Network-Based Integrative Analysis of Genomics, Epigenomics and Transcriptomics in Autism Spectrum Disorders

 , ,
, ,  and
and
Abstract
:
1. Introduction
2. Results
2.1. Genomics Analysis
2.2. Multi-Omics Analysis
3. Discussion
4. Materials and Methods
4.1. Molecular Interactions
4.2. Genomics
4.3. Epigenomics
4.4. Transcriptomics
4.5. Gene Prioritization Based on Network Diffusion
4.6. Functional Characterization of the INT-MODULE
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Schaaf, C.P.; Zoghbi, H.Y. Solving the autism puzzle a few pieces at a time. Neuron 2011, 70, 806–808. [Google Scholar] [CrossRef] [PubMed]
- Barabási, A.L.; Gulbahce, N.; Loscalzo, J. Network medicine: A network-based approach to human disease. Nat. Rev. Genet. 2011, 12, 56–68. [Google Scholar] [CrossRef] [PubMed]
- Boyle, E.A.; Li, Y.I.; Pritchard, J.K. An Expanded View of Complex Traits: From Polygenic to Omnigenic. Cell 2017, 169, 1177–1186. [Google Scholar] [CrossRef] [PubMed]
- Devlin, B.; Scherer, S.W. Genetic architecture in autism spectrum disorder. Curr. Opin. Genet. Dev. 2012, 22, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Mitra, K.; Carvunis, A.R.; Ramesh, S.K.; Ideker, T. Integrative approaches for finding modular structure in biological networks. Nat. Rev. Genet. 2013, 14, 719–732. [Google Scholar] [CrossRef] [PubMed]
- Cowen, L.; Ideker, T.; Raphael, J.B.; Sharan, R. Network propagation: A universal amplifier of genetic associations. Nat. Rev. 2017, 18, 551–562. [Google Scholar] [CrossRef] [PubMed]
- Mosca, E.; Bersanelli, M.; Gnocchi, M.; Moscatelli, M.; Castellani, G.; Milanesi, L.; Mezzelani, A. Network Diffusion-Based Prioritization of Autism Risk Genes Identifies Significantly Connected Gene Modules. Front. Genet. 2017, 8, 129. [Google Scholar] [CrossRef]
- Abrahams, B.S.; Arking, D.E.; Campbell, D.B.; Mefford, H.C.; Morrow, E.M.; Weiss, L.A.; Menashe, I.; Wadkins, T.; Banerjee-Basu, S.; Packer, A. SFARI Gene 2.0: A community-driven knowledgebase for the autism spectrum disorders (ASDs). Mol. Autism 2013, 4, 36. [Google Scholar] [CrossRef]
- Wiśniowiecka-Kowalnik, B.; Nowakowska, B.A. Genetics and epigenetics of autism spectrum disorder—Current evidence in the field. J. Appl. Genet. 2019, 60, 37–47. [Google Scholar] [CrossRef]
- Andrews, S.V.; Sheppard, B.; Windham, G.C.; Schieve, L.A.; Schendel, D.E.; Croen, L.A.; Chopra, P.; Alisch, R.S.; Newschaffer, C.J.; Warren, S.T.; et al. Case-control meta-analysis of blood DNA methylation and autism spectrum disorder. Mol. Autism 2018, 9, 40. [Google Scholar] [CrossRef] [Green Version]
- Luo, R.; Sanders, S.J.; Tian, Y.; Voineagu, I.; Huang, N.; Chu, S.H.; Klei, L.; Cai, C.; Ou, J.; Lowe, J.K.; et al. Genome-wide transcriptome profiling reveals the functional impact of rare de novo and recurrent CNVs in autism spectrum disorders. Am. J. Hum. Genet. 2012, 91, 38–55. [Google Scholar] [CrossRef] [PubMed]
- Codina-Solà, M.; Rodríguez-Santiago, B.; Homs, A.; Santoyo, J.; Rigau, M.; Aznar-Laín, G.; Campo, M.; Gener, B.; Gabau, E.; Botella, M.P.; et al. Integrated analysis of whole-exome sequencing and transcriptome profiling in males with autism spectrum disorders. Mol. Autism 2015, 6, 21. [Google Scholar] [CrossRef] [PubMed]
- Karczewski, K.J.; Snyder, M.P. Integrative omics for health and disease. Nat. Rev. Genet. 2018, 19, 299–310. [Google Scholar] [CrossRef]
- Higdon, R.; Earl, R.K.; Stanberry, L.; Hudac, C.M.; Montague, E.; Stewart, E.; Janko, I.; Choiniere, J.; Broomall, W.; Kolker, N.; et al. The promise of multi-omics and clinical data integration to identify and target personalized healthcare approaches in autism spectrum disorders. OMICS 2015, 19, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Kong, S.W.; Collins, C.D.; Shimizu-Motohashi, Y.; Holm, I.A.; Campbell, M.G.; Lee, I.H.; Brewster, S.J.; Hanson, E.; Harris, H.K.; Lowe, K.R.; et al. Characteristics and Predictive Value of Blood Transcriptome Signature in Males with Autism Spectrum Disorders. PLoS ONE 2012, 7, e49475. [Google Scholar] [CrossRef]
- Tylee, D.S.; Kawaguchi, D.M.; Glatt, S.J. On the Outside, Looking in: A Review and Evaluation of the Comparability of Blood and Brain “-omes”. Am. J. Med. Genet. Part B 2013, 162, 595–603. [Google Scholar] [CrossRef]
- Andrews, S.V.; Ellis, S.E.; Bakulski, K.M.; Sheppard, B.; Croen, L.A.; Hertz-Picciotto, I.; Newschaffer, C.J.; Feinberg, A.P.; Arking, D.E.; Ladd-Acosta, C.; et al. Cross-tissue integration of genetic and epigenetic data offers insight into autism spectrum disorder. Nat. Commun. 2017, 8, 1011. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Shi, M.; Zhihai Ma, Z.; Zhao, S.; Euskirchen, G.; Ziskin, J.; Urban, A.; Hallmayer, J.; Snyder, M. Integrated systems analysis reveals a molecular network underlying autism spectrum disorders. Mol. Syst. Biol. 2014, 10, 774. [Google Scholar] [CrossRef]
- Gilman, S.R.; Iossifov, I.; Levy, D.; Ronemus, M.; Wigler, M.; Vitkup, D. Rare de novo variants associated with autism implicate a large functional network of genes involved in formation and function of synapses. Neuron 2011, 70, 898–907. [Google Scholar] [CrossRef]
- Hormozdiari, F.; Penn, O.; Borenstein, E.; Eichler, E.E. The discovery of integrated gene networks for autism and related disorders. Genome Res. 2015, 25, 142–154. [Google Scholar] [CrossRef]
- Parikshak, N.N.; Luo, R.; Zhang, A.; Won, H.; Lowe, J.K.; Chandran, V.; Horvath, S.; Geschwind, D.H. Integrative functional genomic analyses implicate specific molecular pathways and circuits in autism. Cell 2013, 155, 1008–1021. [Google Scholar] [CrossRef]
- Pinto, D.; Delaby, E.; Merico, D.; Barbosa, M.; Merikangas, A.; Klei, L.; Thiruvahindrapuram, B.; Xu, X.; Ziman, R.; Wang, Z.; et al. Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am. J. Hum. Genet. 2014, 94, 677–694. [Google Scholar] [CrossRef] [PubMed]
- The Autism Spectrum Disorders Working Group of the Psychiatric Genomics Consortium. Meta-analysis of GWAS of over 16,000 individuals with autism spectrum disorder highlights a novel locus at 10q24.32 and a significant overlap with schizophrenia. Mol. Autism 2017, 8, 21. [CrossRef]
- De Rubeis, S.; He, X.; Goldberg, A.P.; Poultney, C.S.; Samocha, K.; Cicek, A.E.; Kou, Y.; Liu, L.; Fromer, M.; Walker, S.; et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 2014, 515, 209–215. [Google Scholar] [CrossRef]
- Hu, V.W.; Sarachana, T.; Kim, K.S.; Nguyen, A.; Kulkarni, S.; Steinberg, M.E.; Luu, T.; Lai, Y.; Lee, N.H. Gene Expression Profiling Differentiates Autism Case–Controls and Phenotypic Variants of Autism Spectrum Disorders: Evidence for Circadian Rhythm Dysfunction in Severe Autism. Autism Res. 2009, 2, 78–97. [Google Scholar] [CrossRef] [PubMed]
- Pramparo, T.; Lombardo, M.V.; Campbell, K.; Barnes, C.C.; Marinero, S.; Solso, S.; Young, J.; Mayo, M.; Dale, A.; Ahrens-Barbeau, C.; et al. Cell cycle networks link gene expression dysregulation, mutation, and brain maldevelopment in autistic toddlers. Mol. Syst. Biol. 2015, 11, 841. [Google Scholar] [CrossRef] [PubMed]
- Gregg, J.P.; Lit, L.; Baron, C.A.; Hertz-Picciotto, I.; Walker, W.; Davis, R.A.; Croen, L.A.; Ozonoff, S.; Hansen, R.; Pessah, I.N.; et al. Gene expression changes in children with autism. Genomics 2008, 91, 22–29. [Google Scholar] [CrossRef] [Green Version]
- Bersanelli, M.; Mosca, E.; Remondini, D.; Castellani, G.; Milanesi, L. Network diffusion-based analysis of high-throughput data for the detection of differentially enriched modules. Sci. Rep. 2016, 6, 34841. [Google Scholar] [CrossRef] [Green Version]
- Yi, F.; Danko, T.; Botelho, S.C.; Patzke, C.; Pak, C.; Wernig, M.; Südhof, T.C. Autism-associated SHANK3 haploinsufficiency causes Ih channelopathy in human neurons. Science 2016, 6, aaf2669. [Google Scholar] [CrossRef]
- Zhu, M.; Idikuda, V.K.; Wang, J.; Wei, F.; Kumar, V.; Shah, N.; Waite, C.B.; Liu, Q.; Zhou, L. Shank3-deficient thalamocortical neurons show HCN channelopathy and alterations in intrinsic electrical properties. J Physiol. 2018, 596, 1259–1276. [Google Scholar] [CrossRef] [Green Version]
- Nava, C.; Dalle, C.; Rastetter, A.; Striano, P.; de Kovel, C.G.; Nabbout, R.; Cancès, C.; Ville, D.; Brilstra, E.H.; Gobbi, G.; et al. De novo mutations in HCN1 cause early infantile epileptic encephalopathy. Nat. Genet. 2014, 46, 640–645. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Schroer, R.; Yan, J.; Song, W.; Yang, C.; Bockholt, A.; Cook, E.H., Jr.; Skinner, C.; Schwartz, C.E.; Sommer, S.S. High frequency of neurexin 1β signal peptide structural variants in patients with autism. Neurosci. Lett. 2006, 409, 10–13. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, J.; Siddiqui, T.J.; Huashan, P.; Yokomaku, D.; Hamdan, F.F.; Champagne, N.; Lapointe, M.; Spiegelman, D.; Noreau, A.; Lafrenière, R.G.; et al. Truncating mutations in NRXN2 and NRXN1 in autism spectrum disorders and schizophrenia. Hum. Genet. 2011, 130, 563–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parente, D.J.; Garriga, C.; Baskin, B.; Douglas, G.; Cho, M.T.; Araujo, G.C.; Shinawi, M. Neuroligin 2 nonsense variant associated with anxiety, autism, intellectual disability, hyperphagia, and obesity. Am. J. Med. Genet. A 2017, 173, 213–216. [Google Scholar] [CrossRef] [PubMed]
- Jamain, S.; Quach, H.; Betancur, C.; Råstam, M.; Colineaux, C.; Gillberg, I.C.; Soderstrom, H.; Giros, B.; Leboyer, M.; Gillberg, C.; et al. Paris Autism Research International Sibpair Study. Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat. Genet. 2003, 34, 27–29. [Google Scholar] [CrossRef] [PubMed]
- Jiang-Xie, L.F.; Liao, H.M.; Chen, C.H.; Chen, Y.T.; Ho, S.Y.; Lu, D.H.; Lee, L.J.; Liou, H.H.; Fu, W.M.; Gau, S.S. Autism-associated gene Dlgap2 mutant mice demonstrate exacerbated aggressive behaviors and orbitofrontal cortex deficits. Mol. Autism 2014, 1, 32. [Google Scholar] [CrossRef]
- Marshall, C.R.; Noor, A.; Vincent, J.B.; Lionel, A.C.; Feuk, L.; Skaug, J.; Shago, M.; Moessner, R.; Pinto, D.; Ren, Y.; et al. Structural variation of chromosomes in autism spectrum disorder. Am. J. Hum. Genet. 2008, 82, 477–488. [Google Scholar] [CrossRef]
- Pinto, D.; Pagnamenta, A.T.; Klei, L.; Anney, R.; Merico, D.; Regan, R.; Conroy, J.; Magalhaes, T.R.; Correia, C.; Abrahams, B.S.; et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature 2010, 466, 368–372. [Google Scholar] [CrossRef] [Green Version]
- Poquet, H.; Faivre, L.; El Chehadeh, S.; Morton, J.; McMullan, D.; Hamilton, S.; Goel, H.; Isidor, B.; Le Caignec, C.; Andrieux, J.; et al. Further Evidence for Dlgap2 as Strong Autism Spectrum Disorders/Intellectual Disability Candidate Gene. Autism Open Access 2017, 6, 197. [Google Scholar] [CrossRef]
- Nakamura, K.; Anitha, A.; Yamada, K.; Tsujii, M.; Iwayama, Y.; Hattori, E.; Toyota, T.; Suda, S.; Takei, N.; Iwata, Y.; et al. Genetic and expression analyses reveal elevated expression of syntaxin 1A (STX1A) in high functioning autism. Int. J. Neuropsychopharmacol. 2008, 11, 1073–1084. [Google Scholar] [CrossRef]
- Nakamura, K.; Iwata, Y.; Anitha, A.; Miyachi, T.; Toyota, T.; Yamada, S.; Tsujii, M.; Tsuchiya, K.J.; Iwayama, Y.; Yamada, K.; et al. Replication study of Japanese cohorts supports the role of STX1A in autism susceptibility. Prog. Neuropsychopharmacol. Biol. Psychiatry 2011, 35, 454–458. [Google Scholar] [CrossRef] [PubMed]
- Kofuji, T.; Hayashi, Y.; Fujiwara, T.; Sanada, M.; Tamaru, M.; Akagawa, K. A part of patients with autism spectrum disorder has haploidy of HPC-1/syntaxin1A gene that possibly causes behavioral disturbance as in experimentally gene ablated mice. Neurosci. Lett. 2017, 644, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Durdiaková, J.; Warrier, V.; Banerjee-Basu, S.; Baron-Cohen, S.; Chakrabarti, B. STX1A and Asperger syndrome: A replication study. Mol. Autism. 2014, 5, 14. [Google Scholar] [CrossRef] [PubMed]
- Fatemi, S.H.; Reutiman, T.J.; Folsom, T.D.; Rooney, R.J.; Patel, D.H.; Thuras, P.D. mRNA and protein levels for GABAAα4, α5, β1 and GABABR1 receptors are altered in brains from subjects with autism. J. Autism Dev. Disord. 2010, 40, 743–750. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Guo, X.; Dong, X.; Han, Y.; Gao, L.; Su, Y.; Dai, W.; Zhang, X. GABA(A) receptor subunit gene polymorphisms predict symptom-based and developmental deficits in Chinese Han children and adolescents with autistic spectrum disorders. Sci. Rep. 2017, 7, 3290. [Google Scholar] [CrossRef] [PubMed]
- Coe, B.P.; Stessman, H.A.F.; Sulovari, A.; Geisheker, M.R.; Bakken, T.E.; Lake, A.M.; Dougherty, J.D.; Lein, E.S.; Hormozdiari, F.; Bernier, R.A.; et al. Neurodevelopmental disease genes implicated by de novo mutation and copy number variation morbidity. Nat. Genet. 2019, 51, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Sudarov, A.; Gooden, F.; Tseng, D.; Gan, W.B.; Ross, M.E. Lis1 controls dynamics of neuronal filopodia and spines to impact synaptogenesis and social behaviour. EMBO Mol. Med. 2013, 5, 591–607. [Google Scholar] [CrossRef] [PubMed]
- O’Roak, B.J.; Vives, L.; Girirajan, S.; Karakoc, E.; Krumm, N.; Coe, B.P.; Levy, R.; Ko, A.; Lee, C.; Smith, J.D.; et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 2012, 485, 246–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iossifov, I.; Ronemus, M.; Levy, D.; Wang, Z.; Hakker, I.; Rosenbaum, J.; Yamrom, B.; Lee, Y.H.; Narzisi, G.; Leotta, A.; et al. De novo gene disruptions in children on the autistic spectrum. Neuron 2012, 2, 285–299. [Google Scholar] [CrossRef]
- Griesi-Oliveira, K.; Acab, A.; Gupta, A.R.; Sunaga, D.Y.; Chailangkarn, T.; Nicol, X.; Nunez, Y.; Walker, M.F.; Murdoch, J.D.; Sanders, S.J.; et al. Modeling non-syndromic autism and the impact of TRPC6 disruption in human neurons. Mol. Psychiatry 2015, 11, 1350–1365. [Google Scholar] [CrossRef]
- Beltrão-Braga, P.C.; Muotri, A.R. Modeling autism spectrum disorders with human neurons. Brain Res. 2017, 1656, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.R.; Hem, V.; Katz, K.S.; Ovetsky, M.; Wallin, C.; Ermolaeva, O.; Tolstoy, I.; Tatusova, T.; Pruitt, K.D.; Maglott, D.R.; et al. Gene: A gene-centered information resource at NCBI. Nucleic Acids Res. 2015, 43, D36–D42. [Google Scholar] [CrossRef] [PubMed]
- Willer, C.J.; Li, Y.; Abecasis, G.R. METAL: Fast and efficient meta-analysis of genomewide association scans. Bioinformatics 2010, 26, 2190–2191. [Google Scholar] [CrossRef] [PubMed]
- Saeliw, T.; Tangsuwansri, C.; Thongkorn, S.; Chonchaiya, W.; Suphapeetiporn, K.; Mutirangura, A.; Tencomnao, T.; Hu, V.W.; Sarachana, T. Integrated genome-wide Alu methylation and transcriptome profiling analyses reveal novel epigenetic regulatory networks associated with autism spectrum disorder. Mol. Autism 2018, 9, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanunu, O.; Magger, O.; Ruppin, E.; Shlomi, T.; Sharan, R. Associating Genes and Protein Complexes with Disease via Network Propagation. PLoS Comput. Biol. 2010, 6, e1000641. [Google Scholar] [CrossRef]
- Mosca, E.; Alfieri, R.; Milanesi, L. Diffusion of Information throughout the Host Interactome Reveals Gene Expression Variations in Network Proximity to Target Proteins of Hepatitis C Virus. PLoS ONE 2014, 9, e113660. [Google Scholar] [CrossRef]
- Hofree, M.; Shen, J.P.; Carter, H.; Gross, A.; Ideker, T. Network-based stratification of tumor mutations. Nat. Methods 2013, 10, 1108–1115. [Google Scholar] [CrossRef]
- Ruffalo, M.; Koyuturk, M.; Sharan, R. Network-Based Integration of Disparate Omic Data to Identify “Silent Players” in Cancer. PLoS Comput. Biol. 2015, 11, e1004595. [Google Scholar] [CrossRef]
- Di Nanni, N.; Gnocchi, M.; Moscatelli, M.; Milanesi, L.; Mosca, E. Gene relevance based on multiple evidences in complex networks. Bioinformatics. under review.
- Newman, M.E.J. Modularity and community structure in networks. Proc. Natl. Acad. Sci. USA 2006, 103, 8577–8582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Csardi, G.; Nepusz, T. The igraph software package for complex network research. InterJ. Complex Syst. 2006, 1695, 1–9. [Google Scholar]
- Geer, L.Y.; Marchler-Bauer, A.; Geer, R.C.; Han, L.; He, J.; He, S.; Liu, C.; Shi, W.; Stephen, H.; Bryant, S.H. The NCBI BioSystems database. Nucleic Acids Res. 2010, 38, D492–D496. [Google Scholar] [CrossRef] [PubMed]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdóttir, H.; Tamayo, P.; Mesirov, J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Evidence | Description | Subjects | Number of Genes | |||
|---|---|---|---|---|---|---|
| Initial | Selected | |||||
| ** | * | ** | * | |||
| G | SFARI [8]. | - | 404 | 1087 | 334 | 799 |
| G | Network diffusion-based prioritization of autism risk genes identifies significantly connected gene modules [7]. | - | ||||
| G | Meta-analysis of GWAS of over 16,000 individuals with autism spectrum disorder [23]. | 15,954 | ||||
| G | Synaptic, transcriptional and chromatin genes disrupted in autism [24]. | 13,808 | ||||
| E | Case-control meta-analysis of blood DNA methylation and autism spectrum disorder [10]. | 1654 | 416 | 1444 | 272 | 955 |
| T | Gene expression profiling differentiates autism case- controls and phenotypic variants of autism spectrum disorders [25]. | 116 | 330 | 3045 | 256 | 2131 |
| T | Blood gene expression signatures distinguish autism spectrum disorders from controls [15]. | 285 | ||||
| T | Disrupted functional networks in autism underlie early brain mal-development and provide accurate classification. [26]. | 147 | ||||
| T | Gene expression in blood of children with autism spectrum disorder [27]. | 47 | ||||
| Symbol | Description | |I| | |Ic| | |core| | p | G | E | T | SFARI Score | Other Modules |
|---|---|---|---|---|---|---|---|---|---|---|
| HCN4 | hyperpolarization activated cyclic nucleotide gated potassium channel 4 | 4 | 2 | 334 | 3.97 × 10−3 | * | * | 0 | - | - |
| DLGAP2 | DLG associated protein 2 | 21 | 8 | 334 | 3.10 × 10−8 | * | 0 | 0 | 4 | [18,21,22] |
| HCN2 | hyperpolarization activated cyclic nucleotide gated potassium and sodium channel 2 | 4 | 1 | 334 | 1.01 × 10−1 | * | 0 | 0 | - | - |
| UBC | ubiquitin C | 1168 | 43 | 334 | 1.41 × 10−2 | * | 0 | 0 | - | [20] |
| NLGN2 | neuroligin 2 | 28 | 8 | 334 | 4.04 × 10−7 | * | 0 | 0 | 4 | [18] |
| WDR37 | WD repeat domain 37 | 2 | 2 | 334 | 6.85 × 10−4 | 0 | 0 | * | - | - |
| MTMR2 | myotubularin related protein 2 | 6 | 1 | 334 | 1.47 × 10−1 | * | 0 | 0 | - | - |
| EPB41L1 | erythrocyte membrane protein band 4.1 like 1 | 34 | 9 | 334 | 1.55 × 10−7 | * | 0 | 0 | - | [21] |
| GABRA5 | gamma-aminobutyric acid type A receptor alpha5 subunit | 17 | 4 | 334 | 8.43 × 10−4 | * | 0 | 0 | 5 | [21] |
| STX1A | syntaxin 1A | 78 | 10 | 334 | 3.47 × 10−5 | * | 0 | 0 | 4 | [20,21] |
| EPB41 | erythrocyte membrane protein band 4.1 | 16 | 5 | 334 | 4.14 × 10−5 | * | 0 | ** | - | [20] |
| CACNA1F | calcium voltage-gated channel subunit alpha1 F | 37 | 6 | 334 | 3.63 × 10−4 | * | 0 | 0 | 4 | [21] |
| PRKCA | protein kinase C alpha | 197 | 11 | 334 | 1.48 × 10−2 | * | * | * | 4 | - |
| A | B | ||||||
|---|---|---|---|---|---|---|---|
| core+13 | core+13(E) | 347 | 1227 | 12739 | 54 | 3.27 | 2.63 × 10−4 |
| core+13 | core+13(T) | 347 | 2387 | 12739 | 88 | 6.37 | 1.22 × 10−3 |
| G | E | 1133 | 1227 | 12739 | 146 | 109 | 1.09 × 10−4 |
| G | T | 1133 | 2387 | 12739 | 235 | 212 | 3.95 × 10−2 |
| E | T | 1227 | 2387 | 12739 | 243 | 230 | 1.66 × 10−1 |
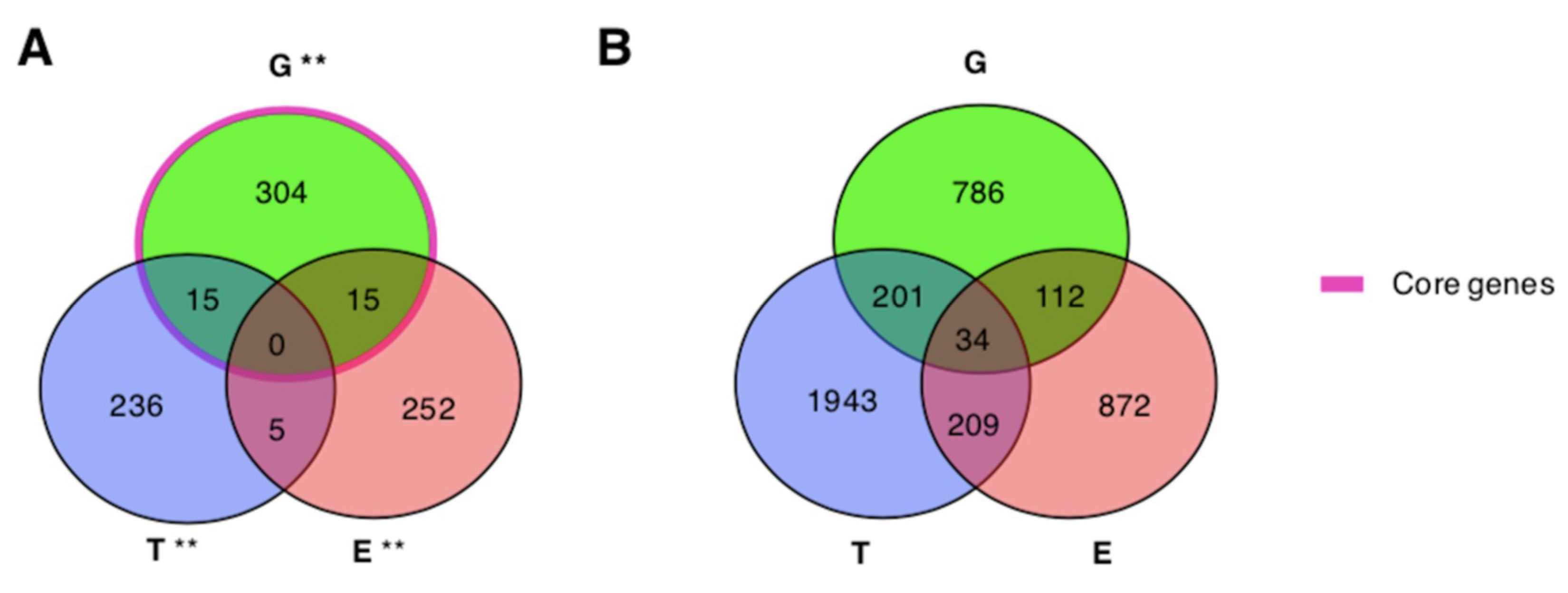
| G ** | E ** | 334 | 272 | 12739 | 15 | 7.13 | 5.47 × 10−3 |
| G ** | T ** | 334 | 256 | 12739 | 15 | 6.71 | 3.12 × 10−3 |
| E ** | T ** | 272 | 256 | 12739 | 5 | 5.47 | 6.42 × 10−1 |
| Symbol | Description | #Im | G | E | T | SFARI Score | Other Modules |
|---|---|---|---|---|---|---|---|
| BAIAP2 | BAI1-associated protein 2 | 4 | * | * | 0 | 5 | [19,21] |
| CACNA1B | calcium voltage-gated channel subunit alpha1 B | 7 | 0 | ** | 0 | 4 | [19,21] |
| CREBBP | CREB binding protein | 43 | 0 | 0 | ** | 5 | [18,20,21] |
| HOXB1 | homeobox B1 | 12 | 0 | * | * | 5 | [18] |
| INPP1 | inositol polyphosphate-1-phosphatase | 1 | 0 | ** | 0 | 4 | [18,21] |
| ITPR1 | inositol 1,4,5-trisphosphate receptor type 1 | 11 | * | * | 0 | 4 | [19,21] |
| KCNMA1 | potassium large conductance calcium-activated channel, subfamily M, alpha member 1 | 1 | 0 | ** | 0 | 4 | [20] |
| RASSF5 | Ras association domain family member 5 | 0 | 0 | ** | ** | 4 | - |
| RBM8A | RNA binding motif protein 8A | 10 | 0 | ** | * | 5 | - |
| SH3KBP1 | SH3-domain kinase binding protein 1 | 12 | * | 0 | ** | 5 | - |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Nanni, N.; Bersanelli, M.; Cupaioli, F.A.; Milanesi, L.; Mezzelani, A.; Mosca, E. Network-Based Integrative Analysis of Genomics, Epigenomics and Transcriptomics in Autism Spectrum Disorders. Int. J. Mol. Sci. 2019, 20, 3363. https://doi.org/10.3390/ijms20133363
Di Nanni N, Bersanelli M, Cupaioli FA, Milanesi L, Mezzelani A, Mosca E. Network-Based Integrative Analysis of Genomics, Epigenomics and Transcriptomics in Autism Spectrum Disorders. International Journal of Molecular Sciences. 2019; 20(13):3363. https://doi.org/10.3390/ijms20133363
Chicago/Turabian StyleDi Nanni, Noemi, Matteo Bersanelli, Francesca Anna Cupaioli, Luciano Milanesi, Alessandra Mezzelani, and Ettore Mosca. 2019. "Network-Based Integrative Analysis of Genomics, Epigenomics and Transcriptomics in Autism Spectrum Disorders" International Journal of Molecular Sciences 20, no. 13: 3363. https://doi.org/10.3390/ijms20133363
APA StyleDi Nanni, N., Bersanelli, M., Cupaioli, F. A., Milanesi, L., Mezzelani, A., & Mosca, E. (2019). Network-Based Integrative Analysis of Genomics, Epigenomics and Transcriptomics in Autism Spectrum Disorders. International Journal of Molecular Sciences, 20(13), 3363. https://doi.org/10.3390/ijms20133363