Age-Dependent Myocardial Dysfunction in Critically Ill Patients: Role of Mitochondrial Dysfunction


Abstract
:1. Epidemiology of Sepsis and Cardiac Arrest
2. Clinical Characteristics
3. Biomarkers
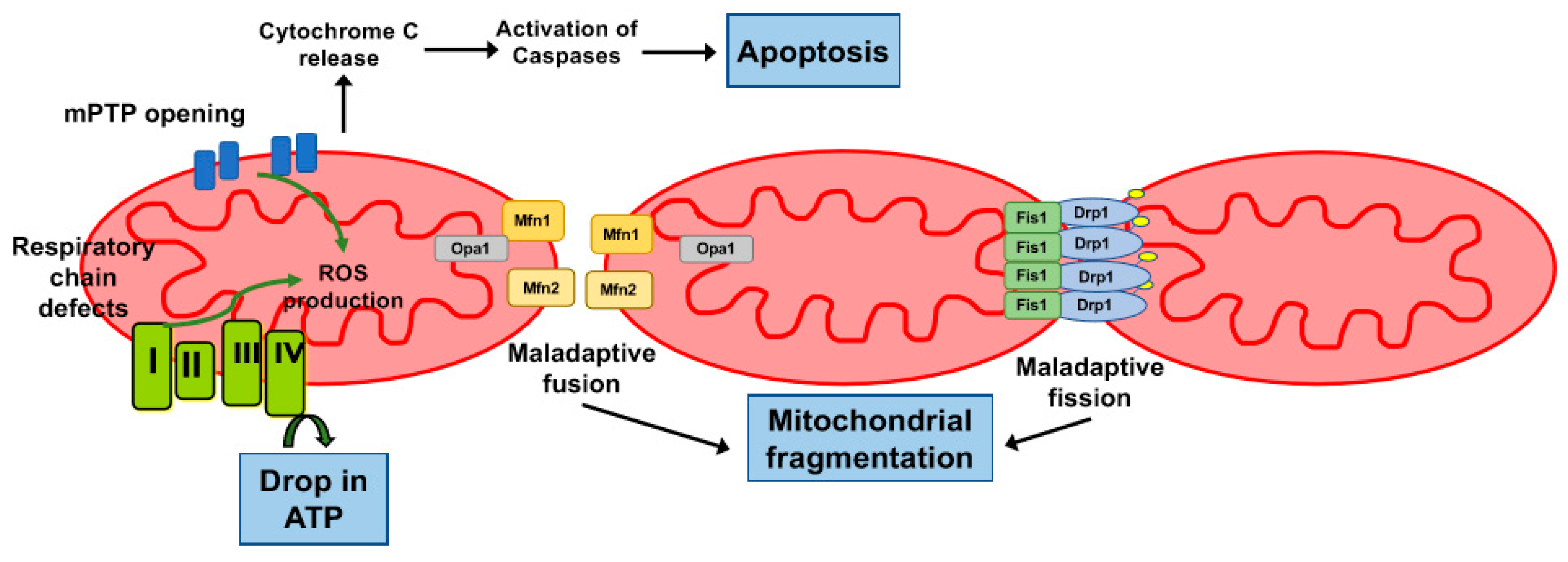
4. Energy Failure and Mitochondrial Dysfunction
5. Role of the Mitochondrial Permeability Transition Pore
6. Mitochondrial Dynamics in the Myocardium
7. Conclusions
Funding
Conflicts of Interest
References
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.D.; Coopersmith, C.M.; et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 2016, 315, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Watson, R.S.; Carcillo, J.A.; Linde-Zwirble, W.T.; Clermont, G.; Lidicker, J.; Angus, D.C. The epidemiology of severe sepsis in children in the United States. Am. J. Respir. Crit. Care Med. 2003, 167, 695–701. [Google Scholar] [CrossRef] [PubMed]
- Angus, D.C.; Linde-Zwirble, W.T.; Lidicker, J.; Clermont, G.; Carcillo, J.; Pinsky, M.R. Epidemiology of severe sepsis in the United States: Analysis of incidence, outcome, and associated costs of care. Crit. Care Med. 2001, 29, 1303–1310. [Google Scholar] [CrossRef] [PubMed]
- Ruth, A.; McCracken, C.E.; Fortenberry, J.D.; Hall, M.; Simon, H.K.; Hebbar, K.B. Pediatric severe sepsis: Current trends and outcomes from the Pediatric Health Information Systems database. Pediatr. Crit. Care Med. 2014, 15, 828–838. [Google Scholar] [CrossRef] [PubMed]
- Balamuth, F.; Weiss, S.L.; Neuman, M.I.; Scott, H.; Brady, P.W.; Paul, R.; Farris, R.W.; McClead, R.; Hayes, K.; Gaieski, D.; et al. Pediatric severe sepsis in U.S. children’s hospitals. Pediatr. Crit. Care Med. 2014, 15, 798–805. [Google Scholar] [CrossRef] [PubMed]
- Martin, G.S.; Mannino, D.M.; Moss, M. The effect of age on the development and outcome of adult sepsis. Crit. Care Med. 2006, 34, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Weiss, S.L.; Fitzgerald, J.C.; Pappachan, J.; Wheeler, D.; Jaramillo-Bustamante, J.C.; Salloo, A.; Singhi, S.C.; Erickson, S.; Roy, J.A.; Bush, J.L.; et al. Global epidemiology of pediatric severe sepsis: The sepsis prevalence, outcomes, and therapies study. Am. J. Respir. Crit. Care Med. 2015, 191, 1147–1157. [Google Scholar] [CrossRef]
- Williams, F.Z.; Sachdeva, R.; Travers, C.D.; Walson, K.H.; Hebbar, K.B. Characterization of Myocardial Dysfunction in Fluid- and Catecholamine-Refractory Pediatric Septic Shock and Its Clinical Significance. J. Intensive Care Med. 2016, 34, 17–25. [Google Scholar] [CrossRef]
- Raj, S.; Killinger, J.S.; Gonzalez, J.A.; Lopez, L. Myocardial dysfunction in pediatric septic shock. J. Pediatr. 2014, 164, 2–77. [Google Scholar] [CrossRef]
- Boissier, F.; Razazi, K.; Seemann, A.; Bedet, A.; Thille, A.W.; de Prost, N.; Lim, P.; Brun-Buisson, C.; Mekontso Dessap, A. Left ventricular systolic dysfunction during septic shock: The role of loading conditions. Intensive Care Med. 2017, 43, 633–642. [Google Scholar] [CrossRef]
- Orde, S.R.; Pulido, J.N.; Masaki, M.; Gillespie, S.; Spoon, J.N.; Kane, G.C.; Oh, J.K. Outcome prediction in sepsis: Speckle tracking echocardiography based assessment of myocardial function. Crit. Care 2014, 18, R149. [Google Scholar] [CrossRef] [PubMed]
- Rolando, G.; Espinoza, E.D.; Avid, E.; Welsh, S.; Pozo, J.D.; Vazquez, A.R.; Arzani, Y.; Masevicius, F.D.; Dubin, A. Prognostic value of ventricular diastolic dysfunction in patients with severe sepsis and septic shock. Rev. Bras. Ter. Intensiva 2015, 27, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Knudson, J.D.; Neish, S.R.; Cabrera, A.G.; Lowry, A.W.; Shamszad, P.; Morales, D.L.; Graves, D.E.; Williams, E.A.; Rossano, J.W. Prevalence and outcomes of pediatric in-hospital cardiopulmonary resuscitation in the United States: An analysis of the Kids’ Inpatient Database. Crit. Care Med. 2012, 40, 2940–2944. [Google Scholar] [CrossRef] [PubMed]
- Berg, R.A.; Sutton, R.M.; Holubkov, R.; Nicholson, C.E.; Dean, J.M.; Harrison, R.; Heidemann, S.; Meert, K.; Newth, C.; Moler, F.; et al. Ratio of PICU versus ward cardiopulmonary resuscitation events is increasing. Crit. Care Med. 2013, 41, 2292–2297. [Google Scholar] [CrossRef] [PubMed]
- Atkins, D.L.; Everson-Stewart, S.; Sears, G.K.; Daya, M.; Osmond, M.H.; Warden, C.R.; Berg, R.A. Resuscitation Outcomes Consortium, I., Epidemiology and outcomes from out-of-hospital cardiac arrest in children: The Resuscitation Outcomes Consortium Epistry-Cardiac Arrest. Circulation 2009, 119, 1484–1491. [Google Scholar] [CrossRef] [PubMed]
- Mozaffarian, D.; Benjamin, E.J.; Go, A.S.; Arnett, D.K.; Blaha, M.J.; Cushman, M.; de Ferranti, S.; Despres, J.P.; Fullerton, H.J.; Howard, V.J.; et al. Heart disease and stroke statistics—2015 update: A report from the American Heart Association. Circulation 2015, 131, 29–322. [Google Scholar] [CrossRef] [PubMed]
- Conlon, T.W.; Falkensammer, C.B.; Hammond, R.S.; Nadkarni, V.M.; Berg, R.A.; Topjian, A.A. Association of left ventricular systolic function and vasopressor support with survival following pediatric out-of-hospital cardiac arrest. Pediatr. Crit. Care Med. 2015, 16, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Bro-Jeppesen, J.; Annborn, M.; Hassager, C.; Wise, M.P.; Pelosi, P.; Nielsen, N.; Erlinge, D.; Wanscher, M.; Friberg, H.; Kjaergaard, J.; et al. Hemodynamics and vasopressor support during targeted temperature management at 33 degrees C Versus 36 degrees C after out-of-hospital cardiac arrest: A post hoc study of the target temperature management trial. Crit. Care Med. 2015, 43, 318–327. [Google Scholar] [CrossRef]
- Dumas, F.; Manzo-Silberman, S.; Fichet, J.; Mami, Z.; Zuber, B.; Vivien, B.; Chenevier-Gobeaux, C.; Varenne, O.; Empana, J.P.; Pene, F.; et al. Can early cardiac troponin I measurement help to predict recent coronary occlusion in out-of-hospital cardiac arrest survivors? Crit. Care Med. 2012, 40, 1777–1784. [Google Scholar] [CrossRef]
- Bergum, D.; Nordseth, T.; Mjolstad, O.C.; Skogvoll, E.; Haugen, B.O. Causes of in-hospital cardiac arrest—Incidences and rate of recognition. Resuscitation 2015, 87, 63–68. [Google Scholar] [CrossRef]
- Neumar, R.W.; Nolan, J.P.; Adrie, C.; Aibiki, M.; Berg, R.A.; Bottiger, B.W.; Callaway, C.; Clark, R.S.; Geocadin, R.G.; Jauch, E.C.; et al. Post-cardiac arrest syndrome: Epidemiology, pathophysiology, treatment, and prognostication. A consensus statement from the International Liaison Committee on Resuscitation (American Heart Association, Australian and New Zealand Council on Resuscitation, European Resuscitation Council, Heart and Stroke Foundation of Canada, InterAmerican Heart Foundation, Resuscitation Council of Asia, and the Resuscitation Council of Southern Africa); the American Heart Association Emergency Cardiovascular Care Committee; the Council on Cardiovascular Surgery and Anesthesia; the Council on Cardiopulmonary, Perioperative, and Critical Care; the Council on Clinical Cardiology; and the Stroke Council. Circulation 2008, 118, 2452–2483. [Google Scholar] [PubMed]
- Jentzer, J.C.; Anavekar, N.S.; Mankad, S.V.; White, R.D.; Kashani, K.B.; Barsness, G.W.; Rabinstein, A.A.; Pislaru, S.V. Changes in left ventricular systolic and diastolic function on serial echocardiography after out-of-hospital cardiac arrest. Resuscitation 2018, 126, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Cha, K.C.; Kim, H.I.; Kim, O.H.; Cha, Y.S.; Kim, H.; Lee, K.H.; Hwang, S.O. Echocardiographic patterns of postresuscitation myocardial dysfunction. Resuscitation 2018, 124, 90–95. [Google Scholar] [CrossRef] [PubMed]
- Deep, A.; Goonasekera, C.D.; Wang, Y.; Brierley, J. Evolution of haemodynamics and outcome of fluid-refractory septic shock in children. Intensive Care Med. 2013, 39, 1602–1609. [Google Scholar] [CrossRef] [PubMed]
- Abdalaziz, F.A.; Algebaly, H.A.F.; Ismail, R.I.; El-Sherbini, S.A.; Behairy, A. The use of bedside echocardiography for measuring cardiac index and systemic vascular resistance in pediatric patients with septic shock. Rev. Bras. Ter. Intensiva 2018, 30, 460–470. [Google Scholar] [CrossRef] [PubMed]
- Sankar, J.; Das, R.R.; Jain, A.; Dewangan, S.; Khilnani, P.; Yadav, D.; Dubey, N. Prevalence and outcome of diastolic dysfunction in children with fluid refractory septic shock—A prospective observational study. Pediatr. Crit. Care Med. 2014, 15, 370–378. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Hady, H.E.; Matter, M.K.; El-Arman, M.M. Myocardial dysfunction in neonatal sepsis: A tissue Doppler imaging study. Pediatr. Crit. Care Med. 2012, 13, 318–323. [Google Scholar] [CrossRef]
- Ranjit, S.; Aram, G.; Kissoon, N.; Ali, M.K.; Natraj, R.; Shresti, S.; Jayakumar, I.; Gandhi, D. Multimodal monitoring for hemodynamic categorization and management of pediatric septic shock: A pilot observational study. Pediatr. Crit. Care Med. 2014, 15, 17–26. [Google Scholar] [CrossRef]
- Conlon, T.W.; Himebauch, A.S.; Fitzgerald, J.C.; Chen, A.E.; Dean, A.J.; Panebianco, N.; Darge, K.; Cohen, M.S.; Greeley, W.J.; Berg, R.A.; et al. Implementation of a pediatric critical care focused bedside ultrasound training program in a large academic PICU. Pediatr. Crit. Care Med. 2015, 16, 219–226. [Google Scholar] [CrossRef]
- Basu, S.; Frank, L.H.; Fenton, K.E.; Sable, C.A.; Levy, R.J.; Berger, J.T. Two-dimensional speckle tracking imaging detects impaired myocardial performance in children with septic shock, not recognized by conventional echocardiography. Pediatr. Crit. Care Med. 2012, 13, 259–264. [Google Scholar] [CrossRef]
- Haileselassie, B.; Su, E.; Pozios, I.; Fiskum, T.; Thompson, R.; Abraham, T. Strain Echocardiography Parameters Correlate With Disease Severity in Children and Infants With Sepsis. Pediatr. Crit. Care Med. 2016, 17, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Parker, M.M.; Shelhamer, J.H.; Natanson, C.; Alling, D.W.; Parrillo, J.E. Serial cardiovascular variables in survivors and nonsurvivors of human septic shock: Heart rate as an early predictor of prognosis. Crit. Care Med. 1987, 15, 923–939. [Google Scholar] [CrossRef] [PubMed]
- Furian, T.; Aguiar, C.; Prado, K.; Ribeiro, R.V.; Becker, L.; Martinelli, N.; Clausell, N.; Rohde, L.E.; Biolo, A. Ventricular dysfunction and dilation in severe sepsis and septic shock: Relation to endothelial function and mortality. J. Crit. Care 2012, 27, 319 e9–319 e15. [Google Scholar] [CrossRef] [PubMed]
- Vallabhajosyula, S.; Kumar, M.; Pandompatam, G.; Sakhuja, A.; Kashyap, R.; Kashani, K.; Gajic, O.; Geske, J.B.; Jentzer, J.C. Prognostic impact of isolated right ventricular dysfunction in sepsis and septic shock: An 8-year historical cohort study. Ann. Intensive Care 2017, 7, 94. [Google Scholar] [CrossRef] [PubMed]
- Harmankaya, A.; Akilli, H.; Gul, M.; Akilli, N.B.; Ergin, M.; Aribas, A.; Cander, B. Assessment of right ventricular functions in patients with sepsis, severe sepsis and septic shock and its prognostic importance: A tissue Doppler study. J. Crit. Care 2013, 28, 1111 e7–1111 e11. [Google Scholar] [CrossRef] [PubMed]
- Lanspa, M.J.; Gutsche, A.R.; Wilson, E.L.; Olsen, T.D.; Hirshberg, E.L.; Knox, D.B.; Brown, S.M.; Grissom, C.K. Application of a simplified definition of diastolic function in severe sepsis and septic shock. Crit. Care 2016, 20, 243. [Google Scholar] [CrossRef] [PubMed]
- Sanfilippo, F.; Corredor, C.; Fletcher, N.; Tritapepe, L.; Lorini, F.L.; Arcadipane, A.; Vieillard-Baron, A.; Cecconi, M. Left ventricular systolic function evaluated by strain echocardiography and relationship with mortality in patients with severe sepsis or septic shock: A systematic review and meta-analysis. Crit. Care 2018, 22, 183. [Google Scholar] [CrossRef]
- Ramjee, V.; Grossestreuer, A.V.; Yao, Y.; Perman, S.M.; Leary, M.; Kirkpatrick, J.N.; Forfia, P.R.; Kolansky, D.M.; Abella, B.S.; Gaieski, D.F. Right ventricular dysfunction after resuscitation predicts poor outcomes in cardiac arrest patients independent of left ventricular function. Resuscitation 2015, 96, 186–191. [Google Scholar] [CrossRef] [Green Version]
- Peberdy, M.A.; Callaway, C.W.; Neumar, R.W.; Geocadin, R.G.; Zimmerman, J.L.; Donnino, M.; Gabrielli, A.; Silvers, S.M.; Zaritsky, A.L.; Merchant, R.; et al. Part 9: Post-cardiac arrest care: 2010 American Heart Association Guidelines for Cardiopulmonary Resuscitation and Emergency Cardiovascular Care. Circulation 2010, 122 (Suppl. S36), 768–786. [Google Scholar] [CrossRef]
- Domico, M.; Liao, P.; Anas, N.; Mink, R.B. Elevation of brain natriuretic peptide levels in children with septic shock. Pediatr. Crit. Care Med. 2008, 9, 478–483. [Google Scholar] [CrossRef]
- Perman, S.M.; Chang, A.M.; Hollander, J.E.; Gaieski, D.F.; Trzeciak, S.; Birkhahn, R.; Otero, R.; Osborn, T.M.; Moretti, E.; Nguyen, H.B.; et al. Relationship between B-type natriuretic peptide and adverse outcome in patients with clinical evidence of sepsis presenting to the emergency department. Acad. Emerg. Med. 2011, 18, 219–222. [Google Scholar] [CrossRef] [PubMed]
- Post, F.; Weilemann, L.S.; Messow, C.M.; Sinning, C.; Munzel, T. B-type natriuretic peptide as a marker for sepsis-induced myocardial depression in intensive care patients. Crit. Care Med. 2008, 36, 3030–3037. [Google Scholar] [CrossRef] [PubMed]
- Rivers, E.P.; McCord, J.; Otero, R.; Jacobsen, G.; Loomba, M. Clinical utility of B-type natriuretic peptide in early severe sepsis and septic shock. J. Intensive Care Med. 2007, 22, 363–373. [Google Scholar] [CrossRef]
- Klouche, K.; Pommet, S.; Amigues, L.; Bargnoux, A.S.; Dupuy, A.M.; Machado, S.; Serveaux-Delous, M.; Morena, M.; Jonquet, O.; Cristol, J.P. Plasma Brain Natriuretic Peptide and Troponin Levels in Severe Sepsis and Septic Shock. J. Intensive Care Med. 2013, 29, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Sodeck, G.H.; Domanovits, H.; Sterz, F.; Schillinger, M.; Losert, H.; Havel, C.; Kliegel, A.; Vlcek, M.; Frossard, M.; Laggner, A.N. Can brain natriuretic peptide predict outcome after cardiac arrest? An observational study. Resuscitation 2007, 74, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Landesberg, G.; Jaffe, A.S.; Gilon, D.; Levin, P.D.; Goodman, S.; Abu-Baih, A.; Beeri, R.; Weissman, C.; Sprung, C.L.; Landesberg, A. Troponin Elevation in Severe Sepsis and Septic Shock. Crit. Care Med. 2014, 42, 790–800. [Google Scholar] [CrossRef] [PubMed]
- De Geer, L.; Engvall, J.; Oscarsson, A. Strain echocardiography in septic shock—A comparison with systolic and diastolic function parameters, cardiac biomarkers and outcome. Crit. Care 2015, 19, 122. [Google Scholar] [CrossRef]
- Kim, J.S.; Kim, M.; Kim, Y.J.; Ryoo, S.M.; Sohn, C.H.; Ahn, S.; Kim, W.Y. Troponin Testing for Assessing Sepsis-Induced Myocardial Dysfunction in Patients with Septic Shock. J. Clin. Med. 2019, 8, 239. [Google Scholar] [CrossRef]
- Rosjo, H.; Masson, S.; Caironi, P.; Stridsberg, M.; Magnoli, M.; Christensen, G.; Moise, G.; Urbano, M.C.; Gattinoni, L.; Pesenti, A.; et al. Prognostic Value of Secretoneurin in Patients With Severe Sepsis and Septic Shock: Data From the Albumin Italian Outcome Sepsis Study. Crit. Care Med. 2018, 46, 404–410. [Google Scholar] [CrossRef]
- Rosjo, H.; Nygard, S.; Kaukonen, K.M.; Karlsson, S.; Stridsberg, M.; Ruokonen, E.; Pettila, V.; Omland, T.; Group, F.S. Prognostic value of chromogranin A in severe sepsis: Data from the FINNSEPSIS study. Intensive Care Med. 2012, 38, 820–829. [Google Scholar] [CrossRef]
- Wang, B.; Chen, G.; Li, J.; Zeng, Y.; Wu, Y.; Yan, X. Neutrophil gelatinase-associated lipocalin predicts myocardial dysfunction and mortality in severe sepsis and septic shock. Int. J. Cardiol. 2017, 227, 589–594. [Google Scholar] [CrossRef]
- Crouser, E.D. Mitochondrial dysfunction in septic shock and multiple organ dysfunction syndrome. Mitochondrion 2004, 4, 729–741. [Google Scholar] [CrossRef] [PubMed]
- Brealey, D.; Brand, M.; Hargreaves, I.; Heales, S.; Land, J.; Smolenski, R.; Davies, N.A.; Cooper, C.E.; Singer, M. Association between mitochondrial dysfunction and severity and outcome of septic shock. Lancet 2002, 360, 219–223. [Google Scholar] [CrossRef] [Green Version]
- Levy, R.J.; Vijayasarathy, C.; Raj, N.R.; Avadhani, N.G.; Deutschman, C.S. Competitive and noncompetitive inhibition of myocardial cytochrome C oxidase in sepsis. Shock 2004, 21, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Piel, D.A.; Deutschman, C.S.; Levy, R.J. Exogenous cytochrome C restores myocardial cytochrome oxidase activity into the late phase of sepsis. Shock 2008, 29, 612–616. [Google Scholar] [CrossRef]
- Takasu, O.; Gaut, J.P.; Watanabe, E.; To, K.; Fagley, R.E.; Sato, B.; Jarman, S.; Efimov, I.R.; Janks, D.L.; Srivastava, A.; et al. Mechanisms of cardiac and renal dysfunction in patients dying of sepsis. Am. J. Respir. Crit. Care Med. 2013, 187, 509–517. [Google Scholar] [CrossRef]
- Doerrier, C.; Garcia, J.A.; Volt, H.; Diaz-Casado, M.E.; Luna-Sanchez, M.; Fernandez-Gil, B.; Escames, G.; Lopez, L.C.; Acuna-Castroviejo, D. Permeabilized myocardial fibers as model to detect mitochondrial dysfunction during sepsis and melatonin effects without disruption of mitochondrial network. Mitochondrion 2016, 27, 56–63. [Google Scholar] [CrossRef]
- Sanchez-Villamil, J.P.; D’Annunzio, V.; Finocchietto, P.; Holod, S.; Rebagliati, I.; Perez, H.; Peralta, J.G.; Gelpi, R.J.; Poderoso, J.J.; Carreras, M.C. Cardiac-specific overexpression of thioredoxin 1 attenuates mitochondrial and myocardial dysfunction in septic mice. Int. J. Biochem. Cell Biol. 2016, 81, 323–334. [Google Scholar] [CrossRef] [Green Version]
- Jarkovska, D.; Markova, M.; Horak, J.; Nalos, L.; Benes, J.; Al-Obeidallah, M.; Tuma, Z.; Sviglerova, J.; Kuncova, J.; Matejovic, M.; et al. Cellular Mechanisms of Myocardial Depression in Porcine Septic Shock. Front. Physiol. 2018, 9, 726. [Google Scholar] [CrossRef] [Green Version]
- Tatsumi, T.; Akashi, K.; Keira, N.; Matoba, S.; Mano, A.; Shiraishi, J.; Yamanaka, S.; Kobara, M.; Hibino, N.; Hosokawa, S.; et al. Cytokine-induced nitric oxide inhibits mitochondrial energy production and induces myocardial dysfunction in endotoxin-treated rat hearts. J. Mol. Cell Cardiol. 2004, 37, 775–784. [Google Scholar] [CrossRef]
- Vanasco, V.; Magnani, N.D.; Cimolai, M.C.; Valdez, L.B.; Evelson, P.; Boveris, A.; Alvarez, S. Endotoxemia impairs heart mitochondrial function by decreasing electron transfer, ATP synthesis and ATP content without affecting membrane potential. J. Bioenerg. Biomembr. 2012, 44, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Watts, J.A.; Kline, J.A.; Thornton, L.R.; Grattan, R.M.; Brar, S.S. Metabolic dysfunction and depletion of mitochondria in hearts of septic rats. J. Mol. Cell Cardiol. 2004, 36, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Inata, Y.; Piraino, G.; Hake, P.W.; O’Connor, M.; Lahni, P.; Wolfe, V.; Schulte, C.; Moore, V.; James, J.M.; Zingarelli, B. Age-Dependent Cardiac Function during Experimental Sepsis: Effect of Pharmacological Activation of AMP-Activated Protein Kinase by AICAR. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, 826–837. [Google Scholar] [CrossRef] [PubMed]
- Checchia, P.A.; Schierding, W.; Polpitiya, A.; Dixon, D.; Macmillan, S.; Muenzer, J.; Stromberg, P.; Coopersmith, C.M.; Buchman, T.G.; Cobb, J.P. Myocardial transcriptional profiles in a murine model of sepsis: Evidence for the importance of age. Pediatr. Crit. Care Med. 2008, 9, 530–535. [Google Scholar] [CrossRef] [PubMed]
- Boheler, K.R.; Volkova, M.; Morrell, C.; Garg, R.; Zhu, Y.; Margulies, K.; Seymour, A.M.; Lakatta, E.G. Sex-and age-dependent human transcriptome variability: Implications for chronic heart failure. Proc. Natl. Acad. Sci. USA 2003, 100, 2754–2759. [Google Scholar] [CrossRef] [PubMed]
- Beal, M.F. Oxidatively modified proteins in aging and disease. Free Radic. Biol. Med. 2002, 32, 797–803. [Google Scholar] [CrossRef]
- Lautz, A.J.; Morgan, R.W.; Karlsson, M.; Mavroudis, C.D.; Ko, T.S.; Licht, D.J.; Nadkarni, V.M.; Berg, R.A.; Sutton, R.M.; Kilbaugh, T.J. Hemodynamic-Directed Cardiopulmonary Resuscitation Improves Neurologic Outcomes and Mitochondrial Function in the Heart and Brain. Crit. Care Med. 2019, 47, e241–e249. [Google Scholar] [CrossRef] [PubMed]
- Han, F.; Da, T.; Riobo, N.A.; Becker, L.B. Early mitochondrial dysfunction in electron transfer activity and reactive oxygen species generation after cardiac arrest. Crit. Care Med. 2008, 36, S447–S453. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.S.; Huang, C.H.; Tsai, S.H.; Tsai, C.Y.; Chen, H.W.; Cheng, H.J.; Hsu, C.Y.; Wang, T.D.; Chang, W.T.; Chen, W.J. The difference in myocardial injuries and mitochondrial damages between asphyxial and ventricular fibrillation cardiac arrests. Am. J. Emerg. Med. 2012, 30, 1540–1548. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Huang, Z.; Zhu, J.; Jiang, L.; Li, H.; Fu, Y.; Sun, S.; Tang, W. Ultrastructural evidence of mitochondrial abnormalities in postresuscitation myocardial dysfunction. Resuscitation 2012, 83, 386–394. [Google Scholar] [CrossRef] [PubMed]
- Brand, M.D. Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic. Biol. Med. 2016, 100, 14–31. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijevic, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.J.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paradies, G.; Paradies, V.; De Benedictis, V.; Ruggiero, F.M.; Petrosillo, G. Functional role of cardiolipin in mitochondrial bioenergetics. Biochim. Biophys. Acta 2014, 1837, 408–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabuel, C.; Samuel, J.L.; Lortat-Jacob, B.; Marotte, F.; Lanone, S.; Keyser, C.; Lessana, A.; Payen, D.; Mebazaa, A. Activation of the ubiquitin proteolytic pathway in human septic heart and diaphragm. Cardiovasc. Pathol. 2010, 19, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Riobo, N.A.; Clementi, E.; Melani, M.; Boveris, A.; Cadenas, E.; Moncada, S.; Poderoso, J.J. Nitric oxide inhibits mitochondrial NADH:ubiquinone reductase activity through peroxynitrite formation. Biochem. J. 2001, 359, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Brand, M.D. Uncoupling to survive? The role of mitochondrial inefficiency in ageing. Exp. Gerontol. 2000, 35, 811–820. [Google Scholar] [CrossRef]
- Jastroch, M.; Divakaruni, A.S.; Mookerjee, S.; Treberg, J.R.; Brand, M.D. Mitochondrial proton and electron leaks. Essays Biochem. 2010, 47, 53–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, G.; Lyu, J.; Liu, S.; Huang, J.; Liu, C.; Xiang, D.; Xie, M.; Zeng, Q. Silencing of uncoupling protein 2 by small interfering RNA aggravates mitochondrial dysfunction in cardiomyocytes under septic conditions. Int. J. Mol. Med. 2015, 35, 1525–1536. [Google Scholar] [CrossRef]
- Huang, J.; Peng, W.; Zheng, Y.; Hao, H.; Li, S.; Yao, Y.; Ding, Y.; Zhang, J.; Lyu, J.; Zeng, Q. Upregulation of UCP2 Expression Protects against LPS-Induced Oxidative Stress and Apoptosis in Cardiomyocytes. Oxid. Med. Cell Longev. 2019, 2019, 2758262. [Google Scholar] [CrossRef]
- He, J.; Carroll, J.; Ding, S.; Fearnley, I.M.; Walker, J.E. Permeability transition in human mitochondria persists in the absence of peripheral stalk subunits of ATP synthase. Proc. Natl. Acad. Sci. USA 2017, 114, 9086–9091. [Google Scholar] [CrossRef] [Green Version]
- Kwong, J.Q.; Molkentin, J.D. Physiological and pathological roles of the mitochondrial permeability transition pore in the heart. Cell Metab. 2015, 21, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Karch, J.; Molkentin, J.D. Regulated necrotic cell death: The passive aggressive side of Bax and Bak. Circ. Res. 2015, 116, 1800–1809. [Google Scholar] [CrossRef] [PubMed]
- Piquereau, J.; Godin, R.; Deschenes, S.; Bessi, V.L.; Mofarrahi, M.; Hussain, S.N.; Burelle, Y. Protective role of PARK2/Parkin in sepsis-induced cardiac contractile and mitochondrial dysfunction. Autophagy 2013, 9, 1837–1851. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, P.; Di Lisa, F. The mitochondrial permeability transition pore: Molecular nature and role as a target in cardioprotection. J. Mol. Cell Cardiol. 2015, 78, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Joseph, L.C.; Kokkinaki, D.; Valenti, M.C.; Kim, G.J.; Barca, E.; Tomar, D.; Hoffman, N.E.; Subramanyam, P.; Colecraft, H.M.; Hirano, M.; et al. Inhibition of NADPH oxidase 2 (NOX2) prevents sepsis-induced cardiomyopathy by improving calcium handling and mitochondrial function. JCI Insight 2017, 2, 94248. [Google Scholar] [CrossRef]
- Fauvel, H.; Marchetti, P.; Obert, G.; Joulain, O.; Chopin, C.; Formstecher, P.; Neviere, R. Protective effects of cyclosporin A from endotoxin-induced myocardial dysfunction and apoptosis in rats. Am. J. Respir. Crit. Care Med. 2002, 165, 449–455. [Google Scholar] [CrossRef] [PubMed]
- Larche, J.; Lancel, S.; Hassoun, S.M.; Favory, R.; Decoster, B.; Marchetti, P.; Chopin, C.; Neviere, R. Inhibition of mitochondrial permeability transition prevents sepsis-induced myocardial dysfunction and mortality. J. Am. Coll. Cardiol. 2006, 48, 377–385. [Google Scholar] [CrossRef]
- Joshi, M.S.; Julian, M.W.; Huff, J.E.; Bauer, J.A.; Xia, Y.; Crouser, E.D. Calcineurin regulates myocardial function during acute endotoxemia. Am. J. Respir. Crit. Care Med. 2006, 173, 999–1007. [Google Scholar] [CrossRef]
- Cour, M.; Abrial, M.; Jahandiez, V.; Loufouat, J.; Belaidi, E.; Gharib, A.; Varennes, A.; Monneret, G.; Thibault, H.; Ovize, M.; et al. Ubiquitous protective effects of cyclosporine A in preventing cardiac arrest-induced multiple organ failure. J. Appl. Physiol. 2014, 117, 930–936. [Google Scholar] [CrossRef] [Green Version]
- Ong, S.B.; Samangouei, P.; Kalkhoran, S.B.; Hausenloy, D.J. The mitochondrial permeability transition pore and its role in myocardial ischemia reperfusion injury. J. Mol. Cell Cardiol. 2015, 78, 23–34. [Google Scholar] [CrossRef]
- Song, F.; Shan, Y.; Cappello, F.; Rappa, F.; Ristagno, G.; Yu, T.; Li Volti, G.; Sun, S.; Weil, M.H.; Tang, W. Apoptosis is not involved in the mechanism of myocardial dysfunction after resuscitation in a rat model of cardiac arrest and cardiopulmonary resuscitation. Crit. Care Med. 2010, 38, 1329–1334. [Google Scholar] [CrossRef] [PubMed]
- Leistner, M.; Sommer, S.; Kanofsky, P.; Leyh, R.; Sommer, S.P. Ischemia time impacts on respiratory chain functions and Ca2+-handling of cardiac subsarcolemmal mitochondria subjected to ischemia reperfusion injury. J. Cardiothorac. Surg. 2019, 14, 92. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.H.; Tsai, M.S.; Hsu, C.Y.; Su, Y.J.; Wang, T.D.; Chang, W.T.; Chen, W.J. Post-cardiac arrest myocardial dysfunction is improved with cyclosporine treatment at onset of resuscitation but not in the reperfusion phase. Resuscitation 2011, 82, S41–S47. [Google Scholar] [CrossRef]
- Oka, N.; Wang, L.; Mi, W.; Caldarone, C.A. Inhibition of mitochondrial remodeling by cyclosporine A preserves myocardial performance in a neonatal rabbit model of cardioplegic arrest. J. Thorac. Cardiovasc. Surg. 2008, 135, 585–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leung, C.H.; Wang, L.; Fu, Y.Y.; Yuen, W.; Caldarone, C.A. Transient mitochondrial permeability transition pore opening after neonatal cardioplegic arrest. J. Thorac. Cardiovasc. Surg. 2011, 141, 975–982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karimi, M.; Wang, L.X.; Hammel, J.M.; Mascio, C.E.; Abdulhamid, M.; Barner, E.W.; Scholz, T.D.; Segar, J.L.; Li, W.G.; Niles, S.D.; et al. Neonatal vulnerability to ischemia and reperfusion: Cardioplegic arrest causes greater myocardial apoptosis in neonatal lambs than in mature lambs. J. Thorac. Cardiovasc. Surg. 2004, 127, 490–497. [Google Scholar] [CrossRef]
- Argaud, L.; Cour, M.; Dubien, P.Y.; Giraud, F.; Jossan, C.; Riche, B.; Hernu, R.; Darmon, M.; Poncelin, Y.; Tchenio, X.; et al. Effect of Cyclosporine in Nonshockable Out-of-Hospital Cardiac Arrest: The CYRUS Randomized Clinical Trial. JAMA Cardiol. 2016, 1, 557–565. [Google Scholar] [CrossRef] [PubMed]
- Glancy, B.; Balaban, R.S. Role of mitochondrial Ca2+ in the regulation of cellular energetics. Biochemistry 2012, 51, 2959–2973. [Google Scholar] [CrossRef]
- Kohlhaas, M.; Maack, C. Calcium release microdomains and mitochondria. Cardiovasc. Res. 2013, 98, 259–268. [Google Scholar] [CrossRef] [Green Version]
- Brookes, P.S.; Yoon, Y.; Robotham, J.L.; Anders, M.W.; Sheu, S.S. Calcium, ATP, and ROS: A mitochondrial love-hate triangle. Am. J. Physiol. Cell Physiol. 2004, 287, C817–C833. [Google Scholar] [CrossRef]
- Arulkumaran, N.; Deutschman, C.S.; Pinsky, M.R.; Zuckerbraun, B.; Schumacker, P.T.; Gomez, H.; Gomez, A.; Murray, P.; Kellum, J.A.; Workgroup, A.X. Mitochondrial Function in Sepsis. Shock 2016, 45, 271–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez, A.S.; Elguero, M.E.; Finocchietto, P.; Holod, S.; Romorini, L.; Miriuka, S.G.; Peralta, J.G.; Poderoso, J.J.; Carreras, M.C. Abnormal mitochondrial fusion-fission balance contributes to the progression of experimental sepsis. Free Radic. Res. 2014, 48, 769–783. [Google Scholar] [CrossRef] [PubMed]
- Suarez-Rivero, J.M.; Villanueva-Paz, M.; de la Cruz-Ojeda, P.; de la Mata, M.; Cotan, D.; Oropesa-Avila, M.; de Lavera, I.; Alvarez-Cordoba, M.; Luzon-Hidalgo, R.; Sanchez-Alcazar, J.A. Mitochondrial Dynamics in Mitochondrial Diseases. Diseases 2016, 5, 1. [Google Scholar] [CrossRef] [PubMed]
- Preau, S.; Delguste, F.; Yu, Y.; Remy-Jouet, I.; Richard, V.; Saulnier, F.; Boulanger, E.; Neviere, R. Endotoxemia Engages the RhoA Kinase Pathway to Impair Cardiac Function By Altering Cytoskeleton, Mitochondrial Fission, and Autophagy. Antioxid. Redox Signal. 2016, 24, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Ouyang, H.; Xiao, X.; Zhong, J.; Dong, M. Irisin ameliorates septic cardiomyopathy via inhibiting DRP1-related mitochondrial fission and normalizing the JNK-LATS2 signaling pathway. Cell Stress Chaperones 2019, 24, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Shang, X.; Li, J.; Yu, R.; Zhu, P.; Zhang, Y.; Xu, J.; Chen, K.; Li, M. Sepsis-related myocardial injury is associated with Mst1 upregulation, mitochondrial dysfunction and the Drp1/F-actin signaling pathway. J. Mol. Histol. 2019, 50, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Haileselassie, B.; Mukherjee, R.; Joshi, A.U.; Napier, B.A.; Massis, L.M.; Ostberg, N.P.; Queliconi, B.B.; Monack, D.; Bernstein, D.; Mochly-Rosen, D. Drp1/Fis1 interaction mediates mitochondrial dysfunction in septic cardiomyopathy. J. Mol. Cell Cardiol. 2019, 130, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Sharp, W.W.; Fang, Y.H.; Han, M.; Zhang, H.J.; Hong, Z.; Banathy, A.; Morrow, E.; Ryan, J.J.; Archer, S.L. Dynamin-related protein 1 (Drp1)-mediated diastolic dysfunction in myocardial ischemia-reperfusion injury: Therapeutic benefits of Drp1 inhibition to reduce mitochondrial fission. FASEB J. 2014, 28, 316–326. [Google Scholar] [CrossRef]
- Sharp, W.W.; Beiser, D.G.; Fang, Y.H.; Han, M.; Piao, L.; Varughese, J.; Archer, S.L. Inhibition of the mitochondrial fission protein dynamin-related protein 1 improves survival in a murine cardiac arrest model. Crit. Care Med. 2015, 43, e38–e47. [Google Scholar] [CrossRef]
- Linton, P.J.; Gurney, M.; Sengstock, D.; Mentzer, R.M., Jr.; Gottlieb, R.A. This old heart: Cardiac aging and autophagy. J. Mol. Cell Cardiol. 2015, 83, 44–54. [Google Scholar] [CrossRef]
- Gross, A.; Katz, S.G. Non-apoptotic functions of BCL-2 family proteins. Cell Death Differ. 2017, 24, 1348–1358. [Google Scholar] [CrossRef] [PubMed]
- Dorn, G.W., II; Vega, R.B.; Kelly, D.P. Mitochondrial biogenesis and dynamics in the developing and diseased heart. Genes Dev 2015, 29, 1981–1991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanasco, V.; Saez, T.; Magnani, N.D.; Pereyra, L.; Marchini, T.; Corach, A.; Vaccaro, M.I.; Corach, D.; Evelson, P.; Alvarez, S. Cardiac mitochondrial biogenesis in endotoxemia is not accompanied by mitochondrial function recovery. Free Radic. Biol. Med. 2014, 77, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hickson-Bick, D.L.; Jones, C.; Buja, L.M. Stimulation of mitochondrial biogenesis and autophagy by lipopolysaccharide in the neonatal rat cardiomyocyte protects against programmed cell death. J. Mol. Cell Cardiol. 2008, 44, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Zhu, P.; Wang, J.; Zhu, H.; Ren, J.; Chen, Y. Pathogenesis of cardiac ischemia reperfusion injury is associated with CK2alpha-disturbed mitochondrial homeostasis via suppression of FUNDC1-related mitophagy. Cell Death Differ. 2018, 25, 1080–1093. [Google Scholar] [CrossRef] [PubMed]
- Chi, L.; Wang, N.; Yang, W.; Wang, Q.; Zhao, D.; Sun, T.; Li, W. Protection of Myocardial Ischemia-Reperfusion by Therapeutic Hypercapnia: A Mechanism Involving Improvements in Mitochondrial Biogenesis and Function. J. Cardiovasc. Transl. Res. 2019. [Google Scholar] [CrossRef]


{kind=link}
| Septic Shock | ||
| Myocardial Dysfunction | Pediatrics | Adult |
| LV systolic | 3/4 of patients | Low EF in 1/4 to 1/3 * |
| RV systolic | 2/3 of patients | 32–55% of patients * |
| Diastolic | 41–48% for LV ~35% for RV | 66–84% of patients |
| Biomarkers | ||
| B-natriuretic peptide | ↑ with LV dysfunction | ↑ with ↓ EF |
| Cardiac troponins | ↑ with LV and RV dysfunction in neonates | ↑ with LV dysfunction |
| Post-Cardiac Arrest | ||
| Myocardial Dysfunction | Pediatrics | Adult |
| LV systolic | 41% (single study) | >50% |
| RV systolic | 18% (single study) | >50% |
| Diastolic | ~2/3 for LV and RV | Common |
| Biomarkers | ||
| B-natriuretic peptide | No clear association | No clear association |
| Cardiac troponins | ↑ with LV dysfunction | Not necessarily indicative of coronary occlusion |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lautz, A.J.; Zingarelli, B. Age-Dependent Myocardial Dysfunction in Critically Ill Patients: Role of Mitochondrial Dysfunction. Int. J. Mol. Sci. 2019, 20, 3523. https://doi.org/10.3390/ijms20143523
Lautz AJ, Zingarelli B. Age-Dependent Myocardial Dysfunction in Critically Ill Patients: Role of Mitochondrial Dysfunction. International Journal of Molecular Sciences. 2019; 20(14):3523. https://doi.org/10.3390/ijms20143523
Chicago/Turabian StyleLautz, Andrew J., and Basilia Zingarelli. 2019. "Age-Dependent Myocardial Dysfunction in Critically Ill Patients: Role of Mitochondrial Dysfunction" International Journal of Molecular Sciences 20, no. 14: 3523. https://doi.org/10.3390/ijms20143523
APA StyleLautz, A. J., & Zingarelli, B. (2019). Age-Dependent Myocardial Dysfunction in Critically Ill Patients: Role of Mitochondrial Dysfunction. International Journal of Molecular Sciences, 20(14), 3523. https://doi.org/10.3390/ijms20143523