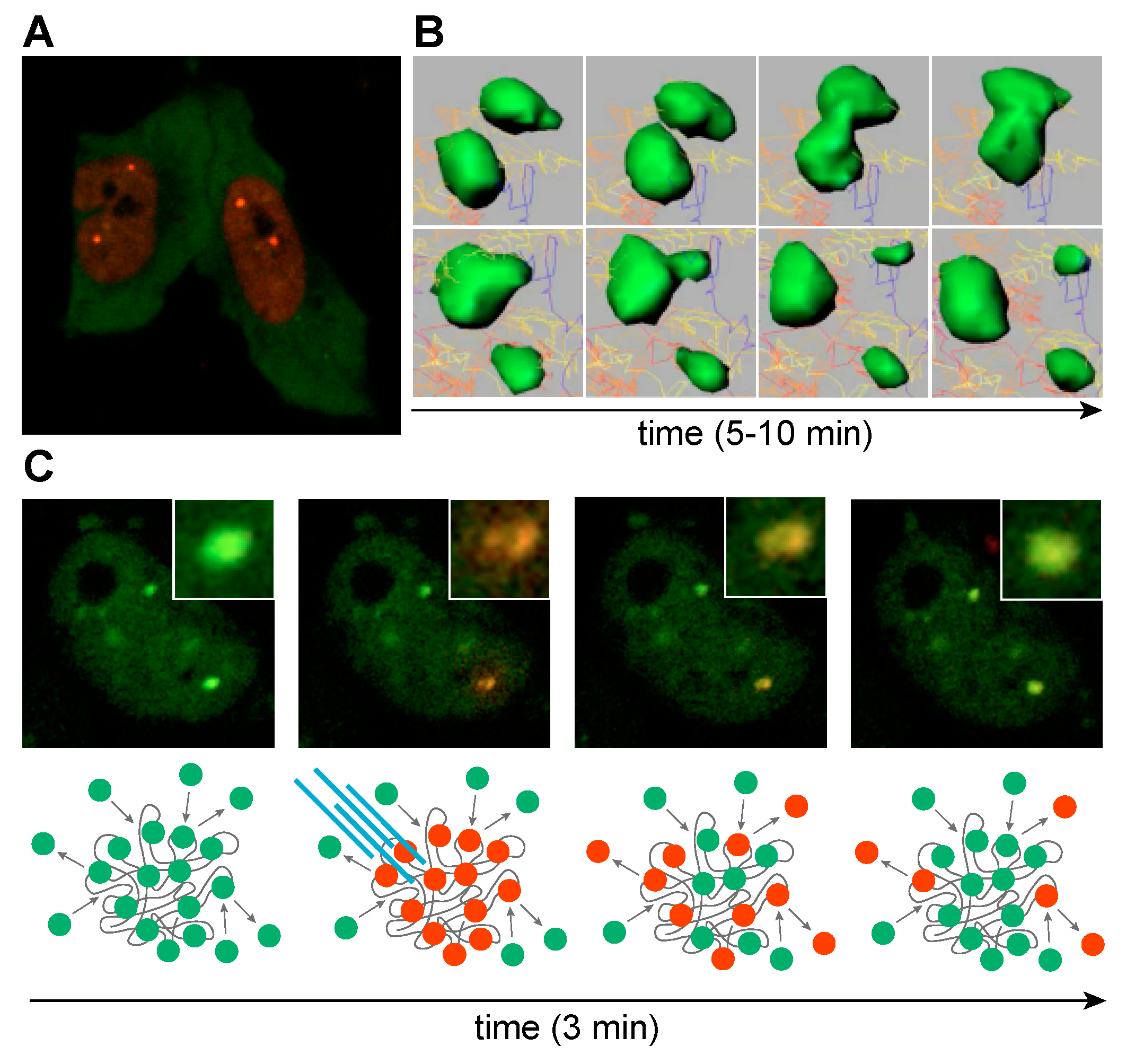{kind=link}
Abstract
:1. Introduction
2. Is Myotonic Dystrophy Caused by Haploinsufficiency?
3. DM1 and DM2 Are RNA-mediated Disorders
4. MBNL Sequestration and Loss-of-function in DM1 and DM2
5. C(C)UGexp RNA-MBNL Interactions in RNA Foci Formation
6. Emerging Roles for RNA-RNA Multivalent Interactions in DM Disease
7. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 3’UTR | 3’ untranslated region |
| 5’UTR | 5’ untranslated region |
| ALS/FTD | Amyotrophic lateral sclerosis and frontotemporal dementia |
| C(C)UGexp | C(C)UG repeat expansion |
| CDM | Congenital myotonic dystrophy |
| DM1 | Myotonic dystrophy type 1 |
| DM2 | Myotonic dystrophy type 2 |
| FECD | Fuchs endothelial corneal dystrophy |
| FXTAS | Fragile X-associated tremor/ataxia syndrome |
| MBNL | Muscleblind-like |
| RAN | repeat-associated non-AUG |
| RBP | RNA-binding protein |
| RNP | Ribonucleoprotein |
| SCA | Spinocerebellar ataxia |
| STR | Short tandem repeat |
| ZnF | Zinc finger |
References
- Bourque, G.; Burns, K.H.; Gehring, M.; Gorbunova, V.; Seluanov, A.; Hammell, M.; Imbeault, M.; Izsvak, Z.; Levin, H.L.; Macfarlan, T.S.; et al. Ten things you should know about transposable elements. Genome. Biol. 2018, 19, 199. [Google Scholar] [CrossRef] [PubMed]
- Padeken, J.; Zeller, P.; Gasser, S.M. Repeat DNA in genome organization and stability. Curr. Opin. Genet. Dev. 2015, 31, 12–19. [Google Scholar] [CrossRef] [PubMed]
- McGinty, R.J.; Mirkin, S.M. Cis- and trans-modifiers of repeat expansions: Blending model systems with human genetics. Trends Genet. 2018, 34, 448–465. [Google Scholar] [CrossRef] [PubMed]
- López Castel, A.; Cleary, J.D.; Pearson, C.E. Repeat instability as the basis for human diseases and as a potential target for therapy. Nat. Rev. Mol. Cell Biol 2010, 11, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Iyer, R.R.; Pluciennik, A.; Napierala, M.; Wells, R.D. DNA triplet repeat expansion and mismatch repair. Annu Rev. Biochem 2015, 84, 199–226. [Google Scholar] [CrossRef] [PubMed]
- LaCroix, A.J.; Stabley, D.; Sahraoui, R.; Adam, M.P.; Mehaffey, M.; Kernan, K.; Myers, C.T.; Fagerstrom, C.; Anadiotis, G.; Akkari, Y.M.; et al. Ggc repeat expansion and exon 1 methylation of xylt1 is a common pathogenic variant in baratela-scott syndrome. Am. J. Hum. Genet. 2019, 104, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Lalioti, M.D.; Mirotsou, M.; Buresi, C.; Peitsch, M.C.; Rossier, C.; Ouazzani, R.; Baldy-Moulinier, M.; Bottani, A.; Malafosse, A.; Antonarakis, S.E. Identification of mutations in cystatin b, the gene responsible for the unverricht-lundborg type of progressive myoclonus epilepsy (epm1). Am. J. Hum. Genet. 1997, 60, 342–351. [Google Scholar]
- van Kuilenburg, A.B.P.; Tarailo-Graovac, M.; Richmond, P.A.; Drögemöller, B.I.; Pouladi, M.A.; Leen, R.; Brand-Arzamendi, K.; Dobritzsch, D.; Dolzhenko, E.; Eberle, M.A.; et al. Glutaminase deficiency caused by short tandem repeat expansion in. N Engl J. Med. 2019, 380, 1433–1441. [Google Scholar] [CrossRef]
- Holmes, S.E.; O’Hearn, E.E.; McInnis, M.G.; Gorelick-Feldman, D.A.; Kleiderlein, J.J.; Callahan, C.; Kwak, N.G.; Ingersoll-Ashworth, R.G.; Sherr, M.; Sumner, A.J.; et al. Expansion of a novel cag trinucleotide repeat in the 5’ region of ppp2r2b is associated with sca12. Nat. Genet. 1999, 23, 391–392. [Google Scholar] [CrossRef]
- Gray, S.J.; Gerhardt, J.; Doerfler, W.; Small, L.E.; Fanning, E. An origin of DNA replication in the promoter region of the human fragile x mental retardation (fmr1) gene. Mol. Cell Biol. 2007, 27, 426–437. [Google Scholar] [CrossRef]
- Barasoain, M.; Barrenetxea, G.; Huerta, I.; Télez, M.; Criado, B.; Arrieta, I. Study of the genetic etiology of primary ovarian insufficiency: Fmr1 gene. Genes (Basel) 2016, 7, 123. [Google Scholar] [CrossRef] [PubMed]
- Knight, S.J.; Flannery, A.V.; Hirst, M.C.; Campbell, L.; Christodoulou, Z.; Phelps, S.R.; Pointon, J.; Middleton-Price, H.R.; Barnicoat, A.; Pembrey, M.E. Trinucleotide repeat amplification and hypermethylation of a cpg island in fraxe mental retardation. Cell 1993, 74, 127–134. [Google Scholar] [CrossRef]
- Parrish, J.E.; Oostra, B.A.; Verkerk, A.J.; Richards, C.S.; Reynolds, J.; Spikes, A.S.; Shaffer, L.G.; Nelson, D.L. Isolation of a gcc repeat showing expansion in fraxf, a fragile site distal to fraxa and fraxe. Nat. Genet. 1994, 8, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Metsu, S.; Rooms, L.; Rainger, J.; Taylor, M.S.; Bengani, H.; Wilson, D.I.; Chilamakuri, C.S.; Morrison, H.; Vandeweyer, G.; Reyniers, E.; et al. Fra2a is a cgg repeat expansion associated with silencing of aff3. PLoS Genet. 2014, 10, e1004242. [Google Scholar] [CrossRef]
- Metsu, S.; Rainger, J.K.; Debacker, K.; Bernhard, B.; Rooms, L.; Grafodatskaya, D.; Weksberg, R.; Fombonne, E.; Taylor, M.S.; Scherer, S.W.; et al. A cgg-repeat expansion mutation in znf713 causes fra7a: Association with autistic spectrum disorder in two families. Hum. Mutat 2014, 35, 1295–1300. [Google Scholar] [PubMed]
- Sarafidou, T.; Kahl, C.; Martinez-Garay, I.; Mangelsdorf, M.; Gesk, S.; Baker, E.; Kokkinaki, M.; Talley, P.; Maltby, E.L.; French, L.; et al. Folate-sensitive fragile site fra10a is due to an expansion of a cgg repeat in a novel gene, fra10ac1, encoding a nuclear protein. Genomics 2004, 84, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Debacker, K.; Winnepenninckx, B.; Longman, C.; Colgan, J.; Tolmie, J.; Murray, R.; van Luijk, R.; Scheers, S.; Fitzpatrick, D.; Kooy, F. The molecular basis of the folate-sensitive fragile site fra11a at 11q13. Cytogenet Genome Res. 2007, 119, 9–14. [Google Scholar] [CrossRef]
- Jones, C.; Penny, L.; Mattina, T.; Yu, S.; Baker, E.; Voullaire, L.; Langdon, W.Y.; Sutherland, G.R.; Richards, R.I.; Tunnacliffe, A. Association of a chromosome deletion syndrome with a fragile site within the proto-oncogene cbl2. Nature 1995, 376, 145–149. [Google Scholar] [CrossRef]
- Winnepenninckx, B.; Debacker, K.; Ramsay, J.; Smeets, D.; Smits, A.; FitzPatrick, D.R.; Kooy, R.F. Cgg-repeat expansion in the dip2b gene is associated with the fragile site fra12a on chromosome 12q13.1. Am. J. Hum. Genet. 2007, 80, 221–231. [Google Scholar] [CrossRef]
- Nancarrow, J.K.; Kremer, E.; Holman, K.; Eyre, H.; Doggett, N.A.; Le Paslier, D.; Callen, D.F.; Sutherland, G.R.; Richards, R.I. Implications of fra16a structure for the mechanism of chromosomal fragile site genesis. Science 1994, 264, 1938–1941. [Google Scholar] [CrossRef]
- Tian, Y.; Wang, J.L.; Huang, W.; Zeng, S.; Jiao, B.; Liu, Z.; Chen, Z.; Li, Y.; Wang, Y.; Min, H.X.; et al. Expansion of human-specific ggc repeat in neuronal intranuclear inclusion disease-related disorders. Am. J. Hum. Genet. 2019, 105, 166–176. [Google Scholar] [CrossRef] [PubMed]
- Renton, A.E.; Majounie, E.; Waite, A.; Simón-Sánchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A hexanucleotide repeat expansion in c9orf72 is the cause of chromosome 9p21-linked als-ftd. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef] [PubMed]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded ggggcc hexanucleotide repeat in noncoding region of c9orf72 causes chromosome 9p-linked ftd and als. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Ishiura, H.; Doi, K.; Mitsui, J.; Yoshimura, J.; Matsukawa, M.K.; Fujiyama, A.; Toyoshima, Y.; Kakita, A.; Takahashi, H.; Suzuki, Y.; et al. Expansions of intronic tttca and tttta repeats in benign adult familial myoclonic epilepsy. Nat. Genet. 2018, 50, 581–590. [Google Scholar] [CrossRef] [PubMed]
- Cortese, A.; Simone, R.; Sullivan, R.; Vandrovcova, J.; Tariq, H.; Yau, W.Y.; Humphrey, J.; Jaunmuktane, Z.; Sivakumar, P.; Polke, J.; et al. Biallelic expansion of an intronic repeat in rfc1 is a common cause of late-onset ataxia. Nat. Genet. 2019, 51, 649–658. [Google Scholar] [CrossRef]
- Liquori, C.L.; Ricker, K.; Moseley, M.L.; Jacobsen, J.F.; Kress, W.; Naylor, S.L.; Day, J.W.; Ranum, L.P. Myotonic dystrophy type 2 caused by a cctg expansion in intron 1 of znf9. Science 2001, 293, 864–867. [Google Scholar] [CrossRef]
- Wieben, E.D.; Aleff, R.A.; Tosakulwong, N.; Butz, M.L.; Highsmith, W.E.; Edwards, A.O.; Baratz, K.H. A common trinucleotide repeat expansion within the transcription factor 4 (tcf4, e2-2) gene predicts fuchs corneal dystrophy. PLoS ONE 2012, 7, e49083. [Google Scholar] [CrossRef]
- Campuzano, V.; Montermini, L.; Moltò, M.D.; Pianese, L.; Cossée, M.; Cavalcanti, F.; Monros, E.; Rodius, F.; Duclos, F.; Monticelli, A.; et al. Friedreich’s ataxia: Autosomal recessive disease caused by an intronic gaa triplet repeat expansion. Science 1996, 271, 1423–1427. [Google Scholar] [CrossRef]
- Matsuura, T.; Yamagata, T.; Burgess, D.L.; Rasmussen, A.; Grewal, R.P.; Watase, K.; Khajavi, M.; McCall, A.E.; Davis, C.F.; Zu, L.; et al. Large expansion of the attct pentanucleotide repeat in spinocerebellar ataxia type 10. Nat. Genet. 2000, 26, 191–194. [Google Scholar] [CrossRef]
- Sato, N.; Amino, T.; Kobayashi, K.; Asakawa, S.; Ishiguro, T.; Tsunemi, T.; Takahashi, M.; Matsuura, T.; Flanigan, K.M.; Iwasaki, S.; et al. Spinocerebellar ataxia type 31 is associated with "inserted" penta-nucleotide repeats containing (tggaa)n. Am. J. Hum. Genet. 2009, 85, 544–557. [Google Scholar] [CrossRef]
- Kobayashi, H.; Abe, K.; Matsuura, T.; Ikeda, Y.; Hitomi, T.; Akechi, Y.; Habu, T.; Liu, W.; Okuda, H.; Koizumi, A. Expansion of intronic ggcctg hexanucleotide repeat in nop56 causes sca36, a type of spinocerebellar ataxia accompanied by motor neuron involvement. Am. J. Hum. Genet. 2011, 89, 121–130. [Google Scholar] [CrossRef]
- Seixas, A.I.; Loureiro, J.R.; Costa, C.; Ordóñez-Ugalde, A.; Marcelino, H.; Oliveira, C.L.; Loureiro, J.L.; Dhingra, A.; Brandão, E.; Cruz, V.T.; et al. A pentanucleotide atttc repeat insertion in the non-coding region of dab1, mapping to sca37, causes spinocerebellar ataxia. Am. J. Hum. Genet. 2017, 101, 87–103. [Google Scholar] [CrossRef] [PubMed]
- Bragg, D.C.; Mangkalaphiban, K.; Vaine, C.A.; Kulkarni, N.J.; Shin, D.; Yadav, R.; Dhakal, J.; Ton, M.L.; Cheng, A.; Russo, C.T.; et al. Disease onset in x-linked dystonia-parkinsonism correlates with expansion of a hexameric repeat within an sva retrotransposon in. Proc. Natl. Acad. Sci. USA 2017, 114, E11020–E11028. [Google Scholar] [CrossRef] [PubMed]
- Koide, R.; Ikeuchi, T.; Onodera, O.; Tanaka, H.; Igarashi, S.; Endo, K.; Takahashi, H.; Kondo, R.; Ishikawa, A.; Hayashi, T. Unstable expansion of cag repeat in hereditary dentatorubral-pallidoluysian atrophy (drpla). Nat. Genet. 1994, 6, 9–13. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, M.E.; Ambrose, C.M.; Duyao, M.P.; Myers, R.H.; Lin, C.; Srinidhi, L.; Barnes, G.; Taylor, S.A.; James, M.; Groot, N.; et al. A novel gene containing a trinucleotide repeat that is expanded and unstable on huntington’s disease chromosomes. The huntington’s disease collaborative research group. Cell 1993, 72, 971–983. [Google Scholar] [CrossRef]
- La Spada, A.R.; Wilson, E.M.; Lubahn, D.B.; Harding, A.E.; Fischbeck, K.H. Androgen receptor gene mutations in x-linked spinal and bulbar muscular atrophy. Nature 1991, 352, 77–79. [Google Scholar] [CrossRef]
- Orr, H.T.; Chung, M.Y.; Banfi, S.; Kwiatkowski, T.J.; Servadio, A.; Beaudet, A.L.; McCall, A.E.; Duvick, L.A.; Ranum, L.P.; Zoghbi, H.Y. Expansion of an unstable trinucleotide cag repeat in spinocerebellar ataxia type 1. Nat. Genet. 1993, 4, 221–226. [Google Scholar] [CrossRef]
- Sanpei, K.; Takano, H.; Igarashi, S.; Sato, T.; Oyake, M.; Sasaki, H.; Wakisaka, A.; Tashiro, K.; Ishida, Y.; Ikeuchi, T.; et al. Identification of the spinocerebellar ataxia type 2 gene using a direct identification of repeat expansion and cloning technique, direct. Nat. Genet. 1996, 14, 277–284. [Google Scholar] [CrossRef]
- Stevanin, G.; Le Guern, E.; Ravisé, N.; Chneiweiss, H.; Dürr, A.; Cancel, G.; Vignal, A.; Boch, A.L.; Ruberg, M.; Penet, C. A third locus for autosomal dominant cerebellar ataxia type i maps to chromosome 14q24.3-qter: Evidence for the existence of a fourth locus. Am. J. Hum. Genet. 1994, 54, 11–20. [Google Scholar]
- Jodice, C.; Mantuano, E.; Veneziano, L.; Trettel, F.; Sabbadini, G.; Calandriello, L.; Francia, A.; Spadaro, M.; Pierelli, F.; Salvi, F.; et al. Episodic ataxia type 2 (ea2) and spinocerebellar ataxia type 6 (sca6) due to cag repeat expansion in the cacna1a gene on chromosome 19p. Hum. Mol. Genet. 1997, 6, 1973–1978. [Google Scholar] [CrossRef]
- David, G.; Dürr, A.; Stevanin, G.; Cancel, G.; Abbas, N.; Benomar, A.; Belal, S.; Lebre, A.S.; Abada-Bendib, M.; Grid, D.; et al. Molecular and clinical correlations in autosomal dominant cerebellar ataxia with progressive macular dystrophy (sca7). Hum. Mol. Genet. 1998, 7, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Koob, M.D.; Moseley, M.L.; Schut, L.J.; Benzow, K.A.; Bird, T.D.; Day, J.W.; Ranum, L.P. An untranslated ctg expansion causes a novel form of spinocerebellar ataxia (sca8). Nat. Genet. 1999, 21, 379–384. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Jeong, S.Y.; Uchihara, T.; Anno, M.; Nagashima, K.; Nagashima, T.; Ikeda, S.; Tsuji, S.; Kanazawa, I. Sca17, a novel autosomal dominant cerebellar ataxia caused by an expanded polyglutamine in tata-binding protein. Hum. Mol. Genet. 2001, 10, 1441–1448. [Google Scholar] [CrossRef] [PubMed]
- De Baere, E.; Beysen, D.; Oley, C.; Lorenz, B.; Cocquet, J.; De Sutter, P.; Devriendt, K.; Dixon, M.; Fellous, M.; Fryns, J.P.; et al. Foxl2 and bpes: Mutational hotspots, phenotypic variability and revision of the genotype-phenotype correlation. Am. J. Hum. Genet. 2003, 72, 478–487. [Google Scholar] [CrossRef]
- Mundlos, S.; Otto, F.; Mundlos, C.; Mulliken, J.B.; Aylsworth, A.S.; Albright, S.; Lindhout, D.; Cole, W.G.; Henn, W.; Knoll, J.H.; et al. Mutations involving the transcription factor cbfa1 cause cleidocranial dysplasia. Cell 1997, 89, 773–779. [Google Scholar] [CrossRef]
- Amiel, J.; Laudier, B.; Attié-Bitach, T.; Trang, H.; de Pontual, L.; Gener, B.; Trochet, D.; Etchevers, H.; Ray, P.; Simonneau, M.; et al. Polyalanine expansion and frameshift mutations of the paired-like homeobox gene phox2b in congenital central hypoventilation syndrome. Nat. Genet. 2003, 33, 459–461. [Google Scholar] [CrossRef]
- Goodman, F.R.; Bacchelli, C.; Brady, A.F.; Brueton, L.A.; Fryns, J.P.; Mortlock, D.P.; Innis, J.W.; Holmes, L.B.; Donnenfeld, A.E.; Feingold, M.; et al. Novel hoxa13 mutations and the phenotypic spectrum of hand-foot-genital syndrome. Am. J. Hum. Genet. 2000, 67, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.A.; Warburton, D.; Brown, L.Y.; Yu, C.Y.; Roeder, E.R.; Stengel-Rutkowski, S.; Hennekam, R.C.; Muenke, M. Holoprosencephaly due to mutations in zic2, a homologue of drosophila odd-paired. Nat. Genet. 1998, 20, 180–183. [Google Scholar] [CrossRef]
- Brais, B.; Bouchard, J.P.; Xie, Y.G.; Rochefort, D.L.; Chrétien, N.; Tomé, F.M.; Lafrenière, R.G.; Rommens, J.M.; Uyama, E.; Nohira, O.; et al. Short gcg expansions in the pabp2 gene cause oculopharyngeal muscular dystrophy. Nat. Genet. 1998, 18, 164–167. [Google Scholar] [CrossRef] [PubMed]
- Muragaki, Y.; Mundlos, S.; Upton, J.; Olsen, B.R. Altered growth and branching patterns in synpolydactyly caused by mutations in hoxd13. Science 1996, 272, 548–551. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Das, S.; Petras, K.; Kitamura, K.; Morohashi, K.; Abuelo, D.N.; Barr, M.; Bonneau, D.; Brady, A.F.; Carpenter, N.J.; et al. Mutations of arx are associated with striking pleiotropy and consistent genotype-phenotype correlation. Hum. Mutat 2004, 23, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Strømme, P.; Mangelsdorf, M.E.; Shaw, M.A.; Lower, K.M.; Lewis, S.M.; Bruyere, H.; Lütcherath, V.; Gedeon, A.K.; Wallace, R.H.; Scheffer, I.E.; et al. Mutations in the human ortholog of aristaless cause x-linked mental retardation and epilepsy. Nat. Genet. 2002, 30, 441–445. [Google Scholar] [CrossRef] [PubMed]
- Laumonnier, F.; Ronce, N.; Hamel, B.C.; Thomas, P.; Lespinasse, J.; Raynaud, M.; Paringaux, C.; Van Bokhoven, H.; Kalscheuer, V.; Fryns, J.P.; et al. Transcription factor sox3 is involved in x-linked mental retardation with growth hormone deficiency. Am. J. Hum. Genet. 2002, 71, 1450–1455. [Google Scholar] [CrossRef] [PubMed]
- Délot, E.; King, L.M.; Briggs, M.D.; Wilcox, W.R.; Cohn, D.H. Trinucleotide expansion mutations in the cartilage oligomeric matrix protein (comp) gene. Hum. Mol. Genet. 1999, 8, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Mahadevan, M.; Tsilfidis, C.; Sabourin, L.; Shutler, G.; Amemiya, C.; Jansen, G.; Neville, C.; Narang, M.; Barceló, J.; O’Hoy, K. Myotonic dystrophy mutation: An unstable ctg repeat in the 3’ untranslated region of the gene. Science 1992, 255, 1253–1255. [Google Scholar] [CrossRef] [PubMed]
- Holmes, S.E.; O’Hearn, E.; Rosenblatt, A.; Callahan, C.; Hwang, H.S.; Ingersoll-Ashworth, R.G.; Fleisher, A.; Stevanin, G.; Brice, A.; Potter, N.T.; et al. A repeat expansion in the gene encoding junctophilin-3 is associated with huntington disease-like 2. Nat. Genet. 2001, 29, 377–378. [Google Scholar] [CrossRef] [PubMed]
- Neueder, A.; Landles, C.; Ghosh, R.; Howland, D.; Myers, R.H.; Faull, R.L.M.; Tabrizi, S.J.; Bates, G.P. The pathogenic exon 1 htt protein is produced by incomplete splicing in huntington’s disease patients. Sci. Rep. 2017, 7, 1307. [Google Scholar] [CrossRef]
- Sznajder, Ł.J.; Thomas, J.D.; Carrell, E.M.; Reid, T.; McFarland, K.N.; Cleary, J.D.; Oliveira, R.; Nutter, C.A.; Bhatt, K.; Sobczak, K.; et al. Intron retention induced by microsatellite expansions as a disease biomarker. Proc. Natl. Acad. Sci. USA 2018, 115, 4234–4239. [Google Scholar] [CrossRef] [Green Version]
- Batra, R.; Charizanis, K.; Swanson, M.S. Partners in crime: Bidirectional transcription in unstable microsatellite disease. Hum. Mol. Genet. 2010, 19, R77–R82. [Google Scholar] [CrossRef]
- Mohan, A.; Goodwin, M.; Swanson, M.S. Rna-protein interactions in unstable microsatellite diseases. Brain Res. 2014, 1584, 3–14. [Google Scholar] [CrossRef]
- Szlachcic, W.J.; Switonski, P.M.; Kurkowiak, M.; Wiatr, K.; Figiel, M. Mouse polyq database: A new online resource for research using mouse models of neurodegenerative diseases. Mol. Brain 2015, 8, 69. [Google Scholar] [CrossRef] [PubMed]
- Amiel, J.; Trochet, D.; Clément-Ziza, M.; Munnich, A.; Lyonnet, S. Polyalanine expansions in human. Hum. Mol. Genet. 2004, 13, R235–R243. [Google Scholar] [CrossRef] [Green Version]
- Cleary, J.D.; Pattamatta, A.; Ranum, L.P.W. Repeat-associated non-atg (ran) translation. J. Biol Chem 2018, 293, 16127–16141. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.P.; Brown, R.H.; Cleveland, D.W. Decoding als: From genes to mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Polak, U.; Li, Y.; Butler, J.S.; Napierala, M. Alleviating gaa repeat induced transcriptional silencing of the friedreich’s ataxia gene during somatic cell reprogramming. Stem Cells Dev. 2016, 25, 1788–1800. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.S.; Wu, H.; Krzisch, M.; Wu, X.; Graef, J.; Muffat, J.; Hnisz, D.; Li, C.H.; Yuan, B.; Xu, C.; et al. Rescue of fragile x syndrome neurons by DNA methylation editing of the fmr1 gene. Cell 2018, 172, 979–992.e976. [Google Scholar] [CrossRef]
- Leroux, A.E.; Schulze, J.O.; Biondi, R.M. Agc kinases, mechanisms of regulation and innovative drug development. Semin Cancer Biol. 2018, 48, 1–17. [Google Scholar] [CrossRef]
- Oude Ophuis, R.J.; Mulders, S.A.; van Herpen, R.E.; van de Vorstenbosch, R.; Wieringa, B.; Wansink, D.G. Dmpk protein isoforms are differentially expressed in myogenic and neural cell lineages. Muscle Nerve 2009, 40, 545–555. [Google Scholar] [CrossRef]
- Benhalevy, D.; Gupta, S.K.; Danan, C.H.; Ghosal, S.; Sun, H.W.; Kazemier, H.G.; Paeschke, K.; Hafner, M.; Juranek, S.A. The human cchc-type zinc finger nucleic acid-binding protein binds g-rich elements in target mrna coding sequences and promotes translation. Cell Rep. 2017, 18, 2979–2990. [Google Scholar] [CrossRef]
- Reddy, S.; Smith, D.B.; Rich, M.M.; Leferovich, J.M.; Reilly, P.; Davis, B.M.; Tran, K.; Rayburn, H.; Bronson, R.; Cros, D.; et al. Mice lacking the myotonic dystrophy protein kinase develop a late onset progressive myopathy. Nat. Genet. 1996, 13, 325–335. [Google Scholar] [CrossRef]
- Jansen, G.; Groenen, P.J.; Bachner, D.; Jap, P.H.; Coerwinkel, M.; Oerlemans, F.; van den Broek, W.; Gohlsch, B.; Pette, D.; Plomp, J.J.; et al. Abnormal myotonic dystrophy protein kinase levels produce only mild myopathy in mice. Nat. Genet. 1996, 13, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Carrell, S.T.; Carrell, E.M.; Auerbach, D.; Pandey, S.K.; Bennett, C.F.; Dirksen, R.T.; Thornton, C.A. Dmpk gene deletion or antisense knockdown does not compromise cardiac or skeletal muscle function in mice. Hum. Mol. Genet. 2016, 25, 4328–4338. [Google Scholar] [CrossRef]
- Santoro, M.; Fontana, L.; Maiorca, F.; Centofanti, F.; Massa, R.; Silvestri, G.; Novelli, G.; Botta, A. Expanded [cctg]n repetitions are not associated with abnormal methylation at the cnbp locus in myotonic dystrophy type 2 (dm2) patients. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 917–924. [Google Scholar] [CrossRef] [PubMed]
- Margolis, J.M.; Schoser, B.G.; Moseley, M.L.; Day, J.W.; Ranum, L.P. Dm2 intronic expansions: Evidence for ccug accumulation without flanking sequence or effects on znf9 mrna processing or protein expression. Hum. Mol. Genet. 2006, 15, 1808–1815. [Google Scholar] [CrossRef]
- Botta, A.; Caldarola, S.; Vallo, L.; Bonifazi, E.; Fruci, D.; Gullotta, F.; Massa, R.; Novelli, G.; Loreni, F. Effect of the [cctg]n repeat expansion on znf9 expression in myotonic dystrophy type ii (dm2). Biochim. Biophys. Acta 2006, 1762, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Massa, R.; Panico, M.B.; Caldarola, S.; Fusco, F.R.; Sabatelli, P.; Terracciano, C.; Botta, A.; Novelli, G.; Bernardi, G.; Loreni, F. The myotonic dystrophy type 2 (dm2) gene product zinc finger protein 9 (znf9) is associated with sarcomeres and normally localized in dm2 patients’ muscles. Neuropathol Appl. Neurobiol. 2010, 36, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Raheem, O.; Olufemi, S.E.; Bachinski, L.L.; Vihola, A.; Sirito, M.; Holmlund-Hampf, J.; Haapasalo, H.; Li, Y.P.; Udd, B.; Krahe, R. Mutant (cctg)n expansion causes abnormal expression of zinc finger protein 9 (znf9) in myotonic dystrophy type 2. Am. J. Pathol. 2010, 177, 3025–3036. [Google Scholar] [CrossRef]
- Huichalaf, C.; Schoser, B.; Schneider-Gold, C.; Jin, B.; Sarkar, P.; Timchenko, L. Reduction of the rate of protein translation in patients with myotonic dystrophy 2. J. Neurosci 2009, 29, 9042–9049. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, R.; Hamel, F.; Beaulieu, D.; Patry, L.; Haineault, C.; Tarnopolsky, M.; Schoser, B.; Puymirat, J. Absence of a differentiation defect in muscle satellite cells from dm2 patients. Neurobiol. Dis. 2009, 36, 181–190. [Google Scholar] [CrossRef]
- Wei, C.; Stock, L.; Schneider-Gold, C.; Sommer, C.; Timchenko, N.A.; Timchenko, L. Reduction of cellular nucleic acid binding protein encoded by a myotonic dystrophy type 2 gene causes muscle atrophy. Mol. Cell Biol. 2018, 38. [Google Scholar] [CrossRef] [PubMed]
- Mankodi, A.; Takahashi, M.P.; Jiang, H.; Beck, C.L.; Bowers, W.J.; Moxley, R.T.; Cannon, S.C.; Thornton, C.A. Expanded cug repeats trigger aberrant splicing of clc-1 chloride channel pre-mrna and hyperexcitability of skeletal muscle in myotonic dystrophy. Mol. Cell 2002, 10, 35–44. [Google Scholar] [CrossRef]
- Charlet-B, N.; Savkur, R.S.; Singh, G.; Philips, A.V.; Grice, E.A.; Cooper, T.A. Loss of the muscle-specific chloride channel in type 1 myotonic dystrophy due to misregulated alternative splicing. Mol. Cell 2002, 10, 45–53. [Google Scholar] [CrossRef]
- Wheeler, T.M.; Sobczak, K.; Lueck, J.D.; Osborne, R.J.; Lin, X.; Dirksen, R.T.; Thornton, C.A. Reversal of rna dominance by displacement of protein sequestered on triplet repeat rna. Science 2009, 325, 336–339. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.Z.; Yarotskyy, V.; Wei, L.; Sobczak, K.; Nakamori, M.; Eichinger, K.; Moxley, R.T.; Dirksen, R.T.; Thornton, C.A. Muscle weakness in myotonic dystrophy associated with misregulated splicing and altered gating of ca(v)1.1 calcium channel. Hum. Mol. Genet. 2012, 21, 1312–1324. [Google Scholar] [CrossRef] [PubMed]
- Fugier, C.; Klein, A.F.; Hammer, C.; Vassilopoulos, S.; Ivarsson, Y.; Toussaint, A.; Tosch, V.; Vignaud, A.; Ferry, A.; Messaddeq, N.; et al. Misregulated alternative splicing of bin1 is associated with t tubule alterations and muscle weakness in myotonic dystrophy. Nat. Med. 2011, 17, 720–725. [Google Scholar] [CrossRef] [PubMed]
- Rau, F.; Lainé, J.; Ramanoudjame, L.; Ferry, A.; Arandel, L.; Delalande, O.; Jollet, A.; Dingli, F.; Lee, K.Y.; Peccate, C.; et al. Abnormal splicing switch of dmd’s penultimate exon compromises muscle fibre maintenance in myotonic dystrophy. Nat. Commun 2015, 6, 7205. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Wang, H.; Wei, B.; Guo, Y.; Gu, L.; Yang, Z.; Zhang, Q.; Wu, Y.; Yuan, Q.; Zhao, G.; et al. Cug-bp1 regulates ryr1 asi alternative splicing in skeletal muscle atrophy. Sci. Rep. 2015, 5, 16083. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Cooper, T.A. Reexpression of pyruvate kinase m2 in type 1 myofibers correlates with altered glucose metabolism in myotonic dystrophy. Proc. Natl. Acad. Sci. USA 2013, 110, 13570–13575. [Google Scholar] [CrossRef] [PubMed]
- Freyermuth, F.; Rau, F.; Kokunai, Y.; Linke, T.; Sellier, C.; Nakamori, M.; Kino, Y.; Arandel, L.; Jollet, A.; Thibault, C.; et al. Splicing misregulation of scn5a contributes to cardiac-conduction delay and heart arrhythmia in myotonic dystrophy. Nat. Commun. 2016, 7, 11067. [Google Scholar] [CrossRef]
- Pang, P.D.; Alsina, K.M.; Cao, S.; Koushik, A.B.; Wehrens, X.H.T.; Cooper, T.A. Crispr -mediated expression of the fetal scn5a isoform in adult mice causes conduction defects and arrhythmias. J. Am. Heart Assoc. 2018, 7, e010393. [Google Scholar] [CrossRef]
- Philips, A.V.; Timchenko, L.T.; Cooper, T.A. Disruption of splicing regulated by a cug-binding protein in myotonic dystrophy. Science 1998, 280, 737–741. [Google Scholar] [CrossRef] [PubMed]
- Sergeant, N.; Sablonnière, B.; Schraen-Maschke, S.; Ghestem, A.; Maurage, C.A.; Wattez, A.; Vermersch, P.; Delacourte, A. Dysregulation of human brain microtubule-associated tau mrna maturation in myotonic dystrophy type 1. Hum. Mol. Genet. 2001, 10, 2143–2155. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Mankodi, A.; Swanson, M.S.; Moxley, R.T.; Thornton, C.A. Myotonic dystrophy type 1 is associated with nuclear foci of mutant rna, sequestration of muscleblind proteins and deregulated alternative splicing in neurons. Hum. Mol. Genet. 2004, 13, 3079–3088. [Google Scholar] [CrossRef] [PubMed]
- Savkur, R.S.; Philips, A.V.; Cooper, T.A. Aberrant regulation of insulin receptor alternative splicing is associated with insulin resistance in myotonic dystrophy. Nat. Genet. 2001, 29, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Renna, L.V.; Bosè, F.; Iachettini, S.; Fossati, B.; Saraceno, L.; Milani, V.; Colombo, R.; Meola, G.; Cardani, R. Receptor and post-receptor abnormalities contribute to insulin resistance in myotonic dystrophy type 1 and type 2 skeletal muscle. PLoS ONE 2017, 12, e0184987. [Google Scholar] [CrossRef] [PubMed]
- Rau, F.; Freyermuth, F.; Fugier, C.; Villemin, J.P.; Fischer, M.C.; Jost, B.; Dembele, D.; Gourdon, G.; Nicole, A.; Duboc, D.; et al. Misregulation of mir-1 processing is associated with heart defects in myotonic dystrophy. Nat. Struct Mol. Biol. 2011, 18, 840–845. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Liang, Y.; Deng, W.; Shimizu, K.; Ashique, A.M.; Li, E.; Li, Y.P. The zinc-finger protein cnbp is required for forebrain formation in the mouse. Development 2003, 130, 1367–1379. [Google Scholar] [CrossRef]
- Chen, W.; Wang, Y.; Abe, Y.; Cheney, L.; Udd, B.; Li, Y.P. Haploinsuffciency for znf9 in znf9+/- mice is associated with multiorgan abnormalities resembling myotonic dystrophy. J. Mol. Biol 2007, 368, 8–17. [Google Scholar] [CrossRef]
- de Peralta, M.S.; Mouguelar, V.S.; Sdrigotti, M.A.; Ishiy, F.A.; Fanganiello, R.D.; Passos-Bueno, M.R.; Coux, G.; Calcaterra, N.B. Cnbp ameliorates treacher collins syndrome craniofacial anomalies through a pathway that involves redox-responsive genes. Cell Death Dis. 2016, 7, e2397. [Google Scholar] [CrossRef]
- Mankodi, A.; Logigian, E.; Callahan, L.; McClain, C.; White, R.; Henderson, D.; Krym, M.; Thornton, C.A. Myotonic dystrophy in transgenic mice expressing an expanded cug repeat. Science 2000, 289, 1769–1773. [Google Scholar] [CrossRef]
- Handa, V.; Yeh, H.J.; McPhie, P.; Usdin, K. The auucu repeats responsible for spinocerebellar ataxia type 10 form unusual rna hairpins. J. Biol. Chem. 2005, 280, 29340–29345. [Google Scholar] [CrossRef]
- Park, H.; González, À.; Yildirim, I.; Tran, T.; Lohman, J.R.; Fang, P.; Guo, M.; Disney, M.D. Crystallographic and computational analyses of auucu repeating rna that causes spinocerebellar ataxia type 10 (sca10). Biochemistry 2015, 54, 3851–3859. [Google Scholar] [CrossRef] [PubMed]
- Napierała, M.; Krzyzosiak, W.J. Cug repeats present in myotonin kinase rna form metastable “slippery” hairpins. J. Biol. Chem. 1997, 272, 31079–31085. [Google Scholar] [CrossRef] [PubMed]
- Childs-Disney, J.L.; Yildirim, I.; Park, H.; Lohman, J.R.; Guan, L.; Tran, T.; Sarkar, P.; Schatz, G.C.; Disney, M.D. Structure of the myotonic dystrophy type 2 rna and designed small molecules that reduce toxicity. ACS Chem. Biol. 2014, 9, 538–550. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Roland, C.; Sagui, C. Structural and dynamical characterization of DNA and rna quadruplexes obtained from the ggggcc and gggcct hexanucleotide repeats associated with c9ftd/als and sca36 diseases. ACS Chem. Neurosci. 2018, 9, 1104–1117. [Google Scholar] [CrossRef]
- Jansen, G.; Willems, P.; Coerwinkel, M.; Nillesen, W.; Smeets, H.; Vits, L.; Höweler, C.; Brunner, H.; Wieringa, B. Gonosomal mosaicism in myotonic dystrophy patients: Involvement of mitotic events in (ctg)n repeat variation and selection against extreme expansion in sperm. Am. J. Hum. Genet. 1994, 54, 575–585. [Google Scholar]
- Wong, L.J.; Ashizawa, T.; Monckton, D.G.; Caskey, C.T.; Richards, C.S. Somatic heterogeneity of the ctg repeat in myotonic dystrophy is age and size dependent. Am. J. Hum. Genet. 1995, 56, 114–122. [Google Scholar]
- Lia, A.S.; Seznec, H.; Hofmann-Radvanyi, H.; Radvanyi, F.; Duros, C.; Saquet, C.; Blanche, M.; Junien, C.; Gourdon, G. Somatic instability of the ctg repeat in mice transgenic for the myotonic dystrophy region is age dependent but not correlated to the relative intertissue transcription levels and proliferative capacities. Hum. Mol. Genet. 1998, 7, 1285–1291. [Google Scholar] [CrossRef]
- Monckton, D.G.; Coolbaugh, M.I.; Ashizawa, K.T.; Siciliano, M.J.; Caskey, C.T. Hypermutable myotonic dystrophy ctg repeats in transgenic mice. Nat. Genet. 1997, 15, 193–196. [Google Scholar] [CrossRef]
- Thornton, C.A.; Johnson, K.; Moxley, R.T. Myotonic dystrophy patients have larger ctg expansions in skeletal muscle than in leukocytes. Ann. Neurol 1994, 35, 104–107. [Google Scholar] [CrossRef]
- Zatz, M.; Passos-Bueno, M.R.; Cerqueira, A.; Marie, S.K.; Vainzof, M.; Pavanello, R.C. Analysis of the ctg repeat in skeletal muscle of young and adult myotonic dystrophy patients: When does the expansion occur? Hum. Mol. Genet. 1995, 4, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Nakamori, M.; Sobczak, K.; Puwanant, A.; Welle, S.; Eichinger, K.; Pandya, S.; Dekdebrun, J.; Heatwole, C.R.; McDermott, M.P.; Chen, T.; et al. Splicing biomarkers of disease severity in myotonic dystrophy. Ann. Neurol. 2013, 74, 862–872. [Google Scholar] [CrossRef] [PubMed]
- Meola, G.; Cardani, R. Myotonic dystrophy type 2 and modifier genes: An update on clinical and pathomolecular aspects. Neurol. Sci. 2017, 38, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Braz, S.O.; Acquaire, J.; Gourdon, G.; Gomes-Pereira, M. Of mice and men: Advances in the understanding of neuromuscular aspects of myotonic dystrophy. Front. Neurol. 2018, 9, 519. [Google Scholar] [CrossRef] [PubMed]
- Matloka, M.; Klein, A.F.; Rau, F.; Furling, D. Cells of matter-in vitro models for myotonic dystrophy. Front. Neurol. 2018, 9, 361. [Google Scholar] [CrossRef] [PubMed]
- Kuyumcu-Martinez, N.M.; Wang, G.S.; Cooper, T.A. Increased steady-state levels of cugbp1 in myotonic dystrophy 1 are due to pkc-mediated hyperphosphorylation. Mol. Cell 2007, 28, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Salisbury, E.; Schoser, B.; Schneider-Gold, C.; Wang, G.L.; Huichalaf, C.; Jin, B.; Sirito, M.; Sarkar, P.; Krahe, R.; Timchenko, N.A.; et al. Expression of rna ccug repeats dysregulates translation and degradation of proteins in myotonic dystrophy 2 patients. Am. J. Pathol 2009, 175, 748–762. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, M.; Swanson, M.S. Rna-binding protein misregulation in microsatellite expansion disorders. Adv. Exp. Med. Biol. 2014, 825, 353–388. [Google Scholar] [PubMed]
- Brinegar, A.E.; Cooper, T.A. Roles for rna-binding proteins in development and disease. Brain Res. 2016, 1647, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Miller, J.W.; Mankodi, A.; Kanadia, R.N.; Yuan, Y.; Moxley, R.T.; Swanson, M.S.; Thornton, C.A. Failure of mbnl1-dependent post-natal splicing transitions in myotonic dystrophy. Hum. Mol. Genet. 2006, 15, 2087–2097. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.T.; Cody, N.A.; Jog, S.; Biancolella, M.; Wang, T.T.; Treacy, D.J.; Luo, S.; Schroth, G.P.; Housman, D.E.; Reddy, S.; et al. Transcriptome-wide regulation of pre-mrna splicing and mrna localization by muscleblind proteins. Cell 2012, 150, 710–724. [Google Scholar] [CrossRef] [PubMed]
- Batra, R.; Charizanis, K.; Manchanda, M.; Mohan, A.; Li, M.; Finn, D.J.; Goodwin, M.; Zhang, C.; Sobczak, K.; Thornton, C.A.; et al. Loss of mbnl leads to disruption of developmentally regulated alternative polyadenylation in rna-mediated disease. Mol. Cell 2014, 56, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Ashwal-Fluss, R.; Meyer, M.; Pamudurti, N.R.; Ivanov, A.; Bartok, O.; Hanan, M.; Evantal, N.; Memczak, S.; Rajewsky, N.; Kadener, S. Circrna biogenesis competes with pre-mrna splicing. Mol. Cell 2014, 56, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Masuda, A.; Andersen, H.S.; Doktor, T.K.; Okamoto, T.; Ito, M.; Andresen, B.S.; Ohno, K. Cugbp1 and mbnl1 preferentially bind to 3’ utrs and facilitate mrna decay. Sci. Rep. 2012, 2, 209. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, M.; Mohan, A.; Batra, R.; Lee, K.Y.; Charizanis, K.; Fernandez Gomez, F.J.; Eddarkaoui, S.; Sergeant, N.; Buee, L.; Kimura, T.; et al. Mbnl sequestration by toxic rnas and rna misprocessing in the myotonic dystrophy brain. Cell Rep. 2015, 12, 1159–1168. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Cline, M.S.; Osborne, R.J.; Tuttle, D.L.; Clark, T.A.; Donohue, J.P.; Hall, M.P.; Shiue, L.; Swanson, M.S.; Thornton, C.A.; et al. Aberrant alternative splicing and extracellular matrix gene expression in mouse models of myotonic dystrophy. Nat. Struct Mol. Biol 2010, 17, 187–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perbellini, R.; Greco, S.; Sarra-Ferraris, G.; Cardani, R.; Capogrossi, M.C.; Meola, G.; Martelli, F. Dysregulation and cellular mislocalization of specific mirnas in myotonic dystrophy type 1. Neuromuscul Disord 2011, 21, 81–88. [Google Scholar] [CrossRef]
- Kanadia, R.N.; Urbinati, C.R.; Crusselle, V.J.; Luo, D.; Lee, Y.J.; Harrison, J.K.; Oh, S.P.; Swanson, M.S. Developmental expression of mouse muscleblind genes mbnl1, mbnl2 and mbnl3. Gene Expr. Patterns 2003, 3, 459–462. [Google Scholar] [CrossRef]
- Thomas, J.D.; Sznajder, Ł.J.; Bardhi, O.; Aslam, F.N.; Anastasiadis, Z.P.; Scotti, M.M.; Nishino, I.; Nakamori, M.; Wang, E.T.; Swanson, M.S. Disrupted prenatal rna processing and myogenesis in congenital myotonic dystrophy. Genes Dev. 2017, 31, 1122–1133. [Google Scholar] [CrossRef]
- Teplova, M.; Patel, D.J. Structural insights into rna recognition by the alternative-splicing regulator muscleblind-like mbnl1. Nat. Struct Mol. Biol. 2008, 15, 1343–1351. [Google Scholar] [CrossRef]
- Zhang, C.; Lee, K.Y.; Swanson, M.S.; Darnell, R.B. Prediction of clustered rna-binding protein motif sites in the mammalian genome. Nucleic Acids Res. 2013, 41, 6793–6807. [Google Scholar] [CrossRef] [PubMed]
- Goers, E.S.; Purcell, J.; Voelker, R.B.; Gates, D.P.; Berglund, J.A. Mbnl1 binds gc motifs embedded in pyrimidines to regulate alternative splicing. Nucleic Acids Res. 2010, 38, 2467–2484. [Google Scholar] [CrossRef] [PubMed]
- Charizanis, K.; Lee, K.Y.; Batra, R.; Goodwin, M.; Zhang, C.; Yuan, Y.; Shiue, L.; Cline, M.; Scotti, M.M.; Xia, G.; et al. Muscleblind-like 2-mediated alternative splicing in the developing brain and dysregulation in myotonic dystrophy. Neuron 2012, 75, 437–450. [Google Scholar] [CrossRef] [PubMed]
- Taylor, K.; Sznajder, L.J.; Cywoniuk, P.; Thomas, J.D.; Swanson, M.S.; Sobczak, K. Mbnl splicing activity depends on rna binding site structural context. Nucleic Acids Res. 2018, 46, 9119–9133. [Google Scholar] [CrossRef] [PubMed]
- Sznajder, Ł.J.; Michalak, M.; Taylor, K.; Cywoniuk, P.; Kabza, M.; Wojtkowiak-Szlachcic, A.; Matloka, M.; Konieczny, P.; Sobczak, K. Mechanistic determinants of mbnl activity. Nucleic Acids Res. 2016, 44, 10326–10342. [Google Scholar] [CrossRef] [PubMed]
- deLorimier, E.; Coonrod, L.A.; Copperman, J.; Taber, A.; Reister, E.E.; Sharma, K.; Todd, P.K.; Guenza, M.G.; Berglund, J.A. Modifications to toxic cug rnas induce structural stability, rescue mis-splicing in a myotonic dystrophy cell model and reduce toxicity in a myotonic dystrophy zebrafish model. Nucleic Acids Res. 2014, 42, 12768–12778. [Google Scholar] [CrossRef]
- Yuan, Y.; Compton, S.A.; Sobczak, K.; Stenberg, M.G.; Thornton, C.A.; Griffith, J.D.; Swanson, M.S. Muscleblind-like 1 interacts with rna hairpins in splicing target and pathogenic rnas. Nucleic Acids Res. 2007, 35, 5474–5486. [Google Scholar] [CrossRef]
- Warf, M.B.; Berglund, J.A. Mbnl binds similar rna structures in the cug repeats of myotonic dystrophy and its pre-mrna substrate cardiac troponin t. RNA 2007, 13, 2238–2251. [Google Scholar] [CrossRef]
- Cass, D.; Hotchko, R.; Barber, P.; Jones, K.; Gates, D.P.; Berglund, J.A. The four zn fingers of mbnl1 provide a flexible platform for recognition of its rna binding elements. BMC Mol. Biol 2011, 12, 20. [Google Scholar] [CrossRef]
- Joseph, J.T.; Richards, C.S.; Anthony, D.C.; Upton, M.; Perez-Atayde, A.R.; Greenstein, P. Congenital myotonic dystrophy pathology and somatic mosaicism. Neurology 1997, 49, 1457–1460. [Google Scholar] [CrossRef]
- Tsilfidis, C.; MacKenzie, A.E.; Mettler, G.; Barceló, J.; Korneluk, R.G. Correlation between ctg trinucleotide repeat length and frequency of severe congenital myotonic dystrophy. Nat. Genet. 1992, 1, 192–195. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Aleff, R.A.; Soragni, E.; Kalari, K.; Nie, J.; Tang, X.; Davila, J.; Kocher, J.P.; Patel, S.V.; Gottesfeld, J.M.; et al. Rna toxicity and missplicing in the common eye disease fuchs endothelial corneal dystrophy. J. Biol. Chem. 2015, 290, 5979–5990. [Google Scholar] [CrossRef] [PubMed]
- Wieben, E.D.; Aleff, R.A.; Tang, X.; Butz, M.L.; Kalari, K.R.; Highsmith, E.W.; Jen, J.; Vasmatzis, G.; Patel, S.V.; Maguire, L.J.; et al. Trinucleotide repeat expansion in the transcription factor 4 (tcf4) gene leads to widespread mrna splicing changes in fuchs’ endothelial corneal dystrophy. Invest. Ophthalmol Vis. Sci 2017, 58, 343–352. [Google Scholar] [CrossRef]
- Daughters, R.S.; Tuttle, D.L.; Gao, W.; Ikeda, Y.; Moseley, M.L.; Ebner, T.J.; Swanson, M.S.; Ranum, L.P. Rna gain-of-function in spinocerebellar ataxia type 8. PLoS Genet. 2009, 5, e1000600. [Google Scholar] [CrossRef] [PubMed]
- Mykowska, A.; Sobczak, K.; Wojciechowska, M.; Kozlowski, P.; Krzyzosiak, W.J. Cag repeats mimic cug repeats in the misregulation of alternative splicing. Nucleic Acids Res. 2011, 39, 8938–8951. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Parkesh, R.; Sznajder, L.J.; Childs-Disney, J.L.; Sobczak, K.; Disney, M.D. Chemical correction of pre-mrna splicing defects associated with sequestration of muscleblind-like 1 protein by expanded r(cag)-containing transcripts. ACS Chem. Biol. 2012, 7, 496–505. [Google Scholar] [CrossRef] [PubMed]
- Sellier, C.; Rau, F.; Liu, Y.; Tassone, F.; Hukema, R.K.; Gattoni, R.; Schneider, A.; Richard, S.; Willemsen, R.; Elliott, D.J.; et al. Sam68 sequestration and partial loss of function are associated with splicing alterations in fxtas patients. Embo J. 2010, 29, 1248–1261. [Google Scholar] [CrossRef] [PubMed]
- Konieczny, P.; Stepniak-Konieczna, E.; Sobczak, K. Mbnl expression in autoregulatory feedback loops. RNA Biol. 2018, 15, 1–8. [Google Scholar] [CrossRef]
- Kino, Y.; Washizu, C.; Kurosawa, M.; Oma, Y.; Hattori, N.; Ishiura, S.; Nukina, N. Nuclear localization of mbnl1: Splicing-mediated autoregulation and repression of repeat-derived aberrant proteins. Hum. Mol. Genet. 2015, 24, 740–756. [Google Scholar] [CrossRef]
- Tabaglio, T.; Low, D.H.; Teo, W.K.L.; Goy, P.A.; Cywoniuk, P.; Wollmann, H.; Ho, J.; Tan, D.; Aw, J.; Pavesi, A.; et al. Mbnl1 alternative splicing isoforms play opposing roles in cancer. Life Sci. Alliance 2018, 1, e201800157. [Google Scholar] [CrossRef]
- Pascual, M.; Vicente, M.; Monferrer, L.; Artero, R. The muscleblind family of proteins: An emerging class of regulators of developmentally programmed alternative splicing. Differentiation 2006, 74, 65–80. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Phukan, P.D.; Zeeb, M.; Martinez-Yamout, M.A.; Dyson, H.J.; Wright, P.E. Structural basis for interaction of the tandem zinc finger domains of human muscleblind with cognate rna from human cardiac troponin t. Biochemistry 2017, 56, 4154–4168. [Google Scholar] [CrossRef] [PubMed]
- Meola, G.; Cardani, R. Myotonic dystrophies: An update on clinical aspects, genetic, pathology and molecular pathomechanisms. Biochim Biophys Acta 2015, 1852, 594–606. [Google Scholar] [CrossRef] [PubMed]
- Sellier, C.; Cerro-Herreros, E.; Blatter, M.; Freyermuth, F.; Gaucherot, A.; Ruffenach, F.; Sarkar, P.; Puymirat, J.; Udd, B.; Day, J.W.; et al. Rbfox1/mbnl1 competition for ccug rna repeats binding contributes to myotonic dystrophy type 1/type 2 differences. Nat. Commun. 2018, 9, 2009. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.Y.; Barefield, D.Y.; Vo, A.H.; Gacita, A.M.; Schuster, E.J.; Wyatt, E.J.; Davis, J.L.; Dong, B.; Sun, C.; Page, P.; et al. Distinct pathological signatures in human cellular models of myotonic dystrophy subtypes. Jci. Insight 2019, 4. [Google Scholar] [CrossRef] [PubMed]
- Zu, T.; Cleary, J.D.; Liu, Y.; Banez-Coronel, M.; Bubenik, J.L.; Ayhan, F.; Ashizawa, T.; Xia, G.; Clark, H.B.; Yachnis, A.T.; et al. Ran translation regulated by muscleblind proteins in myotonic dystrophy type 2. Neuron 2017, 95, 1292–1305 e1295. [Google Scholar] [CrossRef] [PubMed]
- Taneja, K.L.; McCurrach, M.; Schalling, M.; Housman, D.; Singer, R.H. Foci of trinucleotide repeat transcripts in nuclei of myotonic dystrophy cells and tissues. J. Cell Biol. 1995, 128, 995–1002. [Google Scholar] [CrossRef] [PubMed]
- Wojciechowska, M.; Krzyzosiak, W.J. Cellular toxicity of expanded rna repeats: Focus on rna foci. Hum. Mol. Genet. 2011, 20, 3811–3821. [Google Scholar] [CrossRef] [PubMed]
- Van Treeck, B.; Parker, R. Emerging roles for intermolecular rna-rna interactions in rnp assemblies. Cell 2018, 174, 791–802. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.P.; Byron, M.; Johnson, C.; Xing, Y.; Lawrence, J.B. Defining early steps in mrna transport: Mutant mrna in myotonic dystrophy type i is blocked at entry into sc-35 domains. J. Cell Biol. 2007, 178, 951–964. [Google Scholar] [CrossRef]
- Fardaei, M.; Larkin, K.; Brook, J.D.; Hamshere, M.G. In vivo co-localisation of mbnl protein with dmpk expanded-repeat transcripts. Nucleic Acids Res 2001, 29, 2766–2771. [Google Scholar] [CrossRef] [PubMed]
- Querido, E.; Gallardo, F.; Beaudoin, M.; Ménard, C.; Chartrand, P. Stochastic and reversible aggregation of mrna with expanded cug-triplet repeats. J. Cell Sci. 2011, 124, 1703–1714. [Google Scholar] [CrossRef] [PubMed]
- Konieczny, P.; Stepniak-Konieczna, E.; Taylor, K.; Sznajder, L.J.; Sobczak, K. Autoregulation of mbnl1 function by exon 1 exclusion from mbnl1 transcript. Nucleic Acids Res. 2017, 45, 1760–1775. [Google Scholar] [CrossRef] [PubMed]
- Tran, H.; Gourrier, N.; Lemercier-Neuillet, C.; Dhaenens, C.M.; Vautrin, A.; Fernandez-Gomez, F.J.; Arandel, L.; Carpentier, C.; Obriot, H.; Eddarkaoui, S.; et al. Analysis of exonic regions involved in nuclear localization, splicing activity and dimerization of muscleblind-like-1 isoforms. J. Biol. Chem. 2011, 286, 16435–16446. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.; Dansithong, W.; Jog, S.P.; Holt, I.; Mittal, S.; Brook, J.D.; Morris, G.E.; Comai, L.; Reddy, S. Expanded cug repeats dysregulate rna splicing by altering the stoichiometry of the muscleblind 1 complex. J. Biol. Chem. 2011, 286, 38427–38438. [Google Scholar] [CrossRef]
- Laurent, F.X.; Sureau, A.; Klein, A.F.; Trouslard, F.; Gasnier, E.; Furling, D.; Marie, J. New function for the rna helicase p68/ddx5 as a modifier of mbnl1 activity on expanded cug repeats. Nucleic Acids Res. 2012, 40, 3159–3171. [Google Scholar] [CrossRef]
- Pettersson, O.J.; Aagaard, L.; Andrejeva, D.; Thomsen, R.; Jensen, T.G.; Damgaard, C.K. Ddx6 regulates sequestered nuclear cug-expanded dmpk-mrna in dystrophia myotonica type 1. Nucleic Acids Res. 2014, 42, 7186–7200. [Google Scholar] [CrossRef]
- Ho, T.H.; Savkur, R.S.; Poulos, M.G.; Mancini, M.A.; Swanson, M.S.; Cooper, T.A. Colocalization of muscleblind with rna foci is separable from mis-regulation of alternative splicing in myotonic dystrophy. J. Cell Sci. 2005, 118, 2923–2933. [Google Scholar] [CrossRef]
- Xia, G.; Ashizawa, T. Dynamic changes of nuclear rna foci in proliferating dm1 cells. Histochem Cell Biol. 2015, 143, 557–564. [Google Scholar] [CrossRef]
- Dansithong, W.; Paul, S.; Comai, L.; Reddy, S. Mbnl1 is the primary determinant of focus formation and aberrant insulin receptor splicing in dm1. J. Biol. Chem. 2005, 280, 5773–5780. [Google Scholar] [CrossRef]
- Jain, A.; Vale, R.D. Rna phase transitions in repeat expansion disorders. Nature 2017, 546, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Gudde, A.E.; González-Barriga, A.; van den Broek, W.J.; Wieringa, B.; Wansink, D.G. A low absolute number of expanded transcripts is involved in myotonic dystrophy type 1 manifestation in muscle. Hum. Mol. Genet. 2016, 25, 1648–1662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wojciechowska, M.; Sobczak, K.; Kozlowski, P.; Sedehizadeh, S.; Wojtkowiak-Szlachcic, A.; Czubak, K.; Markus, R.; Lusakowska, A.; Kaminska, A.; Brook, J.D. Quantitative methods to monitor rna biomarkers in myotonic dystrophy. Sci. Rep. 2018, 8, 5885. [Google Scholar] [CrossRef] [PubMed]
- Zu, T.; Gibbens, B.; Doty, N.S.; Gomes-Pereira, M.; Huguet, A.; Stone, M.D.; Margolis, J.; Peterson, M.; Markowski, T.W.; Ingram, M.A.; et al. Non-atg-initiated translation directed by microsatellite expansions. Proc. Natl. Acad. Sci. USA 2011, 108, 260–265. [Google Scholar] [CrossRef]
- Nedelsky, N.B.; Taylor, J.P. Bridging biophysics and neurology: Aberrant phase transitions in neurodegenerative disease. Nat. Rev. Neurol 2019, 15, 272–286. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.; Cleary, J.D.; Ranum, L.P.W. Repeat-associated non-atg translation: Molecular mechanisms and contribution to neurological disease. Annu. Rev. Neurosci 2019, 42, 227–247. [Google Scholar] [CrossRef] [PubMed]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sznajder, Ł.J.; Swanson, M.S. Short Tandem Repeat Expansions and RNA-Mediated Pathogenesis in Myotonic Dystrophy. Int. J. Mol. Sci. 2019, 20, 3365. https://doi.org/10.3390/ijms20133365
Sznajder ŁJ, Swanson MS. Short Tandem Repeat Expansions and RNA-Mediated Pathogenesis in Myotonic Dystrophy. International Journal of Molecular Sciences. 2019; 20(13):3365. https://doi.org/10.3390/ijms20133365
Chicago/Turabian StyleSznajder, Łukasz J., and Maurice S. Swanson. 2019. "Short Tandem Repeat Expansions and RNA-Mediated Pathogenesis in Myotonic Dystrophy" International Journal of Molecular Sciences 20, no. 13: 3365. https://doi.org/10.3390/ijms20133365
APA StyleSznajder, Ł. J., & Swanson, M. S. (2019). Short Tandem Repeat Expansions and RNA-Mediated Pathogenesis in Myotonic Dystrophy. International Journal of Molecular Sciences, 20(13), 3365. https://doi.org/10.3390/ijms20133365