Unraveling the Impact of Cysteine-to-Serine Mutations on the Structural and Functional Properties of Cu(I)-Binding Proteins

 , and
, and
Abstract
:1. Introduction
2. Results
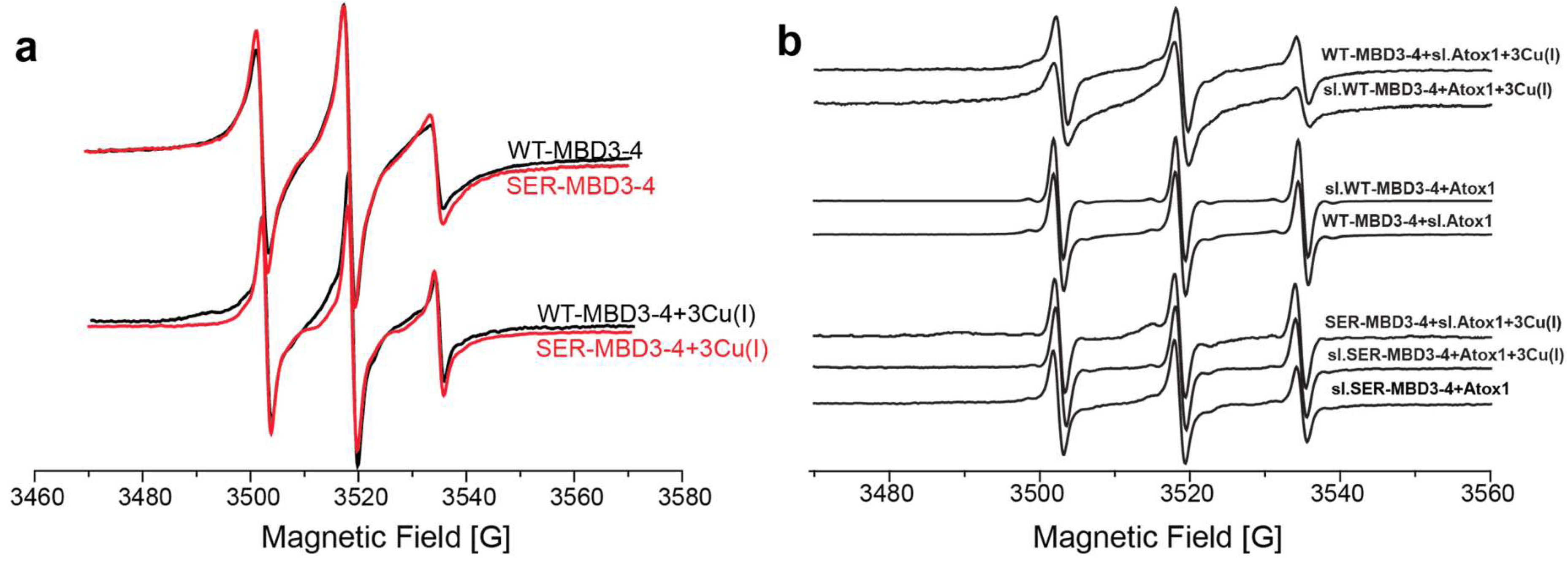
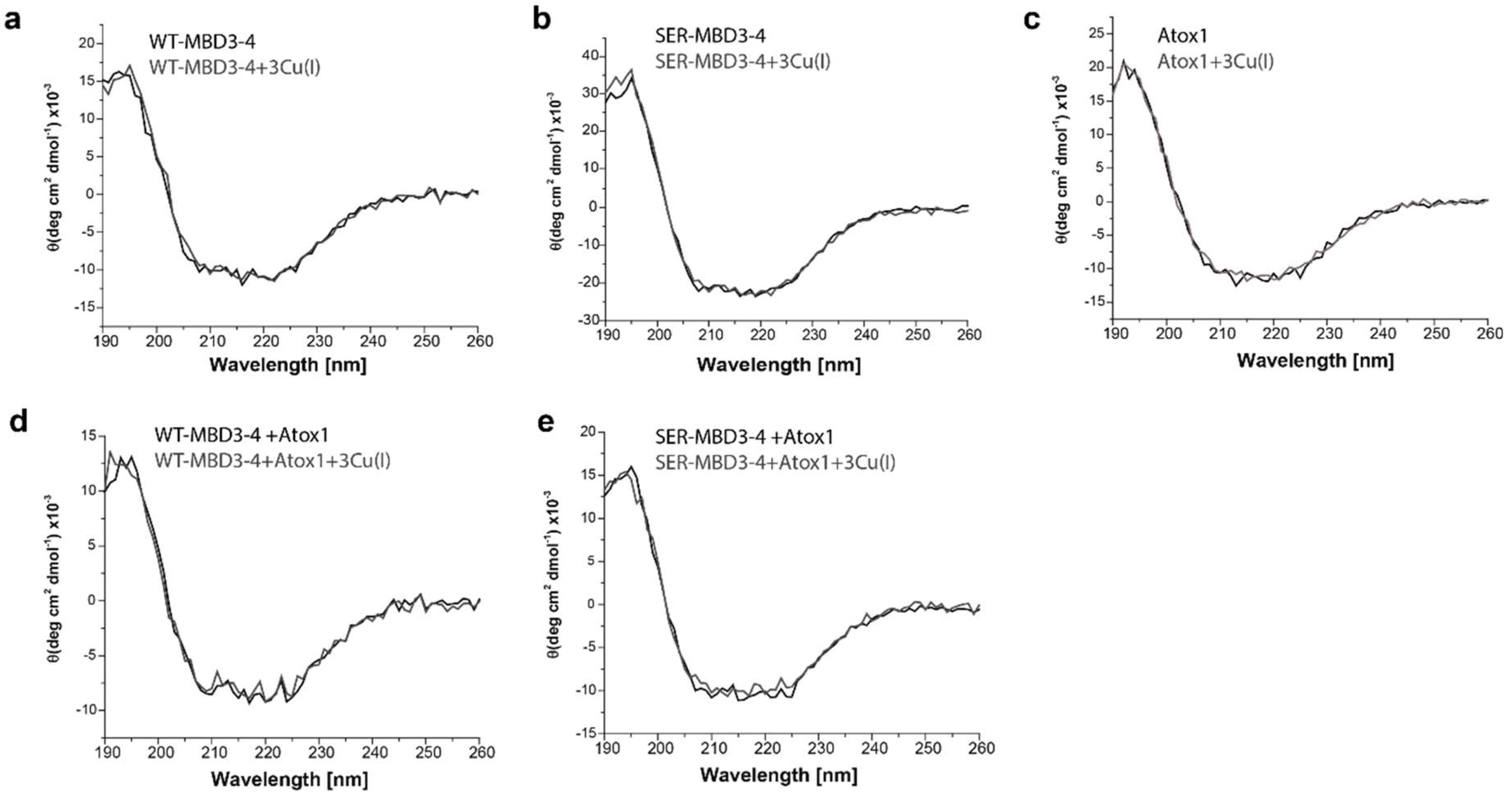
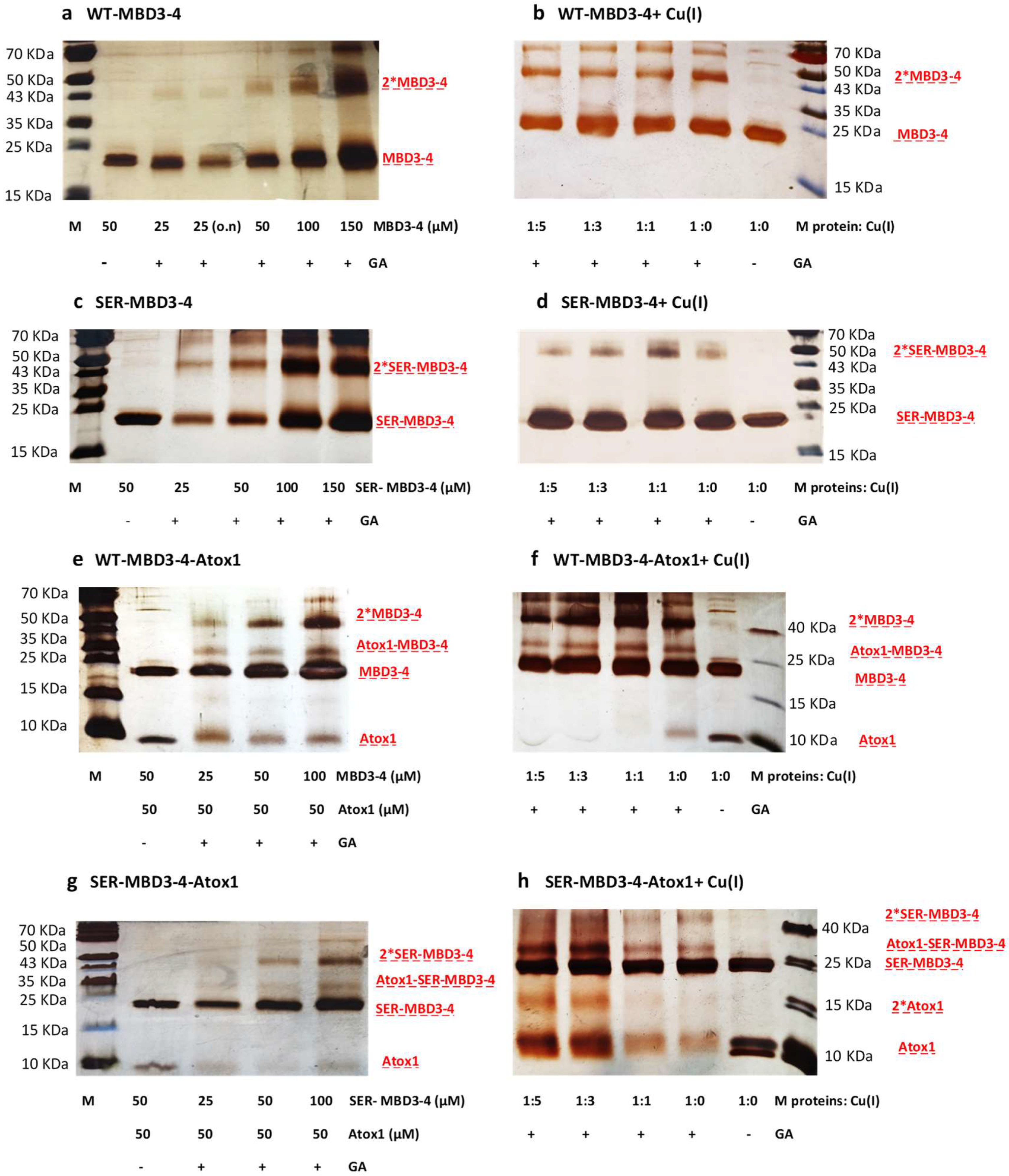
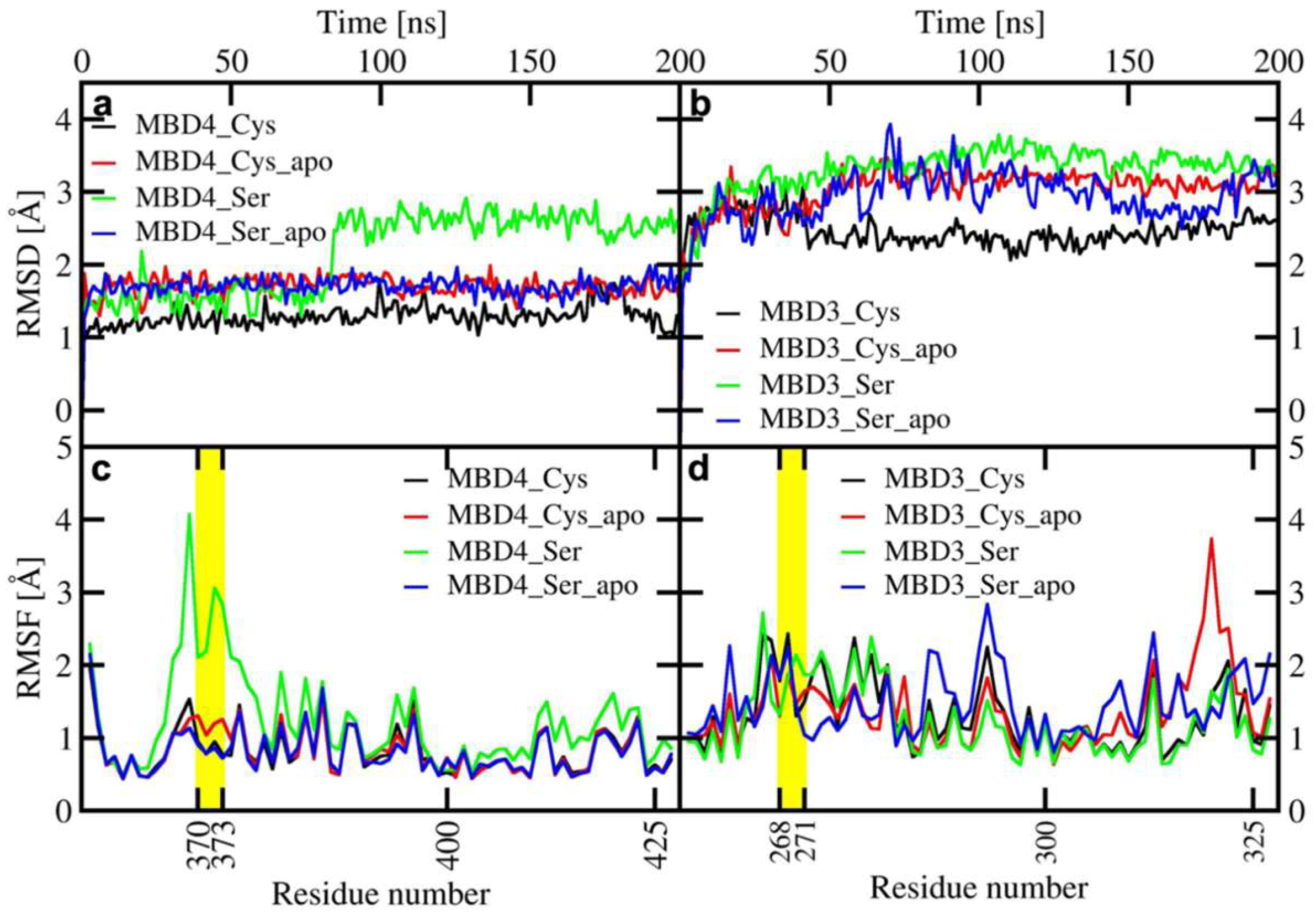
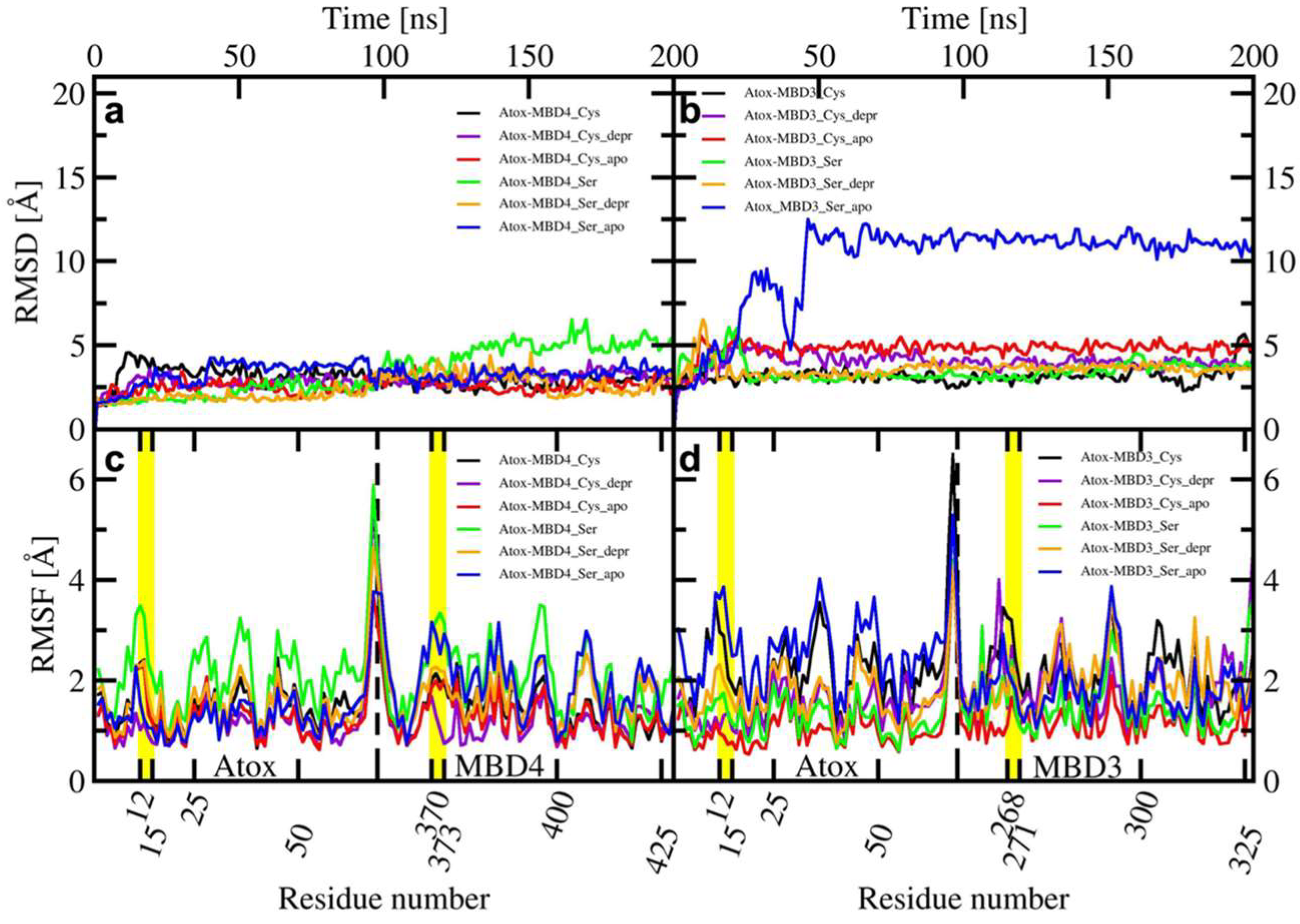
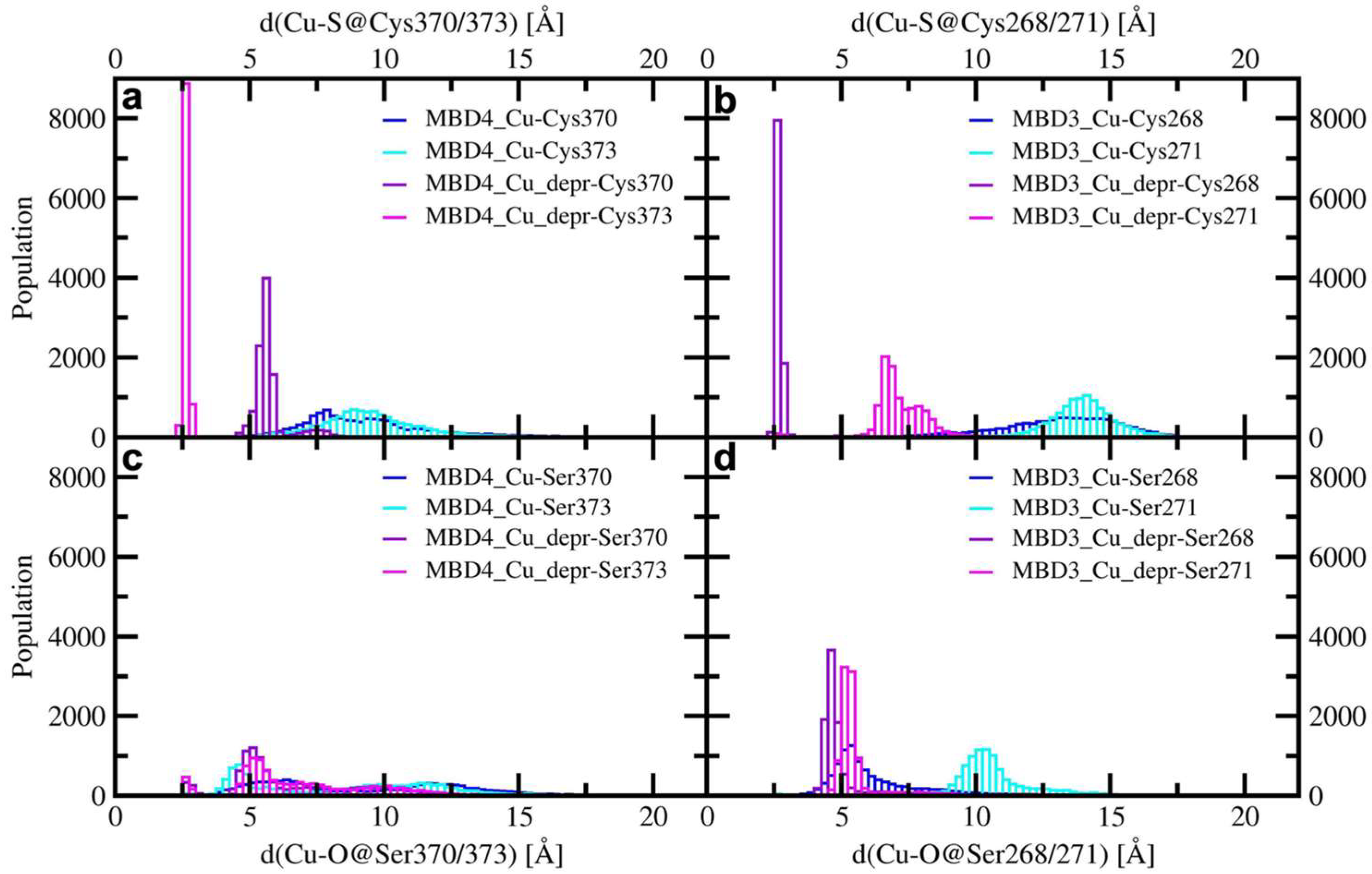
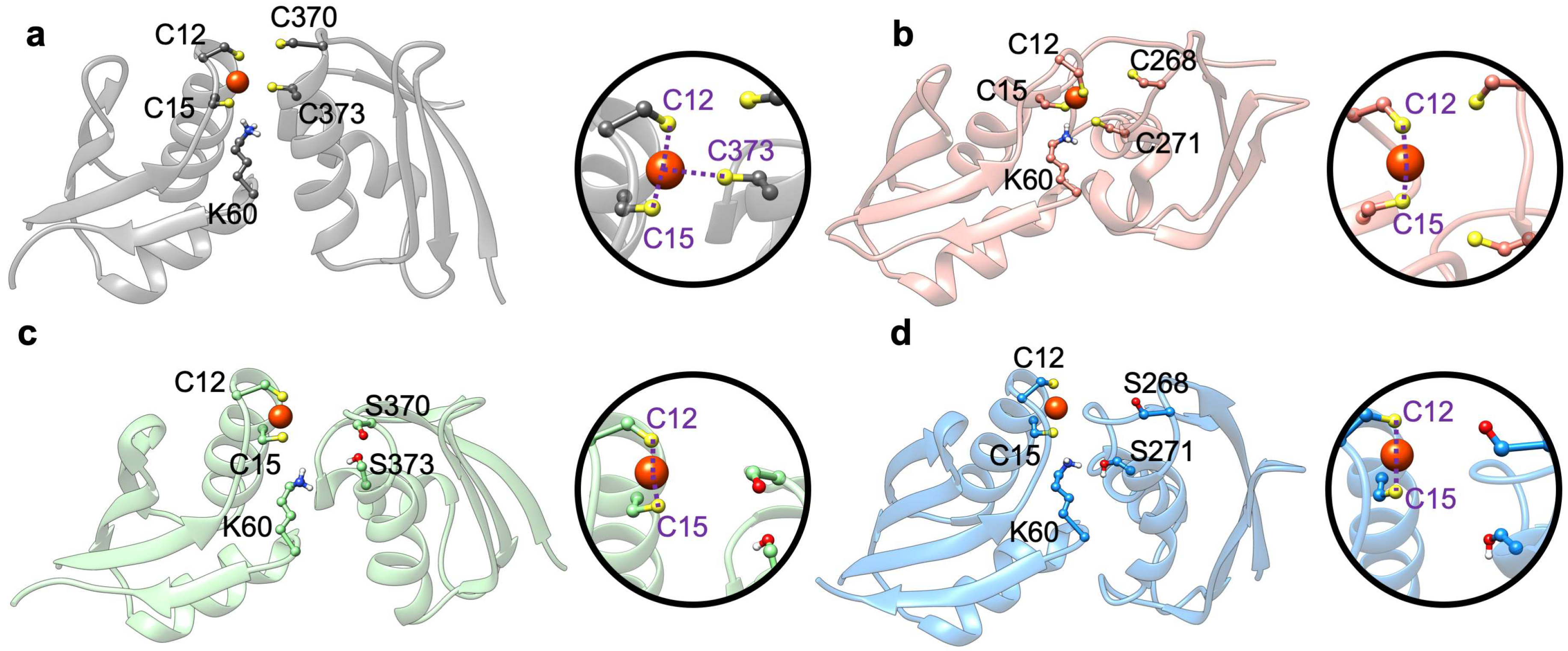
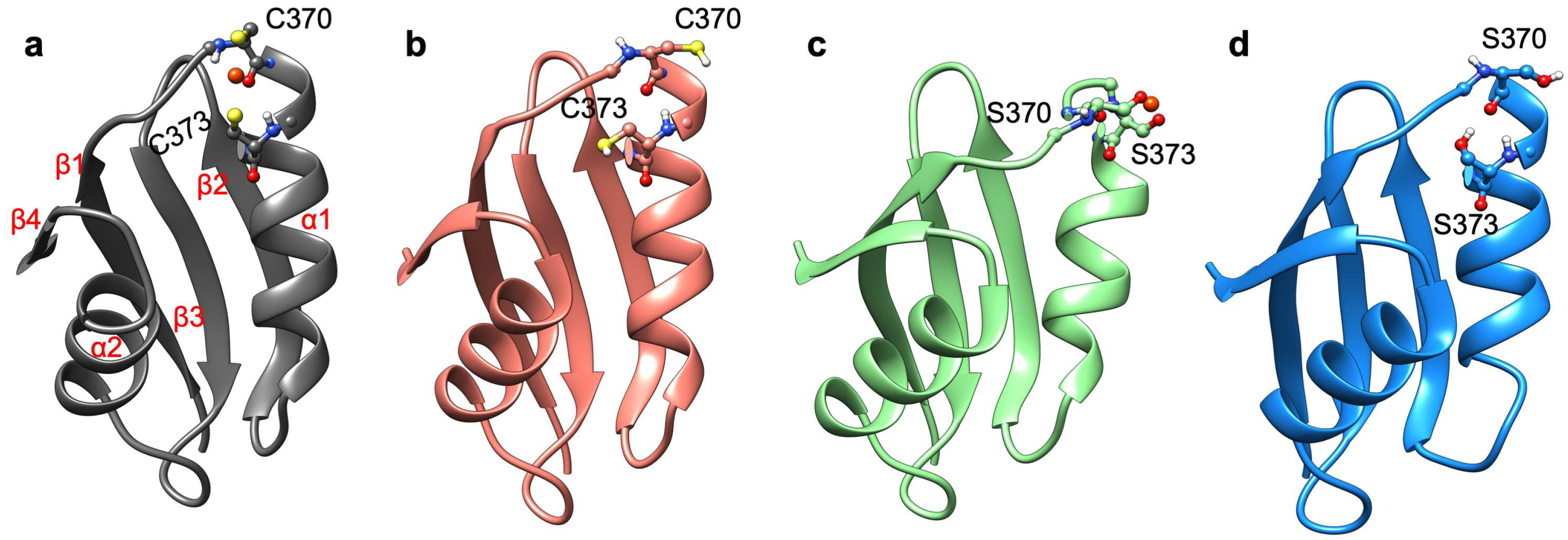
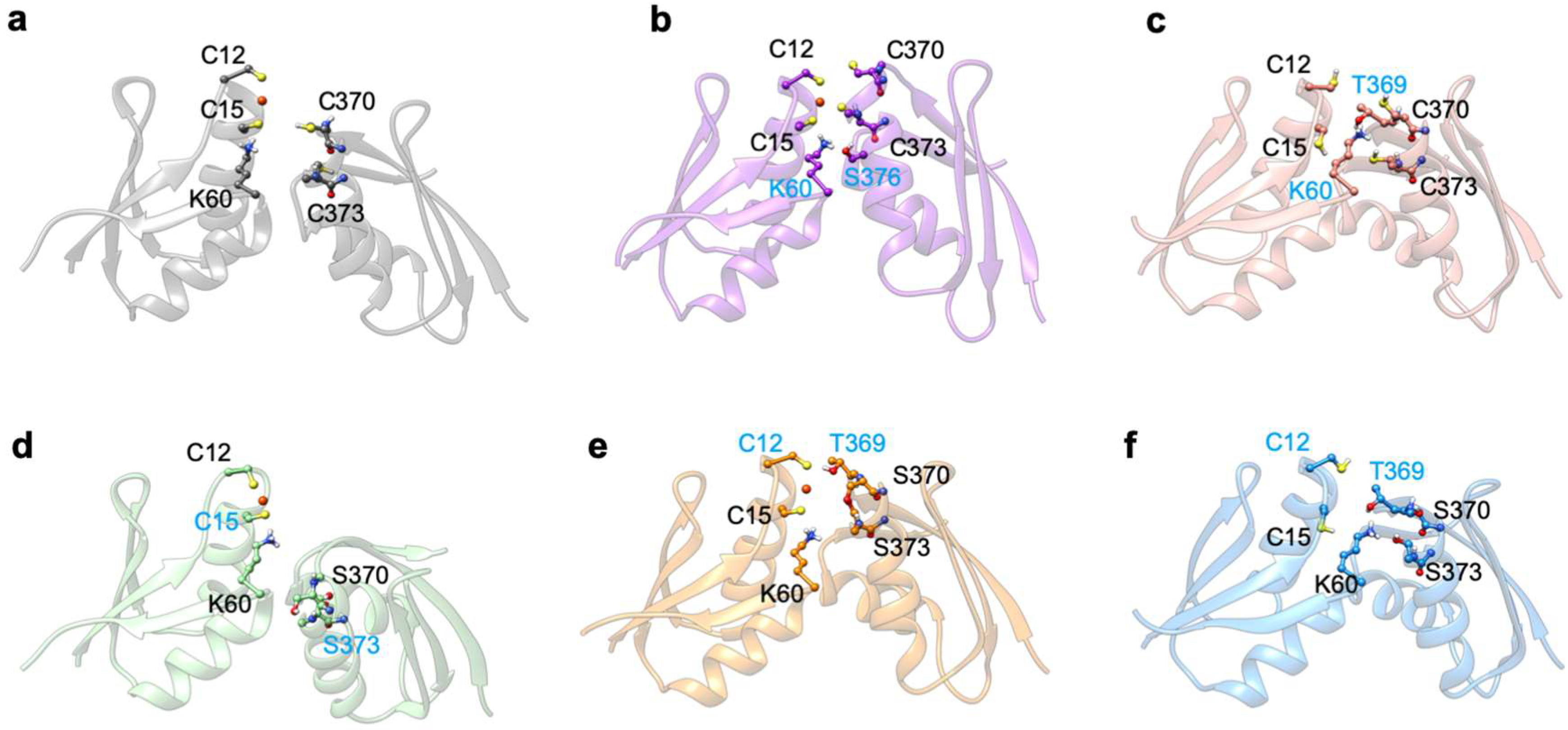
2.1. The Effect of Serine Mutations on ATP7B MBD3-4 and Their Interaction with Atox1
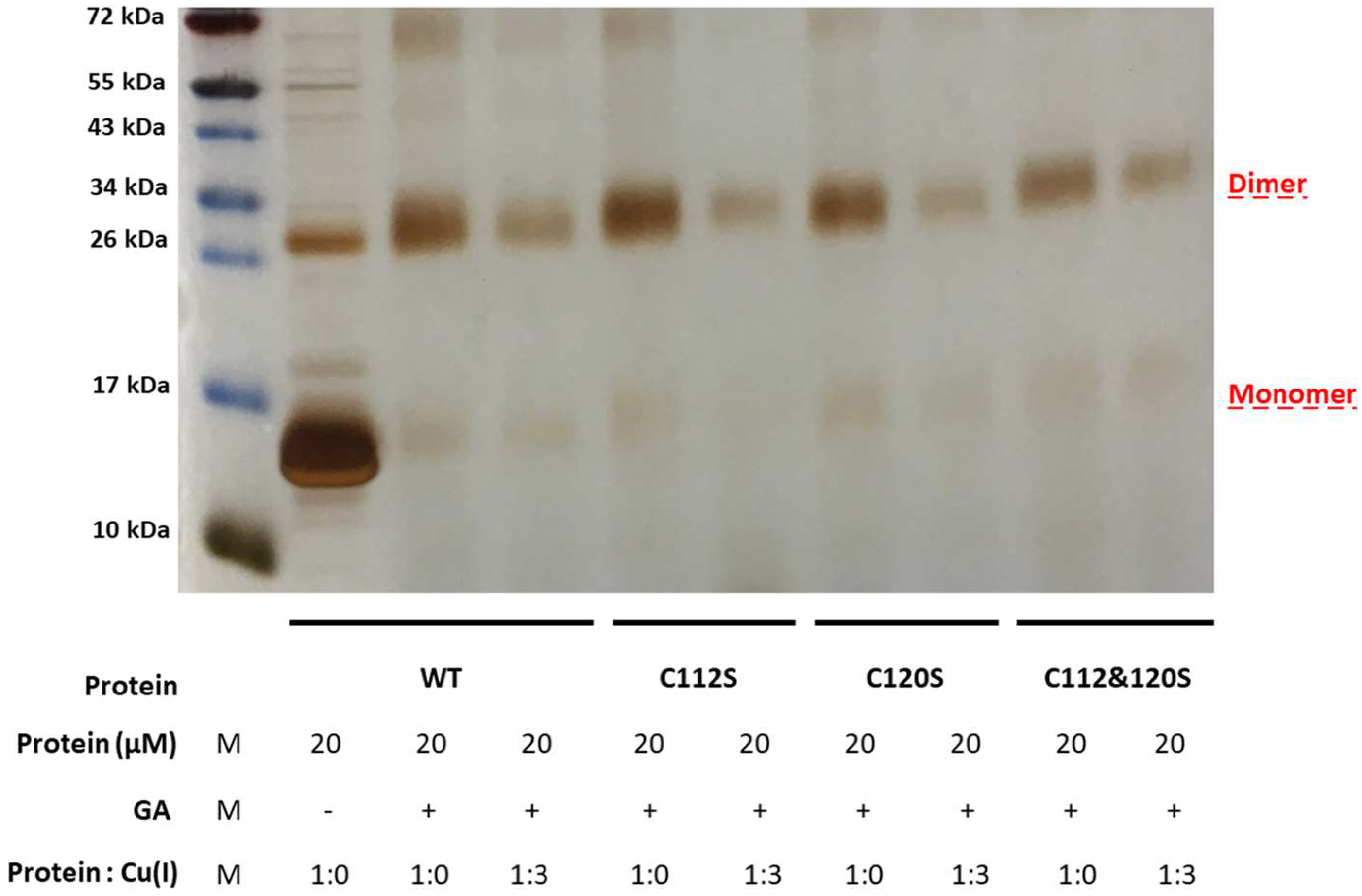
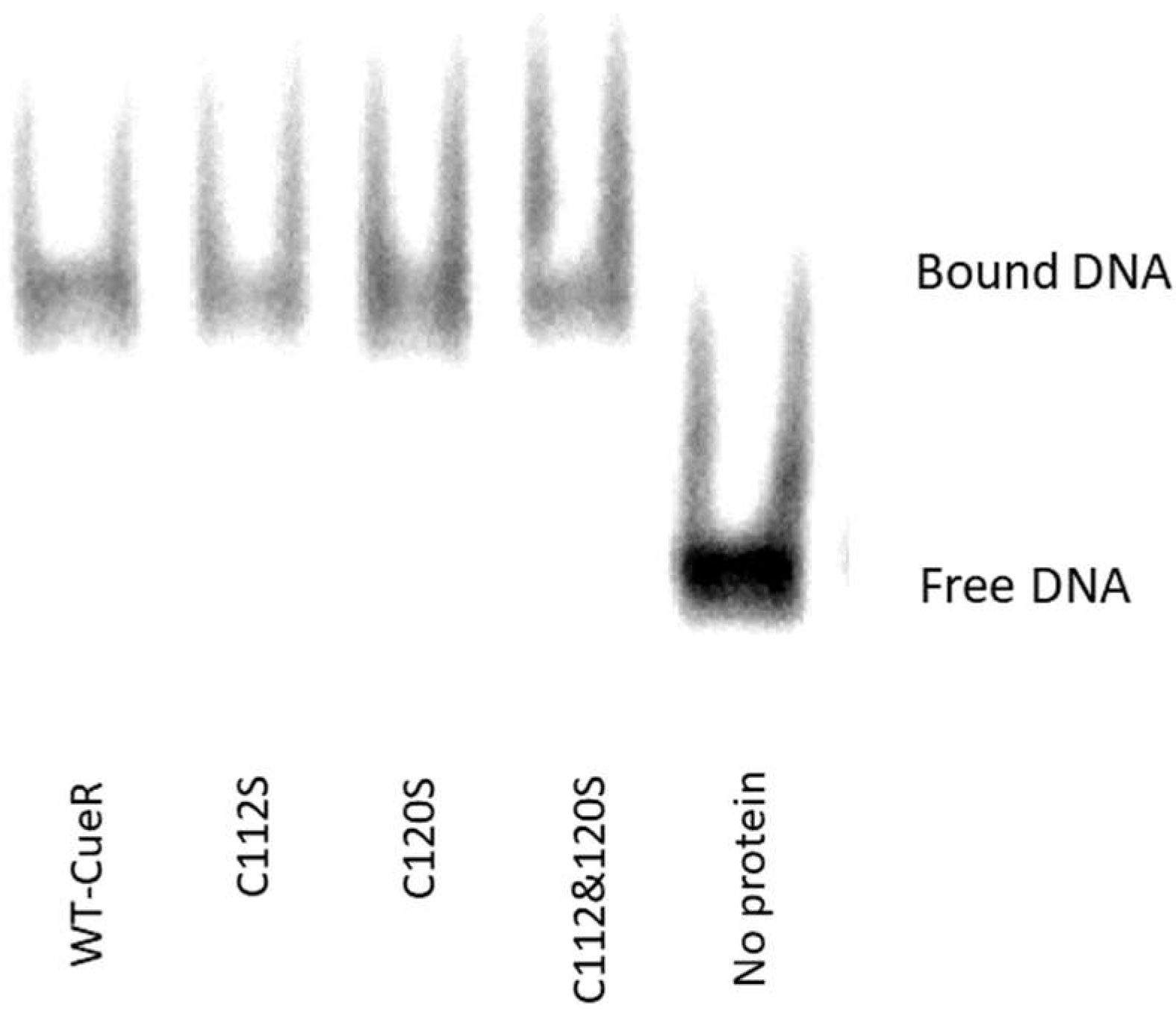
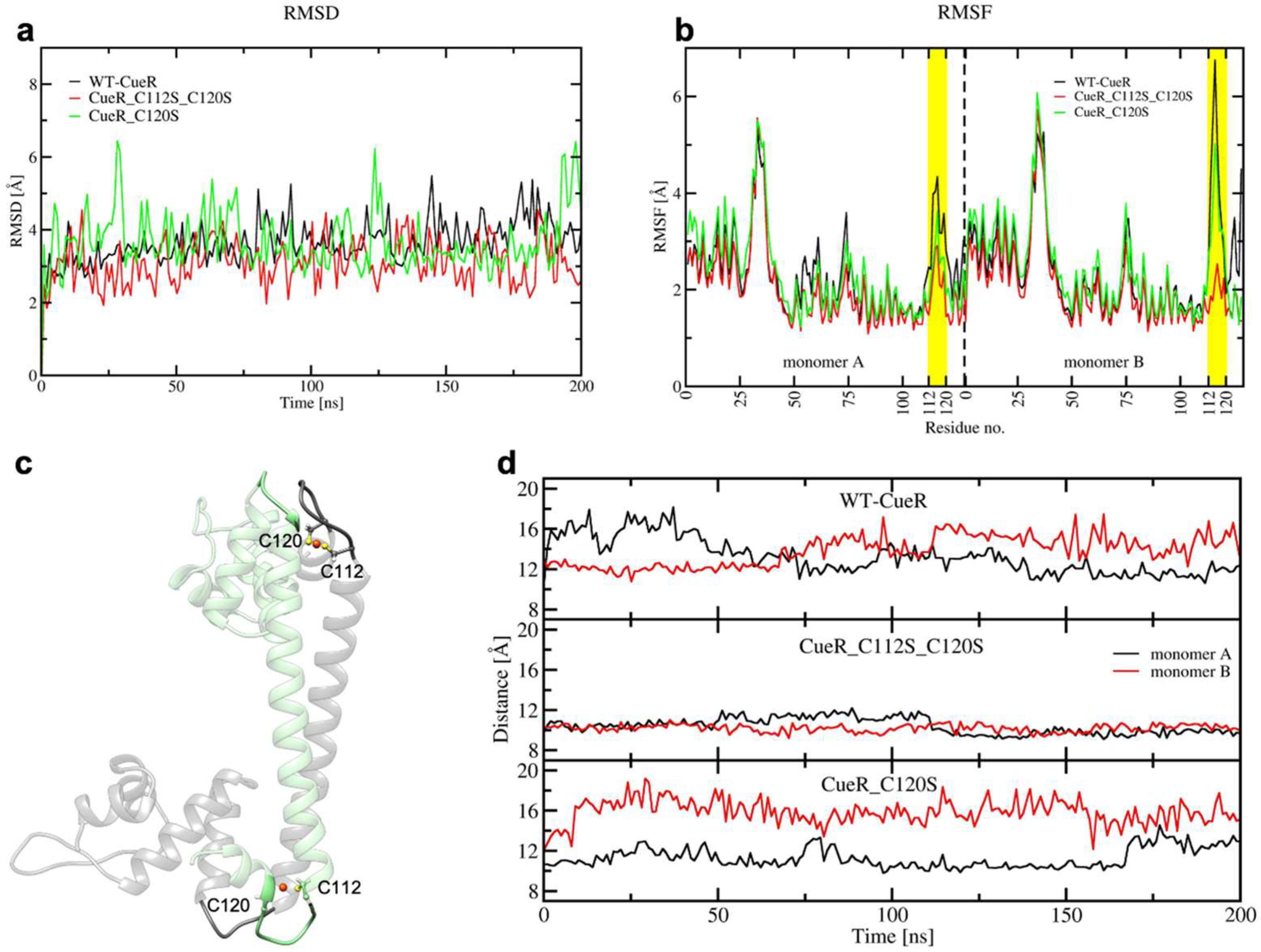
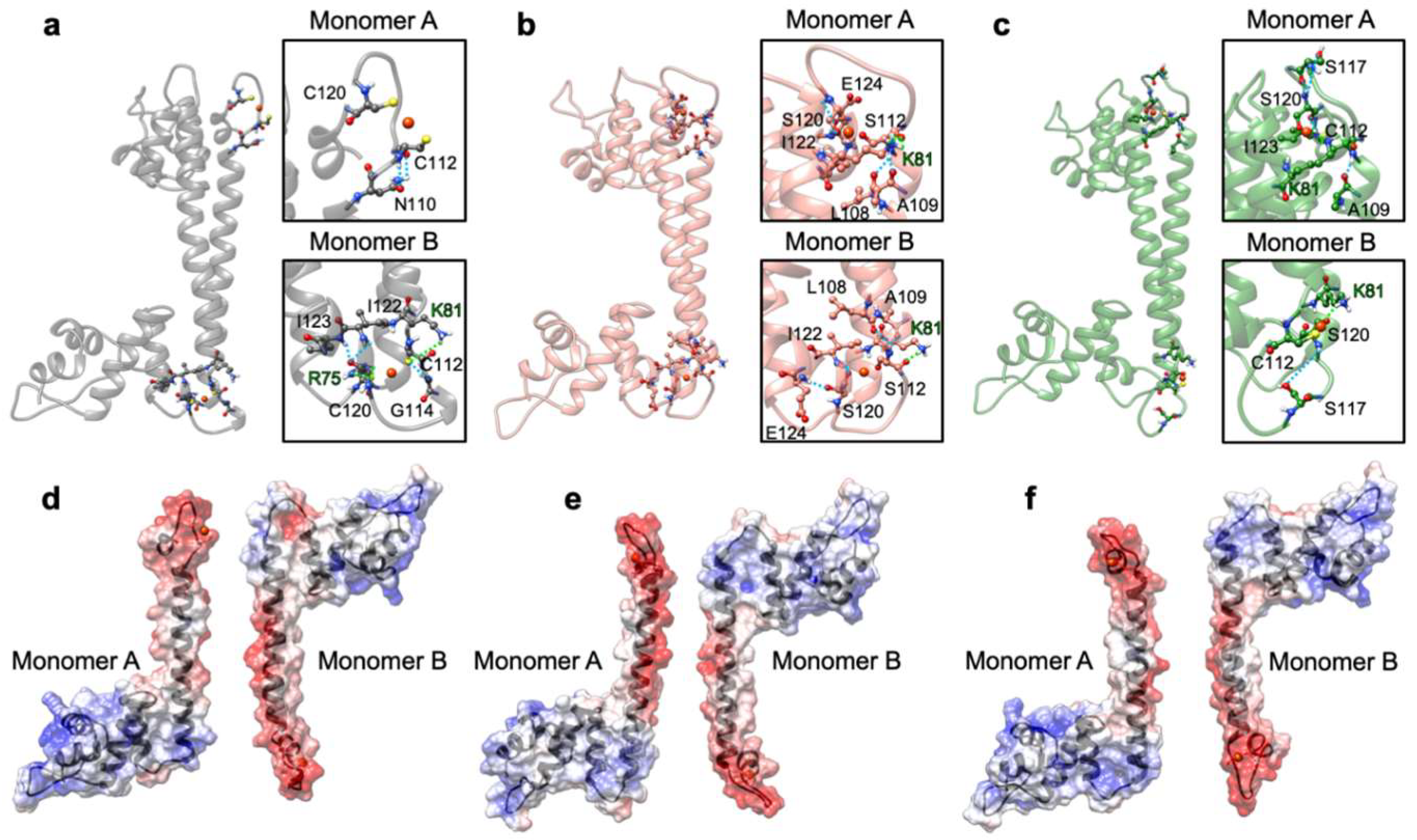
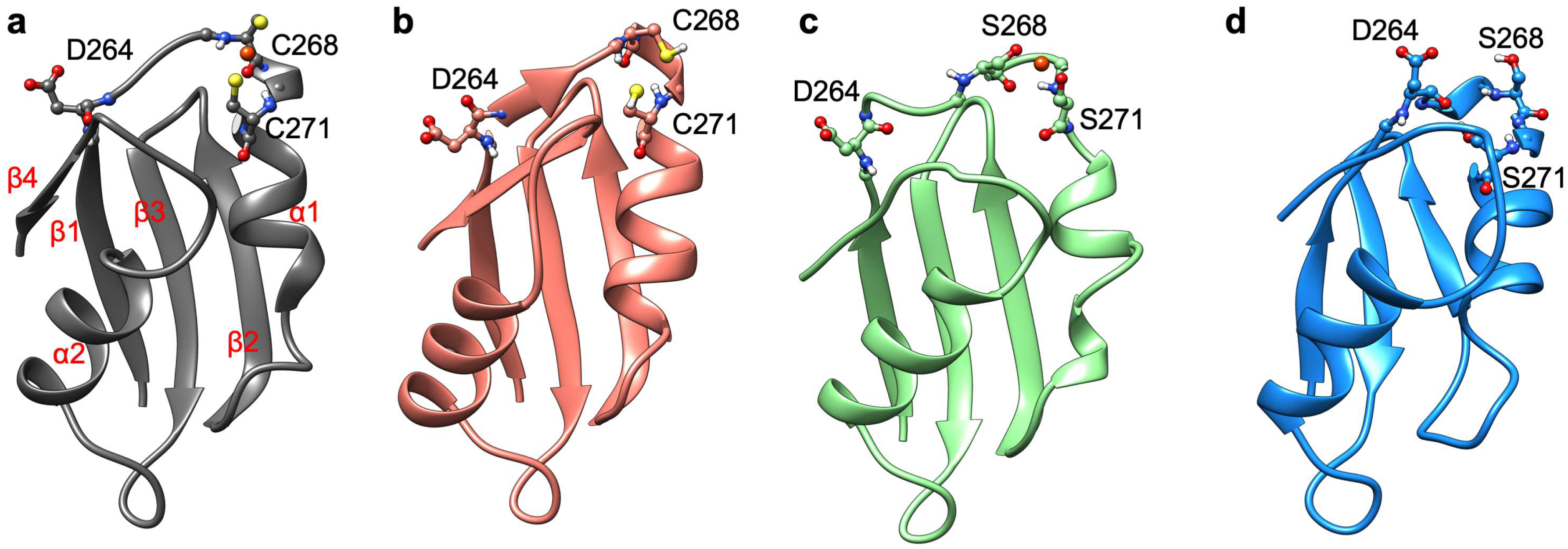
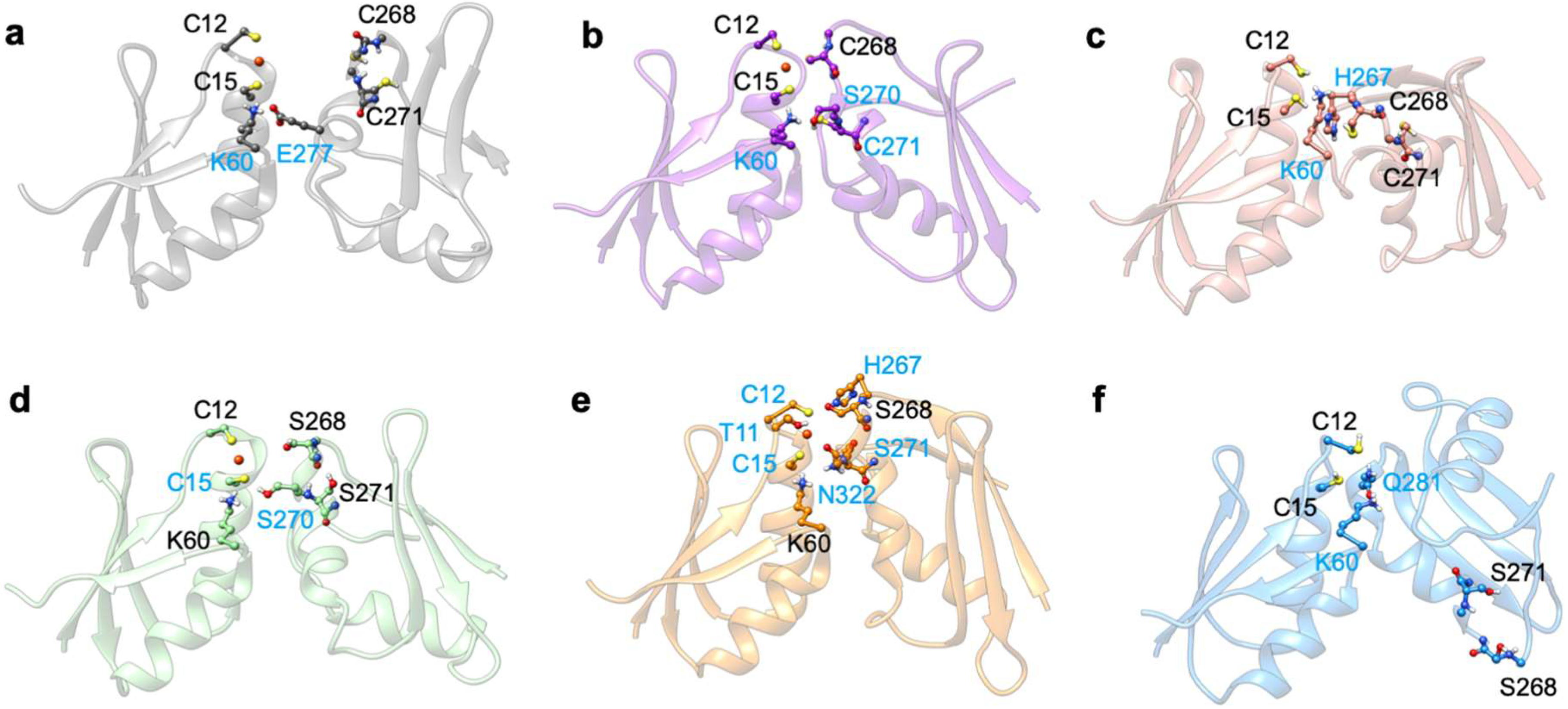
2.2. The Effect of Serine Mutations on CueR Protein
3. Discussion
4. Materials and Methods
4.1. Cloning, Expression, and Purification of ATP7B 3-4 Metal Binding Domains
4.2. CueR Cloning and Expression Protocol
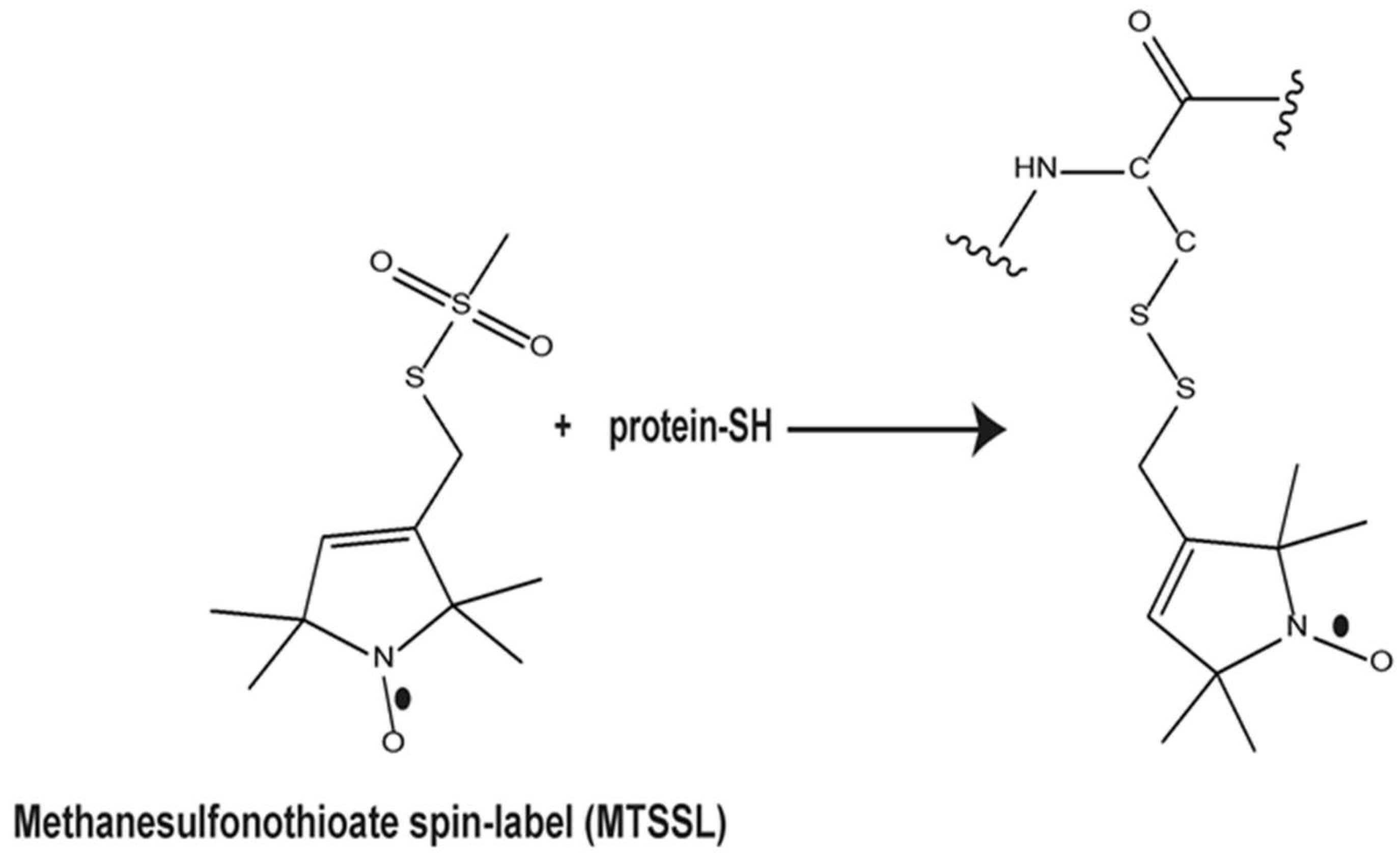
4.3. Spin Labeling
4.4. Cu(I) Addition
4.5. Cross-Linking Experiments
4.6. X-Band CW EPR Experiments
4.7. Circular Dichroism Characterization
4.8. Electrophoresis Mobility Shift Assay by Fluorescence
4.9. Modeling of Systems
4.10. Classical Molecular Dynamics
4.11. QM/MM Simulations
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Cys | Cysteine |
| Ser | Serine |
| WT | Wild type |
| CD | Circular dichroism |
| EPR | Electron paramagnetic resonance |
| CW-EPR | Continuous wave electron paramagnetic resonance |
| RT | Room temperature |
| O.N. | Overnight |
| MBD | Metal binding domain |
| NMR | Nuclear magnetic resonance |
| sm-FRET | Single molecule Förster energy transfer |
| sl | Spin-labeled |
| GA | Glutaraldehyde |
| MTSSL | Methanesulfonothiolate |
| EXAFS | Extended X-ray absorption structure |
| MD | Molecular dynamics |
| cMD | Classical molecular dynamics |
| QM/MM | Quantum Quantum-classical MD |
Appendix A






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | MBD4_CYS_apo | MBD4_CYS | MBD4_CYS_depr |
|---|---|---|---|
| H-Bond | |||
| Lys60–Thr369 ‡ (NZ-HZ*…OG) | 18.0 | / | / |
| Lys60 – Ser376 ‡ (NZ-HZ*…OG) | / | / | 60.9 |
| Cys370 – Cys373 (O…H-N) | 11.6 | / | / |
| System | MBD3_CYS_apo | MBD3_CYS | MBD3_CYS_depr |
|---|---|---|---|
| H-Bond | |||
| Lys60–His267 ‡ (NZ-HZ*…O) | 18.3 | / | / |
| Lys60–Ser270 ‡ (NZ-HZ*…OG) | / | / | 11.4 |
| Lys60–Cys271 ‡ (NZ-HZ*…SG) | / | / | 32.3 |
| Lys60–Glu277 ‡ (NZ-HZ*…OE1/2) | / | 50.7 | / |
| Cys268–Cys271 (O…H-N) | 14.7 | / | / |
| System | MBD4_SER_apo | MBD4_SER | MBD4_SER_depr |
|---|---|---|---|
| H-Bond | |||
| Cys12 – Thr369 ‡ (SG…HG-OG) | / | / | 20.7 |
| Cys15 – Ser373 ‡ (SG…HG-OG) | / | 16.7 | / |
| Lys60 – Thr369 ‡ (NZ-HZ*…OG) | 23.4 | / | / |
| Ser370 – Ser373 (O…H-N) (O…HG-OG) (N-H…OG) (OG…H-N) | 48.2 37.5 / / | 38.3 17.8 / / | / / 70.3 22.3 |
| System | MBD3_SER_apo | MBD3_SER | MBD3_SER_depr |
|---|---|---|---|
| H-Bond | |||
| Cys12–His267 ‡ (SG…HE2-NE2) | / | / | 54.1 |
| Cys15–Ser270 ‡ (SG…HG-OG) | / | 63.4 | / |
| Cys15–Asn322 ‡ (SG…HD22-ND2) | / | / | 15.1 |
| Lys60–Gln281 ‡ (NZ-HZ*…OE1) | 27.6 | / | / |
| Ser268–Ser271 (O…H-N) (O…HG-OG) | / / | 55.9 86.0 | / / |
| Ser271–Thr11 ‡ (OG…HG-OG) | / | / | 72.4 |
| System | WT_CueR (5.0 ps) | CueR_C112S_C120S (5.0 ps) | CueR_C120S (1.5 ps) |
|---|---|---|---|
| d(Cu–S/O@C112/C112S) | 2.18 ± 0.05 (2.20) | 1.88 ± 0.04 Å (1.87 ) | 2.19 ± 0.06 Å (2.20 ) |
| d(Cu–S/O@C120/C120S) | 2.17 ± 0.06 Å (2.20 ) | 1.87 ± 0.06 Å (1.87 ) | 1.83 ± 0.03 Å (1.87) |
| angle(S/O@C112/C112S–Cu–S/O@C120/C120S) | 169.8 ± 5.2 (170.0) | 170.4 ± 5.3 (170.0) | 167.7 ± 6.1 (170.0) |
| Cluster No. | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| System | |||||
| WT_CueR | 49.8 | 47.5 | 1.8 | 0.6 | 0.3 |
| CueR_C112S_C120S | 76.4 | 16.5 | 4.0 | 2.0 | 1.2 |
| CueR_C120S | 72.9 | 12.4 | 11.6 | 2.4 | 0.8 |
| System | WT_CueR | CueR_C112S_C120S | CueR_C120S | |||
|---|---|---|---|---|---|---|
| H-Bond | ||||||
| Cys/Ser112–Lys81 ‡ (O…HZ*-NZ) (SG/OG…HZ*-NZ) | / / | / 17.3 | 27.0 / | 38.9 / | 16.6 66.7 | / / |
| Cys/Ser112–Leu108 (N-H…O) | / | / | 11.6 | 4.9 | / | / |
| Cys/Ser112–Ala109 (N-H…O) | / | / | 33.9 | 44.8 | 30.0 | / |
| Cys/Ser112–Asn110 (O…HD2*-ND2) (N-H…OD1) | 14.2 56.5 | / / | / / | / / | / / | / / |
| Cys/Ser112–Gly114 (SG/OG…H-N) | / | 17.9 | / | / | / | / |
| Cys/Ser120–Arg75 ‡ (SG/OG…HH1*-NH1) (SG/OG…HH2*-NH2) | / / | 74.8% 83.5% | / / | / / | / / | |
| Cys/Ser120–Lys81 ‡ (SG/OG…HZ*-NZ) | / | / | / | / | / | 13.7 |
| Cys/Ser120–Ser117 (N-H…O) (N-H…OG) | / / | / / | / / | / / | 11.9 25.6 | / 14.7 |
| Cys/Ser120–Ile122 (O…H-N) (SG/OG…H-N) | / / | 20.9 / | / 27.2 | / 21.1 | / / | / / |
| Cys/Ser120–Ile123 (O…H-N) | / | 14.6 | / | / | 43.3 | / |
| Cys/Ser120–Glu124 (O…H-N) | / | / | 47.0 | 47.0 | / | / |
References
- Prohaska, J.R. Role of copper transporters in copper homeostasis. Am. J. Clin. Nutr. 2008, 88, 826S–829S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lutsenko, S. Human copper homeostasis: A network of interconnected pathways. Curr. Opin. Chem. Biol. 2010, 14, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Burkhead, J.L.; Gogolin Reynolds, K.A.; Abdel-Ghany, S.E.; Cohu, C.M.; Pilon, M. Copper homeostasis. New Phytol. 2009, 182, 799–816. [Google Scholar] [CrossRef] [PubMed]
- Wernimont, A.; Huffman, D.L.; Lamb, A.L.; O’Halloran, T.V.; Rosenzweig, A.C. Structural basis for copper transfer by the metallochaperone for the menkes/wilson disease proteins. Nat. Struct. Biol. 2000, 7, 766–771. [Google Scholar] [PubMed]
- Badarau, A.; Dennison, C. Copper trafficking mechanism of cxxc-containing domains: Insight from the ph-dependence of their cu(i) affinities. J. Am. Chem. Soc. 2011, 133, 2983–2988. [Google Scholar] [CrossRef]
- Kuo, M.T.; Fu, S.; Savaraj, N.; Chen, H.W. Role of the human high-affinity copper transporter in copper homeostasis regulation and cisplatin sensitivity in cancer chemotherapy. Cancer Res. 2012, 72, 4616–4621. [Google Scholar] [CrossRef]
- Donnelly, P.S.; Xiao, Z.; Wedd, A.G. Copper and alzheimer’s disease. Curr. Opin. Chem. Biol. 2007, 11, 128–133. [Google Scholar] [CrossRef]
- Larson, C.A.; Adams, P.L.; Jandial, D.D.; Blair, B.G.; Safaei, R.; Howell, S.B. The role of the n-terminus of mammalian copper transporter 1 in the cellular accumulation of cisplatin. Biochem. Pharm. 2010, 80, 448–454. [Google Scholar] [CrossRef]
- Du, X.; Wang, X.; Li, H.; Sun, H. Comaprison between copper and cisplatin transport mediated by human copper transport 1 (hctr1). Metallomics 2012, 4, 679–685. [Google Scholar] [CrossRef]
- Boal, A.K.; Rosenzweig, A.C. Crystal structures of cisplatin bound to human copper chaperone. J. Am. Chem. Soc. 2009, 131, 14196–14197. [Google Scholar] [CrossRef]
- Spinello, A.; Magistrato, A. An omics perspective to the molecular mechanisms of anticancer metallo-drugs in the computational microscope era. Expert Opin. Drug Discov. 2017, 12, 813–825. [Google Scholar] [CrossRef] [PubMed]
- Salina, E.G.; Huszar, S.; Zemanova, J.; Keruchenko, J.; Riabova, O.; Kazakova, E.; Grigorov, A.; Azhikina, T.; Kaprelyants, A.; Mikusova, K.; et al. Copper-related toxicity in replicating and dormant mycobacterium tuberculosis caused by 1-hydroxy-5-r-pyridine-2(1h)-thiones. Metallomics 2018, 10, 992–1002. [Google Scholar] [CrossRef] [PubMed]
- Dalecki, A.G.; Crawford, C.L.; Wolschendorf, F. Copper and antibiotics: Discovery, modes of action, and opportunities for medicinal applications. Adv. Microb. Physiol. 2017, 70, 193–260. [Google Scholar] [PubMed]
- Banci, L.; Bertini, I.; Cantini, F.; Chasapis, C.T.; Hadjiliadis, N.; Rosato, A. A nmr study of the interaction of a three-domain construct of atp7a with copper(i) and copper(i)-hah1. J. Biol. Chem. 2005, 280, 38259–38263. [Google Scholar] [CrossRef] [PubMed]
- Luchinat, E.; Gianoncelli, A.; Mello, T.; Galli, A.; Banci, L. Combining in-cell nmr and X-ray fluorescence microscopy to reveal the intracellular maturation states of human superoxide dismutase 1. Chem. Commun. 2015, 51, 584–587. [Google Scholar] [CrossRef] [PubMed]
- Robinson, N.J.; Winge, D.R. Copper metallochaperones. Annu. Rev. Biochem. 2010, 79, 537–562. [Google Scholar] [CrossRef]
- Philips, S.J.; Canalizo-Hernandez, M.; Yildirim, I.; Schatz, G.C.; Mondragon, A.; O’Halloran, T.V. Allosteric transcriptional regulation via changes in the overall topology of the core promotor. Science 2015, 349, 877–881. [Google Scholar] [CrossRef] [PubMed]
- De Feo, C.J.; Aller, S.G.; Siluvai, G.S.; Blackburn, N.J.; Unger, V.M. Three-dimensional structure of the human copper transporter hctr1. Proc. Natl. Acad. Sci. USA 2009, 106, 4237–4242. [Google Scholar] [CrossRef]
- Giedroc, D.P.; Arunkumar, A.I. Metal sensor proteins: Nature’s metalloregulated allosteric switches. Dalton Trans. 2007, 3107–3120. [Google Scholar] [CrossRef]
- Tottey, S.; Harvie, D.R.; Robinson, N.J. Understanding how cells allocate metals using metal sensors and metallochaperone. Acc. Chem. Res. 2005, 38, 775–783. [Google Scholar] [CrossRef]
- Bagai, I.; Rensing, C.; Blackburn, N.J.; McEvoy, M.M. Direct metal transfer between periplasmic proteins identifies a bacterial copper chaperone. Biochemistry-Us 2008, 47, 11408–11414. [Google Scholar] [CrossRef] [PubMed]
- Loftin, I.R.; Franke, S.; Blackburn, N.J.; McEvoy, M.M. Unusual cu(i)/ag(i) coordination of Escherichia coli cusf as revealed by atomic resolution crystallography and X-ray absorption spectroscopy. Protein Sci. 2007, 16, 2287–2293. [Google Scholar] [CrossRef] [PubMed]
- Joshi, C.P.; Panda, D.; Martell, D.J.; Andoy, N.M.; Chen, T.-Y.; Gaballa, A.; Helmann, J.D.; Chen, P. Direct substitution and assisted dossociation pathways for turning off transcription by a merr-family metalloregulator. Proc. Natl. Acad. Sci. USA 2012, 109, 15121–15126. [Google Scholar] [CrossRef] [PubMed]
- Keller, A.M.; Benitez, J.J.; Klarin, D.; Zhong, L.; Goldfogel, M.; Yang, F.; Chen, T.-Y.; Chen, P. Dynamic multi-body protein interactions suggest versatile pathways for copper trafficking. J. Am. Chem. Soc. 2012, 134, 8934–8943. [Google Scholar] [CrossRef]
- Martell, D.J.; Joshi, C.P.; Gaballa, A.; Santiago, A.G.; Chen, T.-Y.; Jung, W.; Helmann, J.D.; Chen, P. Metalloregulator cuer biases rna polymerase’s kinetic sampling of dead-end or open complex to repress or activate transcription. Proc. Natl. Acad. Sci. USA 2015, 112, 13467–13472. [Google Scholar] [CrossRef] [PubMed]
- Levy, A.R.; Nissim, M.; Mendelman, N.; Chill, J.; Ruthstein, S. Ctr1 intracellular loop is involved in the copper transfer mechanism to the atox1 metallochaperone. J. Phys. Chem. B 2016, 120, 12334–12345. [Google Scholar] [CrossRef] [PubMed]
- Levy, A.R.; Turgeman, M.; Gevorkyan-Aiapetov, L.; Ruthstein, S. The structural flexibility of the human copper chaperone atox1: Insights from combined pulsed epr studies and computations. Protein Sci. 2017, 26, 1609–1618. [Google Scholar] [CrossRef]
- Meir, A.; Abdelhai, A.; Moskovitz, Y.; Ruthstein, S. Epr spectroscopy targets conformational and topological changes in the E. coli membrane fusion cusb dimer upon cu(i) binding. Biophys. J. 2017, 112, 2494–2502. [Google Scholar] [CrossRef]
- Sameach, H.; Narunsky, H.; Azoulay Ginsburg, S.; Moskovitz, Y.; Zehavi, Y.; Jueven-Gershon, T.; Ben-Tal, N.; Ruthstein, S. Structural and dynamics characterization of the merr family metalloregulator cuer in its respression and activation states. Structure 2017, 25, 988–996. [Google Scholar] [CrossRef]
- Ucisik, M.N.; Chakravorty, D.K.; Merz, J.K.M. Structure and dynamics of the n-terminal domain of the cu(i) binding protein cusb. Biochemistry 2013, 52, 6911–6923. [Google Scholar] [CrossRef]
- Dhruva, K.; Chakravorty, D.K.; Wang, B.; Ucisik, M.N.; Merz, J.K.M. Insights into the cation-π interaction at the metal binding site of the copper metallochaperone cusf. J. Am. Chem. Soc. 2011, 113, 19330–19333. [Google Scholar]
- Van Dongen, E.M.W.M.; Klomp, L.W.J.; Merkx, M. Copper-dependent protein-protein interactions studied by yeast two-hybrid analysis. Biochem. Biophys. Res. Commun. 2004, 323, 789–795. [Google Scholar] [CrossRef] [PubMed]
- Tsigelny, I.F.; Sharikov, Y.; Greenberg, J.P.; Miller, M.A.; Kouznstsova, V.L.; Larson, C.A.; Howell, S.B. An all-atom model of the structure of human copper transporter 1. Cell. Biochem. Biophys. 2012, 63, 223–234. [Google Scholar] [CrossRef] [PubMed]
- Jung, W.; Chen, P. Biphasic unbinding of zur from DNA for transcription (de)repression in live bacteria. BioRxiv 2018, 434738. [Google Scholar] [CrossRef]
- Heaton, D.; Nittis, T.; Srinivasan, C.; Winge, D.R. Mutational analysis of the mitochondrial copper metallochaperone cox17. J. Biol. Chem. 2000, 275, 37582–37587. [Google Scholar] [CrossRef]
- Chen, K.; Yuldasheva, J.E.; Penner-Hahn, J.E.; O’Halloran, T.V. An atypical linear cu(i)−s2 center constitutes the high-affinity metal-sensing site in the cuer metalloregulatory protein. J. Am. Chem. Soc. 2003, 125, 12088–12089. [Google Scholar] [CrossRef] [PubMed]
- Stasser, J.P.; Eisses, J.F.; Barry, A.N.; Kaplan, J.; Blackburn, N.J. Cysteine-to-serine mutants of the human copper chaperone to superoxide dismutase reveal a copper cluster at a domain iii dimer interface. Biochemistry 2005, 44, 3143–3152. [Google Scholar] [CrossRef]
- Hoyau, S.; Ohanessian, G. Absolute affinities of a-amino acids for cu+ in the gas phase. A theoretical study. J. Am. Chem. Soc. 1997, 119, 2016–2024. [Google Scholar] [CrossRef]
- Portnoy, M.E.; Rosenzweig, A.C.; Rae, T.; Huffman, D.L.; O’Halloran, T.V.; Culotta, V.C. Strcuture-function analyses of the atx1 metallochaperone. J. Biol. Chem. 1999, 274, 15041–15045. [Google Scholar] [CrossRef]
- Rosenzweig, A.C.; Huffman, D.L.; Hou, M.Y.; Wernimont, A.K.; Pufahl, R.A.; O’Halloran, T.V. Crystal structure of the atx1 metallochaperone protein at 1.02 a resolution. Structure 1999, 7, 605–617. [Google Scholar] [CrossRef]
- Changela, A.; Chen, K.; Holschen, J.; Outten, C.E.; O’Halloran, T.V.; Mondragon, A. Molecular basis of metal-ion selectivity and zeptomolar sensitivity by cuer. Science 2003, 301, 1383–1387. [Google Scholar] [CrossRef] [PubMed]
- Banci, L.; Bertini, I.; Cantini, F.; Rosenzweig, A.C.; Yatsunyk, L.A. Metal binding domains 3 and 4 of the wilson disease protein: Solution structure and interaction with the copper(i) chaperone hah1. Biochemistry 2008, 47, 7423–7429. [Google Scholar] [CrossRef] [PubMed]
- Shenberger, Y.; Yarmiayev, V.; Ruthstein, S. Exploring the interaction between the human copper transporter, ctr1, c-terminal domain and a methionine motif in the presence of cu(i) and ag(i) ions, using epr spectrosocopy. Mol. Phys. 2013, 111, 2980–2991. [Google Scholar] [CrossRef]
- Cai, Q.; Kusnetzow, A.K.; Hubbell, W.L.; Haworth, I.S.; Gacho, G.P.C.; Van Eps, N.; Hideg, K.; Chambers, E.J.; Qin, P.Z. Site-directed spin labeling measurements of nanometer distances in nucleic acids using a sequence-independent nitroxide probe. Nucleic Acids Res. 2006, 34, 4722–4730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Columbus, L.; Hubbell, W.L. A new spin on protein dynamics. Trends Biochem. Sci. 2002, 27, 288–295. [Google Scholar] [CrossRef]
- Mchaourab, H.S.; Oh, K.J.; Fang, C.J.; Hubbell, W.L. Conformation of T4 Lysozyme in Solution. Hinge-Bending Motion and the Substrate-Induced Conformational Transition Studied by Site-Directed Spin Labeling. Biochemistry 1997, 36, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Shin, Y.-K.; Levinthal, C.; Levinthal, F.; Hubbell, W.L. Colicin e1 binding to membranes: Time-resolved studies of spin-labeled mutants. Science 1993, 259, 960–963. [Google Scholar] [CrossRef]
- Vamvouka, M.; Cieslak, J.; Van Eps, N.; Hubbell, W.; Gross, A. The structure of the lipid-embedded potassium channel voltage sensor determined by double-electron-electron resonance spectroscopy. Protein Sci. 2008, 17, 506–517. [Google Scholar] [CrossRef] [Green Version]
- Qasem, Z.; Pavlin, M.; Ritacco, I.; Gevorkyan-Airapetov, L.; Magistrato, A.; Ruthstein, S. The pivotal role of mbd4-atp7b in the human cu(i) excretion path as revealed by epr experiments and all-atom simulations. Metallomics 2019, in press. [Google Scholar] [CrossRef]
- Sgrignani, J.; Casalino, L.; Doro, F.; Spinello, A.; Magistrato, A. Can multiscale simulations unravel the function of metallo-enzymes to improve knowledge-based drug discovery? Future Med. Chem. 2019, 11, 771–791. [Google Scholar] [CrossRef]
- Vidossich, P.; Magistrato, A. Qm/mm molecular dynamics studies of metal binding proteins. Biomolecules 2014, 4, 616–645. [Google Scholar] [CrossRef] [PubMed]
- Banci, L.; Bertini, I.; Calderone, V.; Della-Malva, N.; Felli, I.C.; Pavelkova, A.; Rosato, A. Copper(i)-mediated protein-protein interactions result from suboptimal interaction surfaces. Biochem. J. 2009, 422, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Casalino, L.; Palermo, G.; Rothlisberger, U.; Magistrato, A. Who activates the nucleophile in ribozyme catalysis? An answer from the splicing mechanism of group ii introns. J. Am. Chem. Soc. 2016, 138, 10374–10377. [Google Scholar] [CrossRef] [PubMed]
- Hussain, F.; Rodriguez-Granillo, A.; Wittung-Stafshede, P. Lysine-60 in copper chaperone atox1 plays an essential role in adduct formation with a target wilson disease domain. J. Am. Chem. Soc. 2009, 131, 16371–16373. [Google Scholar] [CrossRef] [PubMed]
- Magistrato, A.; Pavlin, M.; Qasem, Z.; Ruthstein, S. Copper trafficking in eukaryotic systems: Current knowledge from experimental and computational efforts. Curr. Opin. Struct. Biol. 2019, 58, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Wernimont, A.K.; Yatsunyk, L.A.; Rosenzweig, A.C. Binding of copper(i) by the wilson disease protein and its copper chaperone. J. Biol. Chem. 2004, 279, 12269–12276. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Granillo, A.; Wittung-Stafshede, P. Structure and dynamics of cu(i) binding in copper chaperone atox1 and copz: A computer simulation study. J. Phys. Chem. B 2008, 112, 4583–4593. [Google Scholar] [CrossRef]
- Erijman, A.; Dantes, A.; Bernheim, R.; Shifman, J.M.; Peleg, Y. Transfer-pcr (tpcr): A highway for DNA cloning and protein engineering. J. Struct. Biol. 2011, 175, 171–177. [Google Scholar] [CrossRef]
- Levy, A.R.; Yarmiayev, V.; Moskovitz, Y.; Ruthstein, S. Probing the structural flexibility of the human copper metallochaperone atox1 dimer and its interaction with the ctr1 c-terminal domain. J. Phys. Chem. B 2014, 118, 5832–5842. [Google Scholar] [CrossRef]
- Stoll, S.; Schweiger, A. Easyspin, a comprehensive software package for spectral simulation and analysis in epr. J. Magn. Reson. 2006, 178, 42–55. [Google Scholar] [CrossRef]
- Olsson, M.H.; Sondergaard, C.R.; Rostkowski, M.; Jensen, J.H. Propka3: Consistent treatment of internal and surface residues in empirical pka predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. Ff14sb: Improving the accuracy of protein side chain and backbone parameters from ff99sb. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed]
- Op’t Holt, B.T.; Merz, K.M., Jr. Insights into cu(i) exchange in hah1 using quantum mechanical and molecular simulations. Biochemistry 2007, 46, 8816–8826. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Case, D.A.; Betz, R.M.; Cerutti, D.S.; Cheatham, I.T.E.; Darden, T.A.; Duke, R.E.; Giese, T.J.; Gohlke, H.; Goetz, A.W.; Homeyer, N.; et al. Amber 2016; University of California: San Francisco, CA, USA, 2016. [Google Scholar]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Grest, G.S.; Kremer, K. Molecular dynamics simulation for polymers in the presence of a heat bath. Phys. Rev. A Gen. Phys. 1986, 33, 3628–3631. [Google Scholar] [CrossRef] [PubMed]
- Dolinsky, T.J.; Nielsen, J.E.; McCammon, J.A.; Baker, N.A. Pdb2pqr: An automated pipeline for the setup of poisson-boltzmann electrostatics calculations. Nucleic Acids Res. 2004, 32, W665–W667. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. Ucsf chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Spinello, A.; Pavlin, M.; Casalino, L.; Magistrato, A. A dehydrogenase dual hydrogen abstraction mechanism promotes estrogen biosynthesis: Can we expand the functional annotation of the aromatase enzyme? Chemistry 2018, 24, 10840–10849. [Google Scholar] [CrossRef]
- Hutter, J.; Iannuzzi, M.; Schiffmann, F.; VandeVondele, J. Cp2k: Atomistic simulations of condensed matter systems. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2014, 4, 15–25. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A Gen. Phys. 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Yang, W.; Parr, R.G. Development of the colle-salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B Condens. Matter 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- VandeVondele, J.; Krack, M.; Mohamed, F.; Parrinello, M.; Chassaing, T.; Hutter, J. Quickstep: Fast and accurate density functional calculations using a mixed gaussian and plane waves approach. Comput. Phys. Commun. 2005, 167, 103–128. [Google Scholar] [CrossRef]
- VandeVondele, J.; Hutter, J. Gaussian basis sets for accurate calculations on molecular systems in gas and condensed phases. J. Chem. Phys. 2007, 127, 114105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goedecker, S.; Teter, M.; Hutter, J. Separable dual-space gaussian pseudopotentials. Phys. Rev. B Condens. Matter 1996, 54, 1703–1710. [Google Scholar] [CrossRef] [PubMed]
- Hartwigsen, C.; Goedecker, S.; Hutter, J. Relativistic separable dual-space gaussian pseudopotentials from h to rn. Phys. Rev. B 1998, 58, 3641–3662. [Google Scholar] [CrossRef]
- Brunk, E.; Rothlisberger, U. Mixed quantum mechanical/molecular mechanical molecular dynamics simulations of biological systems in ground and electronically excited states. Chem. Rev. 2015, 115, 6217–6263. [Google Scholar] [CrossRef]
- Campomanes, P.; Rothlisberger, U.; Alfonso-Prieto, M.; Rovira, C. The molecular mechanism of the catalase-like activity in horseradish peroxidase. J. Am. Chem. Soc. 2015, 137, 11170–11178. [Google Scholar] [CrossRef]
- Quesne, M.G.; Borowski, T.; de Visser, S.P. Quantum mechanics/molecular mechanics modeling of enzymatic processes: Caveats and breakthroughs. Chemistry 2016, 22, 2562–2581. [Google Scholar] [CrossRef]
- Sgrignani, J.; Iannuzzi, M.; Magistrato, A. Role of water in the puzzling mechanism of the final aromatization step promoted by the human aromatase enzyme. Insights from QM/MM MD simulations. J. Chem. Inf. Model. 2015, 55, 2218–2226. [Google Scholar] [CrossRef]











© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pavlin, M.; Qasem, Z.; Sameach, H.; Gevorkyan-Airapetov, L.; Ritacco, I.; Ruthstein, S.; Magistrato, A. Unraveling the Impact of Cysteine-to-Serine Mutations on the Structural and Functional Properties of Cu(I)-Binding Proteins. Int. J. Mol. Sci. 2019, 20, 3462. https://doi.org/10.3390/ijms20143462
Pavlin M, Qasem Z, Sameach H, Gevorkyan-Airapetov L, Ritacco I, Ruthstein S, Magistrato A. Unraveling the Impact of Cysteine-to-Serine Mutations on the Structural and Functional Properties of Cu(I)-Binding Proteins. International Journal of Molecular Sciences. 2019; 20(14):3462. https://doi.org/10.3390/ijms20143462
Chicago/Turabian StylePavlin, Matic, Zena Qasem, Hila Sameach, Lada Gevorkyan-Airapetov, Ida Ritacco, Sharon Ruthstein, and Alessandra Magistrato. 2019. "Unraveling the Impact of Cysteine-to-Serine Mutations on the Structural and Functional Properties of Cu(I)-Binding Proteins" International Journal of Molecular Sciences 20, no. 14: 3462. https://doi.org/10.3390/ijms20143462
APA StylePavlin, M., Qasem, Z., Sameach, H., Gevorkyan-Airapetov, L., Ritacco, I., Ruthstein, S., & Magistrato, A. (2019). Unraveling the Impact of Cysteine-to-Serine Mutations on the Structural and Functional Properties of Cu(I)-Binding Proteins. International Journal of Molecular Sciences, 20(14), 3462. https://doi.org/10.3390/ijms20143462