Collateral Vessels Have Unique Endothelial and Smooth Muscle Cell Phenotypes


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
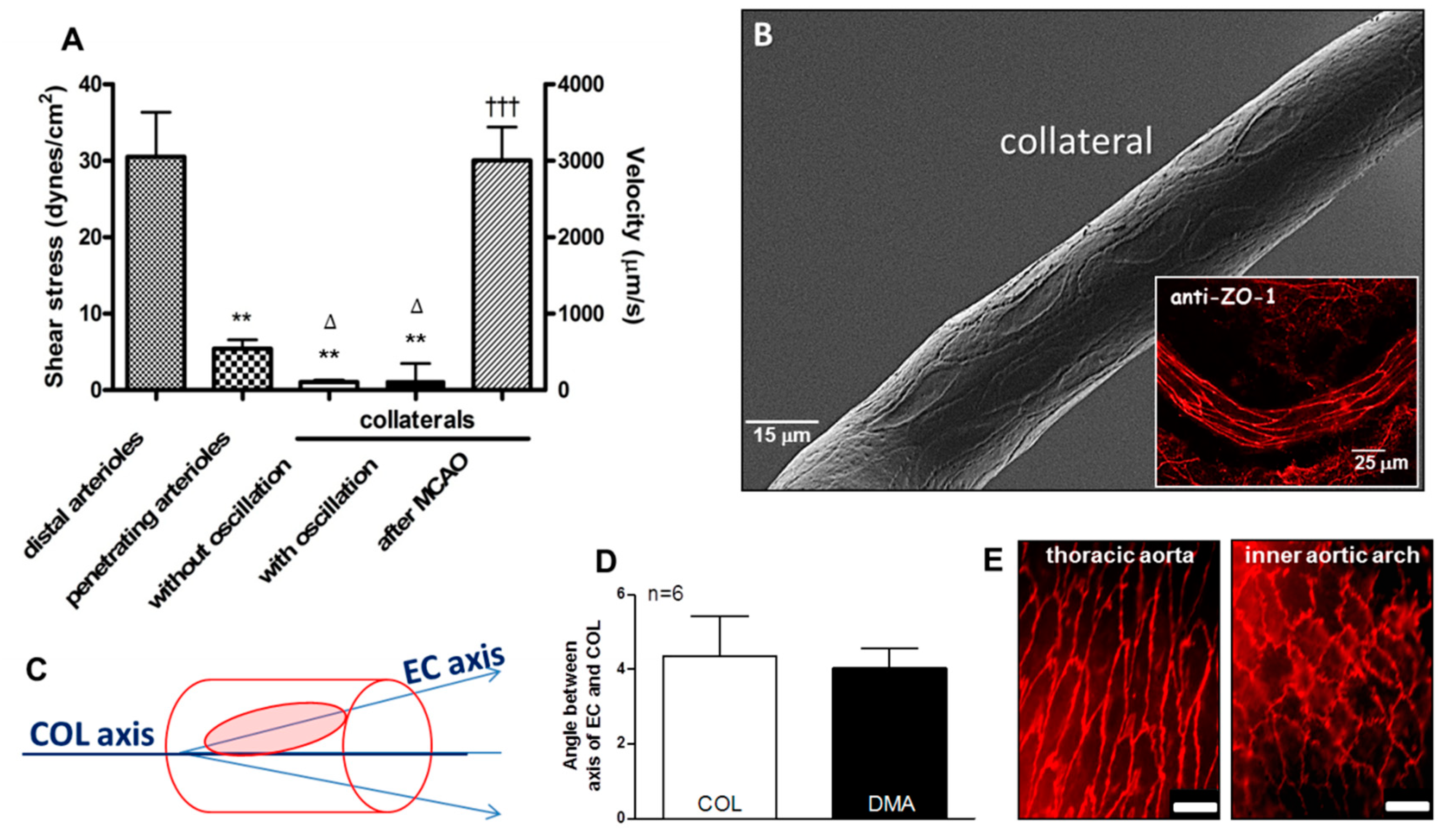
2.1. Collateral Endothelial Cells Are Aligned with the Vessel Axis Despite their Chronic Exposure to Low and Oscillatory Shear Stress
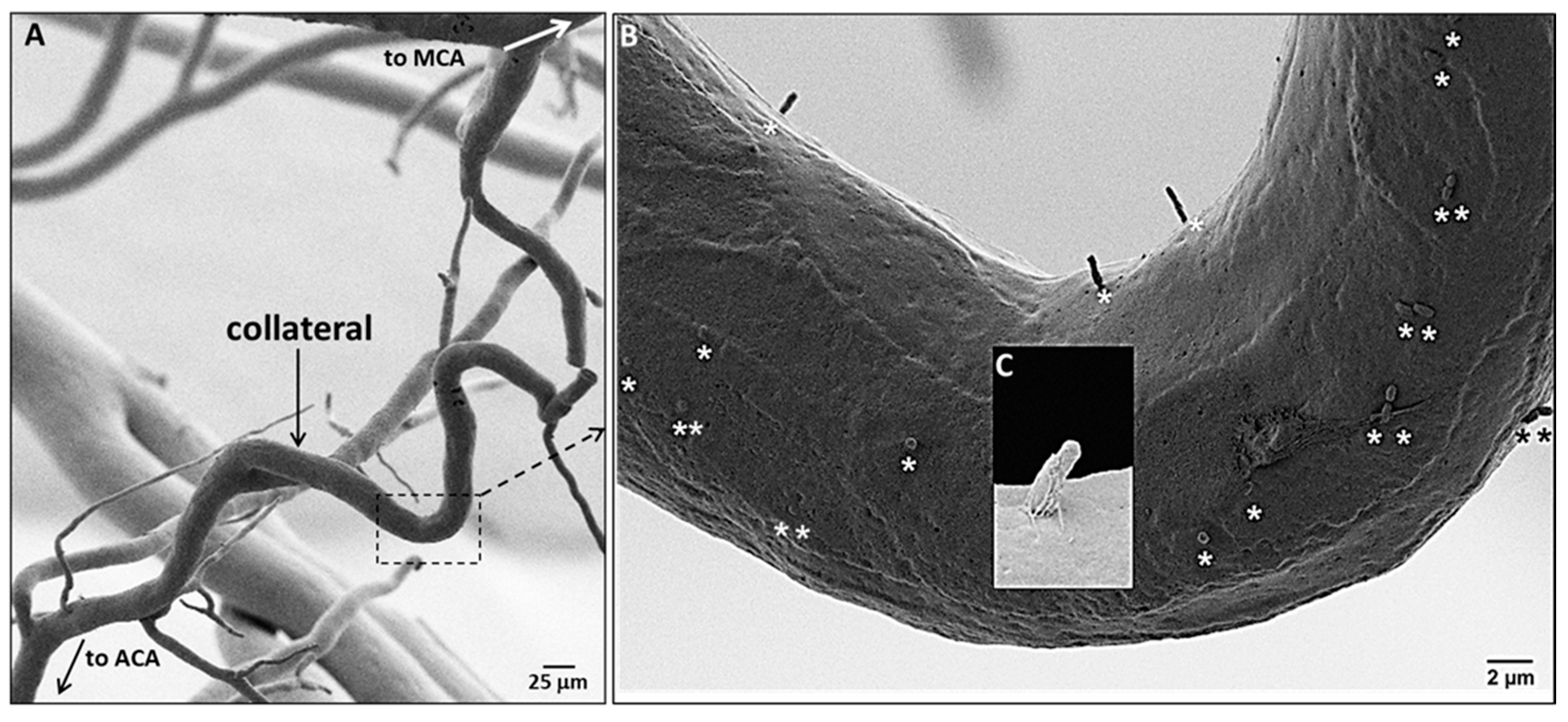
2.2. Endothelial Cells of Collaterals and Distal-Most Arterioles Have Primary Cilia; Collaterals Have Fewer
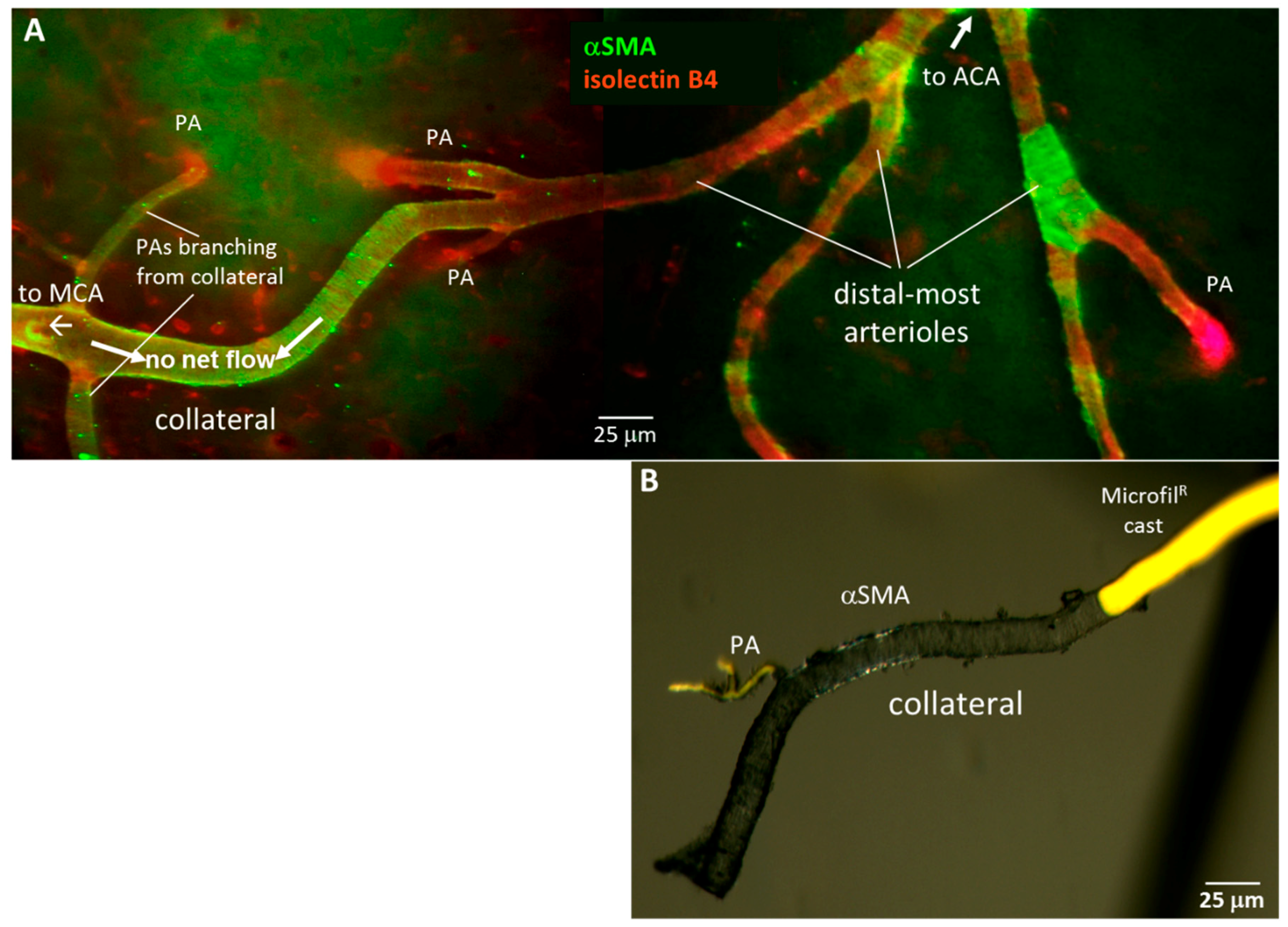
2.3. Collaterals Are Invested with a Continuous Layer of Smooth Muscle Cells, unlike Distal-Most Arterioles Whose Smooth Muscle Cells Are Discontinuous
2.4. Gene Expression Differs for Collaterals Versus Distal-Most Arterioles
2.5. Changes in Tortuosity over a Collateral’s “Lifetime” Suggests Accelerated Proliferative Senescence of their Mural Cells
3. Discussion
4. Materials and Methods
4.1. Angiography and Morphometry
4.2. Permanent Middle Cerebral Artery Occlusion (pMCAO)
4.3. Laser Doppler Perfusion Imaging and Hindlimb Ischemia Model
4.4. Quantitative NanoString Expression Analysis
4.5. Immunohistochemistry
4.6. Collateral Primary Cilia and Endothelial Orientation Assessed by Scanning Electron Microscopy
4.7. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ACA | anterior cerebral artery |
| B6 | C57BL/6 mouse strain |
| COL | collateral |
| DMA | distal-most arteriole |
| EC | endothelial cell |
| eNOS | endothelial nitric oxide synthase |
| Flt 1 | VEGF receptor 1 |
| Flk1 | VEGF receptor 2 |
| MCA | middle cerebral artery |
| NO | nitric oxide |
| PA | penetrating arteriole |
| Pkd1 | gene that encodes polycystin-1 |
| PrC | primary cilia |
| SMC | smooth muscle cell |
| VECAD | vascular endothelial cell adhesion protein, selectively expressed by ECs |
| WT | wildtype littermate controls |
References
- Faber, J.E.; Chilian, W.M.; Deindl, E.; van Royen, N.; Simons, M. A brief etymology of the collateral circulation. Atherioscler. Thromb. Vasc. Biol. 2014, 34, 1854–1859. [Google Scholar] [CrossRef] [PubMed]
- Nishijima, Y.; Akamatsu, Y.; Weinstein, P.R.; Liu, J. Collaterals: Implications in cerebral ischemic diseases and therapeutic interventions. Brain Res. 2015, 1623, 18–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bang, O.Y.; Goyal, M.; Liebeskind, D.S. Collateral circulation in ischemic stroke: Assessment tools and therapeutic strategies. Stroke 2015, 46, 3302–3309. [Google Scholar] [CrossRef]
- Ginsberg, M.D. The cerebral collateral circulation: Relevance to pathophysiology and treatment of stroke. Neuropharmacology 2018, 134, 280–292. [Google Scholar] [CrossRef] [PubMed]
- Schaper, W. Collateral circulation: Past and present. Basic Res. Cardiol. 2009, 104, 5–21. [Google Scholar] [CrossRef] [PubMed]
- Chalothorn, D.; Faber, J.E. Formation and maturation or the murine native cerebral collateral circulation. J. Mol. Cell. Cardiol. 2010, 49, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Lucitti, J.L.; Mackey, J.K.; Morrison, J.C.; Haigh, J.J.; Adams, R.H.; Faber, J.E. Formation of the collateral circulation is regulated by vascular endothelial growth factor-A and A disintegrin and metalloprotease family members 10 and 17. Circ. Res. 2012, 111, 1539–1550. [Google Scholar] [CrossRef] [PubMed]
- Lucitti, J.L.; Tarte, N.J.; Faber, J.E. Chloride intracellular channel 4 is required for maturation of the cerebral collateral circulation. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H1141–H1150. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Prabhakar, P.; Sealock, R.; Faber, J.E. Wide genetic variation in the native pial collateral circulation is a major determinant of variation in severity of stroke. J. Cereb. Blood Flow Metab. 2010, 30, 923–934. [Google Scholar] [CrossRef]
- Chalothorn, D.; Faber, J.E. Strain-dependent variation in native collateral function in mouse hindlimb. Physiol. Genom. 2010, 42, 469–479. [Google Scholar] [CrossRef]
- Lucitti, J.L.; Sealock, R.; Buckley, B.K.; Zhang, H.; Xiao, L.; Dudley, A.C.; Faber, J.E. Variants of Rab GTPase-effector binding protein-2 cause variation in the collateral circulation and severity of stroke. Stroke 2016, 47, 3022–3031. [Google Scholar] [CrossRef] [PubMed]
- Seiler, C.; Stoller, M.; Pitt, B.; Meier, P. The human coronary collateral circulation: Development and clinical importance. Eur. Heart J. 2013, 34, 2674–2682. [Google Scholar] [CrossRef] [PubMed]
- Traupe, T.; Ortmann, J.; Stoller, M.; Baumgartner, I.; de Marchi, S.F.; Seiler, C. Direct quantitative assessment of the peripheral artery collateral circulation in patients undergoing angiography. Circulation 2013, 128, 737–744. [Google Scholar] [CrossRef] [PubMed]
- Rost, N.S.; Giese, A.K.; Worrall, B.B.; Williams, S.; Malik, R.; Cloonan, L.; Furie, K.L.; Lindgren, A.; Frid, P.; Wasselius, J.; et al. RABEP2 (Rab GTPase-effector binding protein-2) is associated with ischemic stroke phenotypes: a translational replication study. Eur. Stroke J. 2018, 3, 42–43. [Google Scholar]
- Toriumi, H.; Tatarishvili, J.; Tomita, M.; Tomita, Y.; Unekawa, M.; Suzuki, N. Dually supplied T-junctions in arteriolo-arteriolar anastomosis in mice: Key to local hemodynamic homeostasis in normal and ischemic states? Stroke 2009, 40, 3378–3383. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Faber, J.E. eNOS deficiency causes collateral vessel rarefaction and impairs activation of a cell cycle gene network during arteriogenesis. Circ. Res. 2010, 106, 1870–1881. [Google Scholar] [CrossRef] [PubMed]
- Faber, J.E.; Zhang, H.; Lassance-Soares, R.M.; Prabhakar, P.; Najafi, A.H.; Burnett, M.S.; Epstein, S.E. Aging causes collateral rarefaction and increased severity of ischemic injury in multiple tissues. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1748–1756. [Google Scholar] [CrossRef] [PubMed]
- Moore, S.M.; Zhang, H.; Maeda, N.; Doerschuk, C.; Faber, J.E. Cardiovascular risk factors cause premature rarefaction of the collateral circulation and greater ischemic tissue injury. Angiogenesis 2015, 18, 265–281. [Google Scholar] [CrossRef] [Green Version]
- Rzechorzek, W.; Zhang, H.; Buckley, B.K.; Hua, H.; Pomp, D.; Faber, J.E. Exercise training prevents rarefaction of pial collaterals and increased severity of stroke with aging. J. Cereb. Blood Flow Metab. 2017, 37, 3544–3555. [Google Scholar] [CrossRef]
- Zhang, H.; Jin, B.; Faber, J.E. Mouse models of Alzheimer’s disease cause loss of pial collaterals and increased severity of ischemic stroke. Angiogenesis 2018, 22, 263–279. [Google Scholar] [CrossRef]
- Hecht, N.; He, J.; Kremenetskaia, I.; Nieminen, M.; Vajkoczy, P.; Woitzik, J. Cerebral hemodynamic reserve and vascular remodeling in C57/BL6 mice are influenced by age. Stroke 2012, 43, 3052–3062. [Google Scholar] [CrossRef] [PubMed]
- Menon, B.K.; Smith, E.E.; Coutts, S.B.; Welsh, D.G.; Faber, J.E.; Damani, Z.; Goyal, M.; Hill, M.D.; Demchuk, A.M.; Hee Cho, K.H.; et al. Leptomeningeal collaterals are associated with modifiable metabolic risk factors. Ann. Neurol. 2013, 74, 241–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malik, N.; Hou, Q.; Vagal, A.; Patrie, J.; Xin, W.; Michel, P.; Eskandari, A.; Jovin, T.; Wintermark, M. Demographic and clinical predictors of leptomeningeal collaterals in stroke patients. J. Stroke Cerebrovasc. Dis. 2014, 23, 2018–2022. [Google Scholar] [CrossRef] [PubMed]
- Arsava, E.M.; Vural, A.; Akpinar, E.; Gocmen, R.; Akcalar, S.; Oguz, K.K.; Topcuoglu, M.A. The detrimental effect of aging on leptomeningeal collaterals in ischemic stroke. J. Stroke Cerebrovasc. Dis. 2014, 23, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Peng, X.; Lassance-Soares, R.M.; Najafi, A.H.; Alderman, L.O.; Sood, S.; Xue, Z.; Chan, R.; Faber, J.E.; Epstein, S.E.; et al. Aging-induced collateral dysfunction: Impaired responsiveness of collaterals and susceptibility to apoptosis via dysfunctional eNOS signaling. J. Cardiovasc. Transl. Res. 2011, 4, 779–789. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.L.; Sweet, J.G.; Bishop, N.; Cipolla, M.J. Pial collateral reactivity during hypertension and aging: Understanding the function of collaterals for stroke therapy. Stroke 2016, 47, 1618–1625. [Google Scholar] [CrossRef] [PubMed]
- Beard, D.J.; Murtha, L.A.; McLeod, D.D.; Spratt, N.J. Intracranial pressure and collateral blood flow. Stroke 2016, 47, 1695–1700. [Google Scholar] [CrossRef] [PubMed]
- Davies, P.F.; Civelek, M.; Fang, Y.; Fleming, I. The atherosusceptible endothelium: Endothelial phenotypes in complex haemodynamic shear stress regions in vivo. Cardiovasc. Res. 2013, 99, 315–327. [Google Scholar] [CrossRef]
- Abe, J.; Berk, B.C. Novel mechanisms of endothelial mechanotransduction. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2378–2386. [Google Scholar] [CrossRef]
- Zhou, J.; Li, Y.S.; Chien, S. Shear stress-initiated signaling and its regulation of endothelial function. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2191–2198. [Google Scholar] [CrossRef]
- Zakkar, M.; Angelini, G.D.; Emanueli, C. Regulation of vascular endothelium inflammatory signaling by shear stress. Curr. Vasc. Pharmacol. 2016, 14, 181–186. [Google Scholar] [CrossRef]
- Briffeuil, P.; Thibaut-Vercruyssen, R.; Ronveaux-Dupal, M.F. Ciliation of bovine aortic endothelial cells in culture. Atherosclerosis 1994, 106, 75–81. [Google Scholar] [CrossRef]
- Van der Heiden, K.; Hierck, B.P.; Krams, R.; de Crom, R.; Cheng, C.; Baiker, M.; Pourquie, J.; Alkemade, F.E.; DeRuiter, M.C.; Gittenberger-de Groot, A.C.; et al. Endothelial primary cilia in areas of disturbed flow are at the base of atherosclerosis. Atherosclerosis 2008, 196, 542–550. [Google Scholar] [CrossRef] [PubMed]
- Geerts, W.J.; Vocking, K.; Schoonen, N.; Haarbosch, L.; van Donselaar, E.G.; Regan-Klapisz, E.; Post, J.A. Cobblestone HUVECs: A human model system for studying primary ciliogenesis. J. Struct. Biol. 2011, 176, 350–359. [Google Scholar] [CrossRef]
- Satir, P. Cilia: Before and after. Cilia 2017, 6. [Google Scholar] [CrossRef]
- Egorova, A.D.; van der Heiden, K.; Poelmann, R.E.; Hierck, B.P. Primary cilia as biomechanical sensors in regulating endothelial function. Differentiation 2012, 83, S56–S61. [Google Scholar] [CrossRef]
- Pala, R.; Jamal, M.; Alshammari, Q.; Nauli, S.M. The roles of primary cilia in cardiovascular diseases. Cells 2018, 7, 233. [Google Scholar] [CrossRef] [PubMed]
- Nauli, S.M.; Kawanabe, Y.; Kaminski, J.J.; Pearce, W.J.; Ingber, D.E.; Zhou, J. Endothelial cilia are fluid shear sensors that regulate calcium signaling and nitric oxide production through polycystin-1. Circulation 2008, 117, 1161–1171. [Google Scholar] [CrossRef] [PubMed]
- AbouAlaiwi, W.A.; Ratnam, S.; Booth, R.L.; Shah, J.V.; Nauli, S.M. Endothelial cells from humans and mice with polycystic kidney disease are characterized by polyploidy and chromosome segregation defects through survivin down-regulation. Hum. Mol. Genet. 2011, 20, 354–367. [Google Scholar] [CrossRef]
- Abdul-Majeed, S.; Moloney, B.C.; Nauli, S.M. Mechanisms regulating cilia growth and cilia function in endothelial cells. Cell. Mol. Life Sci. 2012, 69, 165–173. [Google Scholar] [CrossRef]
- Price, R.J.; Owens, G.K.; Skalak, T.C. Immunohistochemical identification of arteriolar development using markers of smooth muscle differentiation. Evidence that capillary arterialization proceeds from terminal arterioles. Circ. Res. 1994, 75, 520–527. [Google Scholar] [CrossRef] [PubMed]
- Murfee, W.L.; Skalak, T.C.; Peirce, S.M. Differential arterial/venous expression of NG2 proteoglycan in perivascular cells along microvessels: Identifying a venule-specific phenotype. Microcirculation 2005, 12, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Reagan, A.M.; Gu, X.; Paudel, S.; Ashpole, N.M.; Zalles, M.; Sonntag, W.E.; Ungvari, Z.; Csiszar, A.; Otalora, L.; Freeman, W.M.; et al. Age-related focal loss of contractile vascular smooth muscle cells in retinal arterioles is accelerated by caveolin-1 deficiency. Neurobiol. Aging. 2018, 71. [Google Scholar] [CrossRef] [PubMed]
- Niu, N.; Xu, S.; Xu, Y.; Little, P.J.; Jin, Z.G. Targeting mechanosensitive transcription factors in atherosclerosis. Trends Pharmacol. Sci. 2019, 40, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Baeyens, N.; Bandyopadhyay, C.; Coon, B.G.; Yun, S.; Schwartz, M.A. Endothelial fluid shear stress sensing in vascular health and disease. J Clin. Invest. 2016, 126, 821–828. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zhang, H.; Wiltshire, T.; Sealock, R.; Faber, J.E. Genetic dissection of the Canq1 locus governing variation in extent of the collateral circulation. PLOS One 2012, 7, e31910. [Google Scholar] [CrossRef] [PubMed]
- Flurkey, K.; Currer, J.M.; Harrison, D.E. The Mouse in Biomedical Research, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2007; pp. 637–672. [Google Scholar]
- Haust, M.D. Endothelial cilia in human aortic atherosclerotic lesions. Virchows Arch. A 1987, 410, 317–326. [Google Scholar] [CrossRef]
- Yamamoto, K.; Fujimoto, S. Endothelial cilium in the capillaries of the human fetal pineal gland. Microscopy 1980, 29, 256–258. [Google Scholar]
- Toomer, K.A.; Fulmer, D.; Guo, L.; Drohan, A.; Peterson, N.; Swanson, P.; Brooks, B.; Mukherjee, R.; Body, S.; Lipschutz, J.H.; et al. A role for primary cilia in aortic valve development and disease. Dev. Dyn. 2017, 246, 625–634. [Google Scholar] [CrossRef] [Green Version]
- Blom, J.N.; Feng, Q. Cardiac repair by epicardial EMT: Current targets and a potential role for the primary cilium. Pharmacol. Ther. 2018, 186, 114–129. [Google Scholar] [CrossRef]
- Bystrevskaya, V.B.; Lichkun, V.V.; Antonov, A.S.; Perov, N.A. An ultrastructural study of centriolar complexes in adult and embryonic human aortic endothelial cells. Tissue Cell 1988, 20, 493–503. [Google Scholar] [CrossRef]
- Lim, Y.C.; McGlashan, S.R.; Cooling, M.T.; Long, D.S. Culture and detection of primary cilia in endothelial cell models. Cilia 2015, 4, 11. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Snell, W. The primary cilium: Keeper of the key to cell division. Cell 2007, 129, 1255–1257. [Google Scholar] [CrossRef] [PubMed]
- Boselli, F.; Goetz, J.G.; Charvin, G.; Vermot, J. A quantitative approach to study endothelial cilia bending stiffness during blood flow mechanodetection in vivo. Methods Cell Biol. 2015, 127, 161–173. [Google Scholar] [PubMed]
- Wang, N.; Miao, H.; Li, Y.S.; Zhang, P.; Haga, J.H.; Hu, Y.; Young, A.; Yuan, S.; Nguyen, P.; Wu, C.C.; et al. Shear stress regulation of Krüppellike factor 2 expression is flow pattern-specific. Biochem. Biophys. Res. Commun. 2006, 341, 1244–1251. [Google Scholar] [CrossRef] [PubMed]
- Hierck, B.P.; Van der Heiden, K.; Alkemade, F.E.; Van de Pas, S.; Van Thienen, J.V.; Groenendijk, B.C.; Bax, W.H.; Van der Laarse, A.; Deruiter, M.C.; Horrevoets, A.J.; et al. Primary cilia sensitize endothelial cells for fluid shear stress. Dev. Dyn. 2008, 237, 725–735. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; De Silva, T.M.; Chen, J.; Faraci, F.M. Cerebral vascular disease and neurovascular injury in ischemic stroke. Circ. Res. 2017, 120, 449–471. [Google Scholar] [CrossRef] [PubMed]
- Dinsmore, C.; Reiter, J.F. Endothelial primary cilia inhibit atherosclerosis. EMBO Rep. 2016, 17, 156–166. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Duffhues, G.; de Vinuesa, A.G.; Lindeman, J.H.; Mulder-Stapel, A.; DeRuiter, M.C.; Van Munsteren, C.; Goumans, M.J.; Hierck, B.P.; Ten Dijke, P. SLUG is expressed in endothelial cells lacking primary cilia to promote cellular calcification. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 616–627. [Google Scholar] [CrossRef]
- Vion ACAlt, S.; Klaus-Bergmann, A.; Szymborska, A.; Zheng, T.; Perovic, T.; Hammoutene, A.; Oliveira, M.B.; Bartels-Klein, E.; Hollfinger, I.; Rautou, P.E.; et al. Primary cilia sensitize endothelial cells to BMP and prevent excessive vascular regression. J. Cell. Biol. 2018, 217, 1651–1665. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Gays, D.; Milia, C.; Santoro, M.M. Cilia control vascular mural cell recruitment in vertebrates. Cell Rep. 2017, 18, 1033–1047. [Google Scholar] [CrossRef] [PubMed]
- Goetz, J.G.; Steed, E.; Ferreira, R.R.; Roth, S.; Ramspacher, C.; Boselli, F.; Charvin, G.; Liebling, M.; Wyart, C.; Schwab, Y.; et al. Endothelial cilia mediate low flow sensing during zebrafish vascular development. Cell Rep. 2014, 6, 799–808. [Google Scholar] [CrossRef] [PubMed]
- Devuyst, O. Variable renal disease progression in autosomal dominant polycystic kidney disease: A role for nitric oxide? J. Nephrol. 2003, 16, 449–452. [Google Scholar] [PubMed]
- Thoma, C.R.; Frew, I.J.; Krek, W. The VHL tumor suppressor: Riding tandem with GSK3beta in primary cilium maintenance. Cell Cycle 2007, 6, 1809–1813. [Google Scholar] [CrossRef] [PubMed]
- Logie, L.; Van Aalten, L.; Knebel, A.; Force, T.; Hastie, C.J.; MacLauchlan, H.; Campbell, D.G.; Gourlay, R.; Prescott, A.; Davidson, J.; et al. Rab-GTPase binding effector protein 2 (RABEP2) is a primed substrate for glycogen synthase kinase-3 (GSK3). Sci. Rep. 2017, 7, 17682. [Google Scholar] [CrossRef] [PubMed]
- Airik, R.; Schueler, M.; Airik, M.; Cho, J.; Ulanowicz, K.A.; Porath, J.D.; Hurd, T.W.; Bekker-Jensen, S.; Schrøder, J.M.; Andersen, J.S.; et al. SDCCAG8 interacts with RAB effector proteins RABEP2 and ERC1 and is required for hedgehog signaling. PLOS One 2016, 11, e0156081. [Google Scholar] [CrossRef] [PubMed]
- Katusic, Z.S.; Austin, S.A. Neurovascular protective function of endothelial nitric oxide—Recent advances. Circ. J. 2016, 80, 1499–1503. [Google Scholar] [CrossRef] [PubMed]
- Zhong, F.; Lee, K.; He, J.C. Role of Krüppel-like factor-2 in kidney disease. Nephrology 2018, 23, 53–56. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Zou, X.; Jin, Y.; Gao, S.; Lv, J.; Li, B.; Cui, R. The role of KLF4 in Alzheimer’s disease. Front. Cell. Neurosci. 2018, 12, 325. [Google Scholar] [CrossRef] [PubMed]
- Sealock, R.; Zhang, H.; Lucitti, J.L.; Moore, S.M.; Faber, J.E. Congenic fine-mapping identifies a major causal locus for variation in the native collateral circulation and ischemic injury in brain and lower extremity. Circ. Res. 2014, 114, 660–671. [Google Scholar] [CrossRef]
- RIGOR. Improving the Quality of NINDS-Supported Preclinical and Clinical Research through Rigorous Study Design and Transparent Reporting. 2012. Available online: http://www.ninds.nih.gov/funding/transparency_in_ reporting_guidance.pdf (accessed on 1 May 2019).









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, H.; Chalothorn, D.; Faber, J.E. Collateral Vessels Have Unique Endothelial and Smooth Muscle Cell Phenotypes. Int. J. Mol. Sci. 2019, 20, 3608. https://doi.org/10.3390/ijms20153608
Zhang H, Chalothorn D, Faber JE. Collateral Vessels Have Unique Endothelial and Smooth Muscle Cell Phenotypes. International Journal of Molecular Sciences. 2019; 20(15):3608. https://doi.org/10.3390/ijms20153608
Chicago/Turabian StyleZhang, Hua, Dan Chalothorn, and James E Faber. 2019. "Collateral Vessels Have Unique Endothelial and Smooth Muscle Cell Phenotypes" International Journal of Molecular Sciences 20, no. 15: 3608. https://doi.org/10.3390/ijms20153608
APA StyleZhang, H., Chalothorn, D., & Faber, J. E. (2019). Collateral Vessels Have Unique Endothelial and Smooth Muscle Cell Phenotypes. International Journal of Molecular Sciences, 20(15), 3608. https://doi.org/10.3390/ijms20153608