Coexisting Molecular Determinants of Acquired Oxaliplatin Resistance in Human Colorectal and Ovarian Cancer Cell Lines

 and
and 
Abstract
:1. Introduction
2. Results
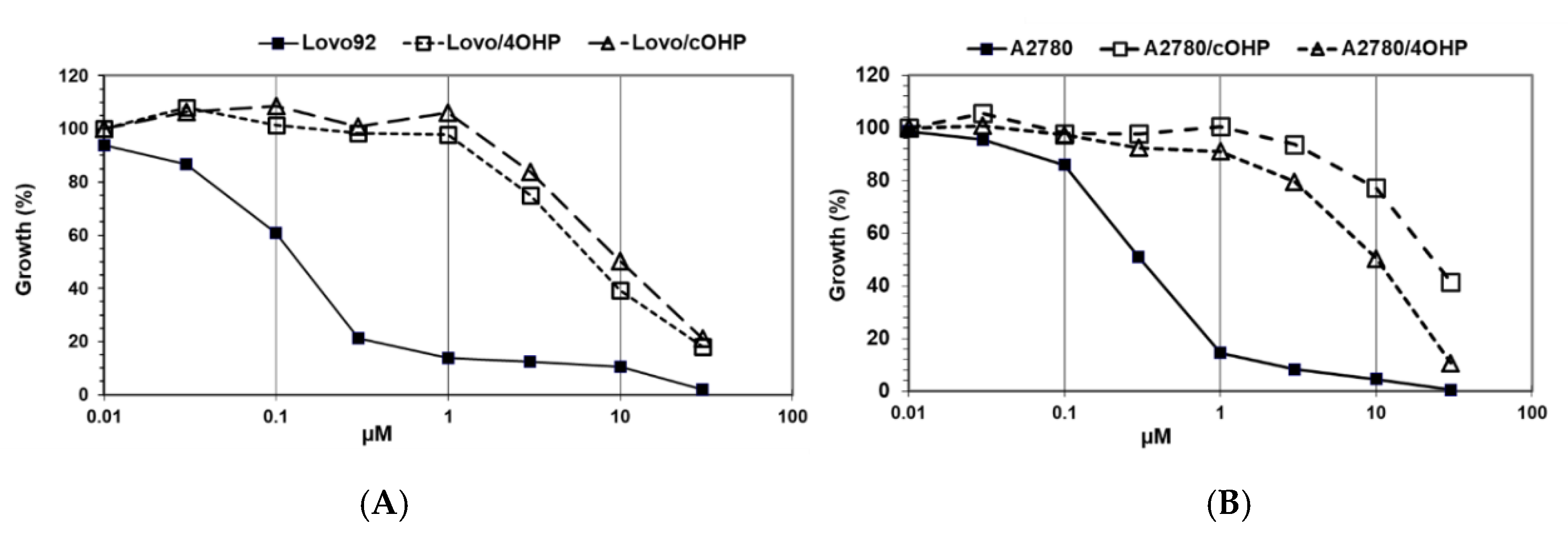
2.1. Growth Inhibition
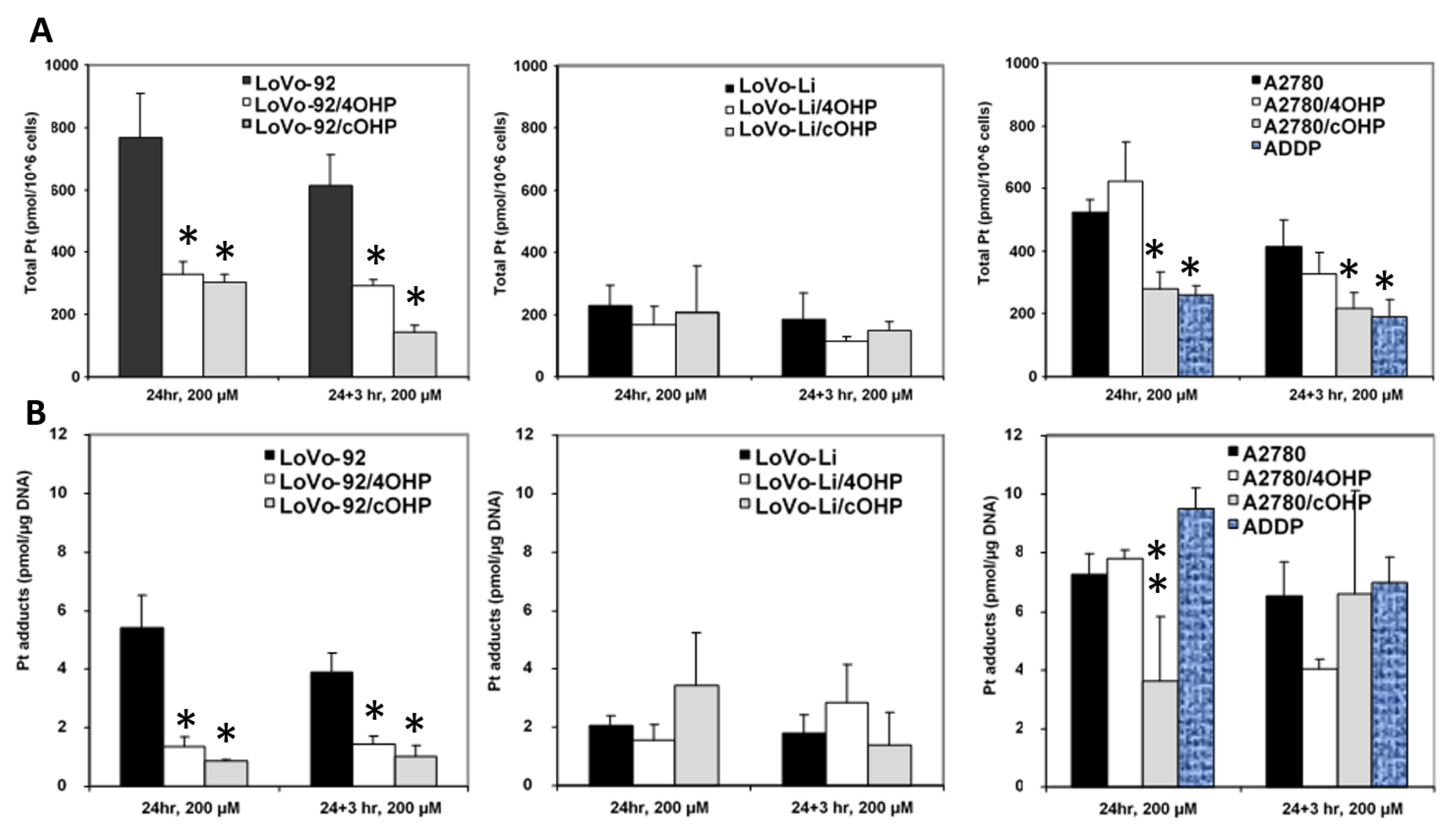
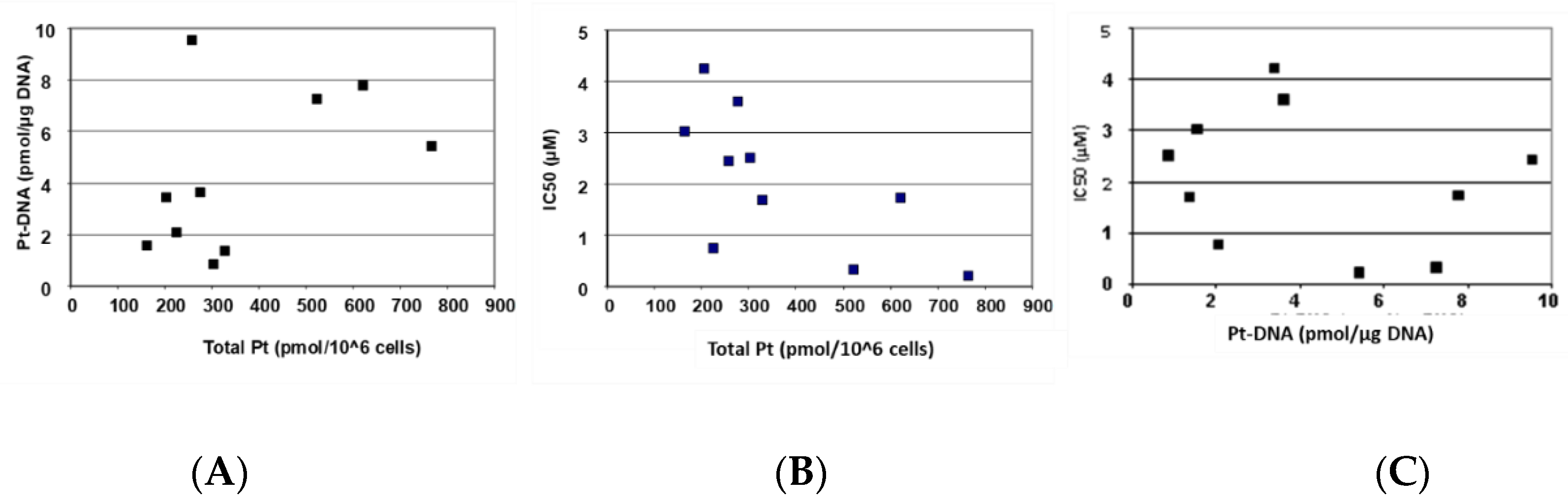
2.2. Cellular Oxaliplatin Accumulation and Formation of DNA Adducts
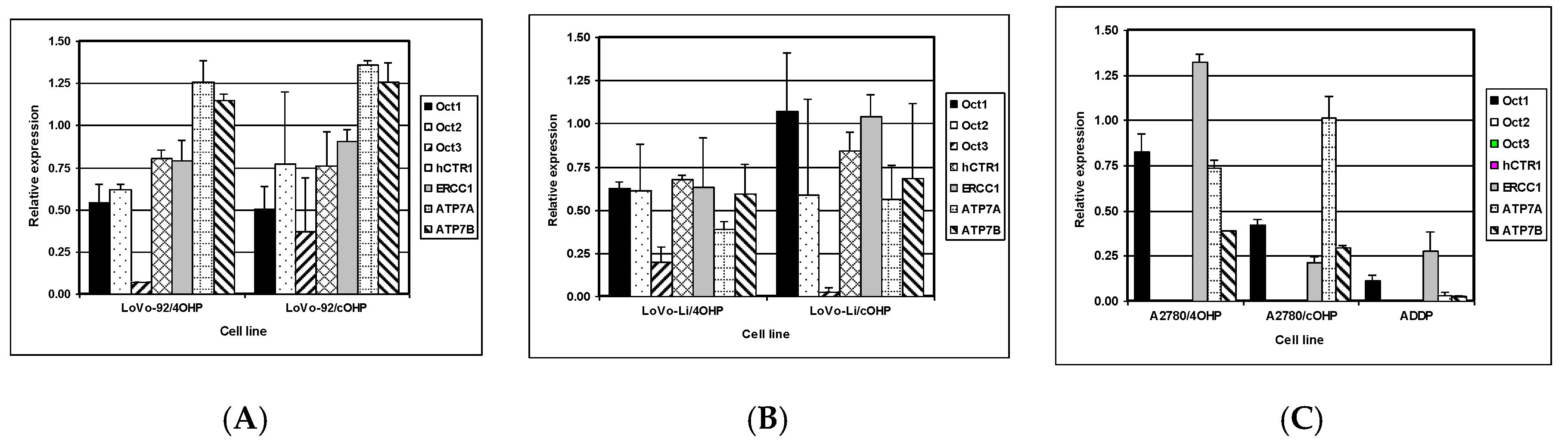
2.3. Gene Expression of Relevant Transporters and DNA Repair Genes
2.4. Genome-Wide Expression Array Analysis
2.4.1. Axonal Guidance Signaling and Aryl Hydrocarbon Receptor Signaling
2.4.2. p53 Signaling
2.4.3. Role of BCRA1 in DNA Damage Response
2.4.4. Xenobiotic Metabolism Signaling Pathways and G1/S Checkpoint Regulation
2.5. Oxaliplatin Induces Chromosomal Aberrations
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Cell Culture
4.3. Establishment of Oxaliplatin Resistance
4.4. Growth Inhibition Experiments
4.5. Oxaliplatin Accumulation and Formation of DNA Adducts
4.6. Quantitative Gene Expression Measurement
4.7. Microarray Analysis of RNA and DNA
4.8. Microarray Data Analysis
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| aCGH | Array comparative genomic hybridization |
| CDDP | Cisplatin |
| hCTR | Human copper transporter |
| FBS | Fetal bovine serum |
| l-OHP | Oxaliplatin |
| MMR | Mismatch repair |
| NER | Nucleotide excision repair |
| OCT | Organic cation transporter |
| Pt | Platinum |
| SRB | Sulforhodamine B |
References
- Carrato, A.; Gallego, J.; Diaz-Rubio, E. Oxaliplatin: Results in colorectal carcinoma. Crit. Rev. Oncol. Hematol. 2002, 44, 29–44. [Google Scholar] [CrossRef]
- Caparello, C.; Meijer, L.L.; Garajova, I.; Falcone, A.; Le Large, T.Y.; Funel, N.; Kazemier, G.; Peters, G.J.; Vasile, E.; Giovannetti, E. FOLFIRINOX and translational studies: Towards personalized therapy in pancreatic cancer. World J. Gastroenterol. 2016, 22, 6987–7005. [Google Scholar] [CrossRef] [PubMed]
- Faivre, S.; Chan, D.; Salinas, R.; Woynarowska, B.; Woynarowski, J.M. DNA strand breaks and apoptosis induced by oxaliplatin in cancer cells. Biochem. Pharmacol. 2003, 66, 225–237. [Google Scholar] [CrossRef]
- Dilruba, S.; Kalayda, G.V. Platinum-based drugs: Past, present and future. Cancer Chemother. Pharmacol. 2016, 77, 1103–1124. [Google Scholar] [CrossRef] [PubMed]
- Perego, P.; Robert, J. Oxaliplatin in the era of personalized medicine: From mechanistic studies to clinical efficacy. Cancer Chemother. Pharmacol. 2016, 77, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.P.; Hamilton, T.C.; Schilder, R.J. Platinum resistance: The role of DNA repair pathways. Clin. Cancer Res. 2008, 14, 1291–1295. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.W.; Laub, P.B.; Beesley, J.S.; Ozols, R.F.; Hamilton, T.C. Increased platinum-DNA damage tolerance is associated with cisplatin resistance and cross-resistance to various chemotherapeutic agents in unrelated human ovarian cancer cell lines. Cancer Res. 1997, 57, 850–856. [Google Scholar] [PubMed]
- Virag, P.; Fischer-Fodor, E.; Perde-Schrepler, M.; Brie, I.; Tatomir, C.; Balacescu, L.; Berindan-Neagoe, I.; Victor, B.; Balacescu, O. Oxaliplatin induces different cellular and molecular chemoresistance patterns in colorectal cancer cell lines of identical origins. BMC Genom. 2013, 14, 480. [Google Scholar] [CrossRef]
- Martinez-Balibrea, E.; Martínez-Cardús, A.; Ginés, A.; Ruiz de Porras, V.; Moutinho, C.; Layos, L.; Manzano, J.L.; Bugés, C.; Bystrup, S.; Esteller, M.; et al. Tumor-Related Molecular Mechanisms of Oxaliplatin Resistance. Mol. Cancer Ther. 2015, 14, 1767–1776. [Google Scholar] [CrossRef] [Green Version]
- Safaei, R.; Howell, S.B. Copper transporters regulate the cellular pharmacology and sensitivity to Pt drugs. Crit. Rev. Oncol. Hematol. 2005, 53, 13–23. [Google Scholar] [CrossRef]
- Holzer, A.K.; Manorek, G.H.; Howell, S.B. Contribution of the major copper influx transporter CTR1 to the cellular accumulation of cisplatin, carboplatin, and oxaliplatin. Mol. Pharmacol. 2006, 70, 1390–1394. [Google Scholar] [CrossRef] [PubMed]
- Yonezawa, A.; Masuda, S.; Yokoo, S.; Katsura, T.; Inui, K. Cisplatin and oxaliplatin, but not carboplatin and nedaplatin, are substrates for human organic cation transporters (SLC22A1-3 and multidrug and toxin extrusion family). J. Pharmacol. Exp. Ther. 2006, 19, 879–886. [Google Scholar] [CrossRef] [PubMed]
- Samimi, G.; Safaei, R.; Katano, K.; Holzer, A.K.; Rochdi, M.; Tomioka, M.; Howell, S.B. Increased expression of the copper efflux transporter ATP7A mediates resistance to cisplatin, carboplatin, and oxaliplatin in ovarian cancer cells. Clin. Cancer Res. 2004, 10, 4661–4669. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Balibrea, E.; Martinez-Cardus, A.; Musulen, E.; Ginés, A.; Manzano, J.L.; Aranda, E.; Abad, A. Increased levels of copper efflux transporter ATP7B are associated with poor outcome in colorectal cancer patients receiving oxaliplatin-based chemotherapy. Int. J. Cancer 2009, 124, 2905–2910. [Google Scholar] [CrossRef]
- Zdraveski, Z.Z.; Mello, J.A.; Farinelli, C.K.; Essigmann, J.M.; Marinus, M.G. MutS preferentially recognizes cisplatin- over oxaliplatin-modified DNA. J. Biol. Chem. 2002, 277, 1255–1260. [Google Scholar] [CrossRef] [PubMed]
- Avan, A.; Narayan, R.; Giovannetti, E.; Peters, G.J. Role of Akt signaling in resistance to DNA-targeted therapy. World J. Clin. Oncol. 2016, 7, 352–369. [Google Scholar] [CrossRef]
- Fink, D.; Zheng, H.; Nebel, S.; Norris, P.S.; Aebi, S.; Lin, T.P.; Howell, S.B. In vitro and in vivo resistance to cisplatin in cells that have lost DNA mismatch repair. Cancer Res. 1997, 57, 1841–1845. [Google Scholar]
- Fink, D.; Nebel, S.; Aebi, S.; Zheng, H.; Cenni, B.; Nehmé, A.; Howell, S.B. The role of DNA mismatch repair in platinum drug resistance. Cancer Res. 1996, 56, 4881–4886. [Google Scholar]
- Vaisman, A.; Varchenko, M.; Umar, A.; Kunkel, T.A.; Risinger, J.I.; Barrett, J.C.; Chaney, S.G. The role of hMLH1, hMSH3, and hMSH6 defects in cisplatin and oxaliplatin resistance: Correlation with replicative bypass of platinum-DNA adducts. Cancer Res. 1998, 58, 3579–3585. [Google Scholar]
- Kwon, H.C.; Roh, M.S.; Oh, S.Y.; Kim, S.H.; Kim, M.C.; Kim, J.S.; Kim, H.J. Prognostic value of expression of ERCC1, thymidylate synthase, and glutathione S-transferase P1 for 5-fluorouracil/oxaliplatin chemotherapy in advanced gastric cancer. Ann. Oncol. 2007, 18, 504–509. [Google Scholar] [CrossRef]
- Shirota, Y.; Stoehlmacher, J.; Brabender, J.; Xiong, Y.P.; Uetake, H.; Danenberg, K.D.; Lenz, H.J. ERCC1 and thymidylate synthase mRNA levels predict survival for colorectal cancer patients receiving combination oxaliplatin and fluorouracil chemotherapy. J. Clin. Oncol. 2001, 19, 4298–4304. [Google Scholar] [CrossRef] [PubMed]
- Gourdier, I.; Crabbe, L.; Andreau, K.; Pau, B.; Kroemer, G. Oxaliplatin-induced mitochondrial apoptotic response of colon carcinoma cells does not require nuclear DNA. Oncogene 2004, 23, 7449–7457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gourdier, I.; Del Rio, M.; Crabbe, L.; Candeil, L.; Copois, V.; Ychou, M.; Pau, B. Drug specific resistance to oxaliplatin is associated with apoptosis defect in a cellular model of colon carcinoma. FEBS Lett. 2002, 529, 232–236. [Google Scholar] [CrossRef]
- Branch, P.; Masson, M.; Aquilina, G.; Bignami, M.; Karran, P. Spontaneous development of drug resistance: Mismatch repair and p53 defects in resistance to cisplatin in human tumor cells. Oncogene 2000, 19, 3138–3145. [Google Scholar] [CrossRef] [PubMed]
- Reles, A.; Wen, W.H.; Schmider, A.; Gee, C.; Runnebaum, I.B.; Kilian, U.; Reich, O. Correlation of p53 mutations with resistance to platinum-based chemotherapy and shortened survival in ovarian cancer. Clin. Cancer Res. 2001, 7, 2984–2997. [Google Scholar] [PubMed]
- Siddik, Z.H.; Mims, B.; Lozano, G.; Thai, G. Independent pathways of p53 induction by cisplatin and X-rays in a cisplatin-resistant ovarian tumor cell line. Cancer Res. 1998, 58, 698–703. [Google Scholar] [PubMed]
- Arango, D.; Wilson, A.J.; Shi, Q.; Corner, G.A.; Aranes, M.J.; Nicholas, C.; Augenlicht, L.H. Molecular mechanisms of action and prediction of response to oxaliplatin in colorectal cancer cells. Br. J. Cancer 2004, 91, 1931–1946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noordhuis, P.; Laan, A.C.; van de Born, K.; Losekoot, N.; Kathmann, I.; Peters, G.J. Oxaliplatin activity in selected and unselected human ovarian and colorectal cancer cell lines. Biochem. Pharmacol. 2008, 76, 53–61. [Google Scholar] [CrossRef]
- Topping, R.P.; Wilkinson, J.C.; Scarpinato, K.D. Mismatch Repair Protein Deficiency Compromises Cisplatin-induced Apoptotic Signaling. J. Biol. Chem. 2009, 284, 14029–14039. [Google Scholar] [CrossRef] [Green Version]
- Corte-Rodríguez, M.; Espina, M.; Sierra, L.M.; Blanco, E.; Ames, T.; Montes-Bayón, M.; Sanz-Medel, A. Quantitative evaluation of cellular uptake, DNA incorporation and adduct formation in cisplatin sensitive and resistant cell lines: Comparison of different Pt-containing drugs. Biochem. Pharmacol. 2015, 98, 69–77. [Google Scholar] [CrossRef]
- Zhang, S.; Lovejoy, K.S.; Shima, J.E.; Lagpacan, L.L.; Shu, Y.; Lapuk, A.; Chen, Y.; Komori, T.; Gray, J.W.; Chen, X.; et al. Organic Cation Transporters Are Determinants of Oxaliplatin Cytotoxicity. Cancer Res. 2006, 66, 8847–8857. [Google Scholar] [CrossRef]
- Yokoo, S.; Yonezawa, A.; Masuda, S.; Fukatsu, A.; Katsura, T.; Inui, K. Differential contribution of organic cation transporters, OCT2 and MATE1, in platinum agent-induced nephrotoxicity. Biochem. Pharmacol. 2007, 74, 477–487. [Google Scholar] [CrossRef]
- Burger, H.; Zoumaro-Djayoon, A.; Boersma, A.W.; Helleman, J.; Berns, E.M.; Mathijssen, R.H.; Loos, W.J.; Wiemer, E.A. Differential transport of platinum compounds by the human organic cation transporter hOCT2 (hSLC22A2). Br. J. Pharmacol. 2010, 159, 898–908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Q.; Yu, L.; Qin, Z.; Chen, L.; Hu, H.; Zheng, X.; Zeng, S. Regulation of OCT2 transcriptional repression by histone acetylation in renal cell carcinoma. Epigenetics 2019, 15, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Buß, I.; Hamacher, A.; Sarin, N.; Kassack, M.U.; Kalayda, G.V. Relevance of copper transporter 1 and organic cation transporters 1-3 for oxaliplatin uptake and drug resistance in colorectal cancer cells. Metallomics 2018, 10, 414–425. [Google Scholar] [CrossRef] [PubMed]
- Yokoo, S.; Masuda, S.; Yonezawa, A.; Terada, T.; Katsura, T.; Inui, K. Significance of organic cation transporter 3 (SLC22A3) expression for the cytotoxic effect of oxaliplatin in colorectal cancer. Drug Metab. Dispos. 2008, 36, 2299–2306. [Google Scholar] [CrossRef]
- Ghose, S.; Oleinik, N.V.; Krupenko, N.I.; Krupenko, S.A. 10-formyltetrahydrofolate dehydrogenase-induced c-Jun-NH2-kinase pathways diverge at the c-Jun-NH2-kinase substrate level in cells with different p53 status. Mol. Cancer Res. 2009, 7, 99–107. [Google Scholar] [CrossRef]
- Krupenko, S.A. FDH: An aldehyde dehydrogenase fusion enzyme in folate metabolism. Chem. Biol. Interact. 2009, 178, 84–93. [Google Scholar] [CrossRef] [Green Version]
- Tanei, T.; Morimoto, K.; Shimazu, K.; Kim, S.J.; Tanji, Y.; Taguchi, T.; Tamaki, Y.; Noguchi, S. Association of breast cancer stem cells identified by aldehyde dehydrogenase 1 expression with resistance to sequential Paclitaxel and epirubicin-based chemotherapy for breast cancers. Clin. Cancer Res. 2009, 15, 4234–4241. [Google Scholar] [CrossRef]
- Wang, X.; Li, M.; Wang, J.; Yeung, C.M.; Zhang, H.; Kung, H.F.; Jiang, B.; Lin, M.C. The BH3-only protein, PUMA, is involved in oxaliplatin-induced apoptosis in colon cancer cells. Biochem. Pharmacol. 2006, 71, 1540–1550. [Google Scholar] [CrossRef]
- Fujie, Y.; Yamamoto, H.; Ngan, C.Y.; Takagi, A.; Hayashi, T.; Suzuki, R.; Ezumi, K.; Takemasa, I.; Ikeda, M.; Sekimoto, M.; et al. Oxaliplatin, a potent inhibitor of survivin, enhances paclitaxel-induced apoptosis and mitotic catastrophe in colon cancer cells. Jpn. J. Clin. Oncol. 2005, 35, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Hsiao, P.W.; Chiu, T.H.; Chao, J.I. Combination of cyclooxygenase-2 inhibitors and oxaliplatin increases the growth inhibition and death in human colon cancer cells. Biochem. Pharmacol. 2005, 70, 658–667. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Yin, J.; Zhang, Y.; Zhang, S. Annexin A3 depletion overcomes resistance to oxaliplatin in colorectal cancer via the MAPK signaling pathway. J. Cell. Biochem. 2019, 120, 14585–14593. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; He, G.; Siddik, Z.H. Functional Activation of Mutant p53 by Platinum Analogues in Cisplatin-Resistant Cells Is Dependent on Phosphorylation. Mol. Cancer Res. 2017, 15, 328–339. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Z.; Kang, Q.J.; Xiang, X. Gene and protein expression in the oxaliplatin-resistant HT29/L-OHP human colon cancer cell line. Genet. Mol. Res. 2015, 14, 11013–11022. [Google Scholar] [CrossRef] [PubMed]
- Stordal, B.; Davey, R. ERCC1 expression and RAD51B activity correlate with cell cycle response to platinum drug treatment not DNA repair. Cancer Chemother. Pharmacol. 2009, 63, 661–672. [Google Scholar] [CrossRef] [PubMed]
- Selga, E.; Noe, V.; Ciudad, C.J. Transcriptional regulation of aldo-keto reductase 1C1 in HT29 human colon cancer cells resistant to methotrexate: Role in the cell cycle and apoptosis. Biochem. Pharmacol. 2008, 75, 414–426. [Google Scholar] [CrossRef]
- Wang, H.W.; Lin, C.P.; Chiu, J.H.; Chow, K.C.; Kuo, K.T.; Lin, C.S.; Wang, L.S. Reversal of inflammation-associated dihydrodiol dehydrogenases (AKR1C1 and AKR1C2) overexpression and drug resistance in nonsmall cell lung cancer cells by wogonin and chrysin. Int. J. Cancer 2007, 120, 2019–2027. [Google Scholar] [CrossRef]
- Wang, C.; Yan, R.; Luo, D.; Watabe, K.; Liao, D.F.; Cao, D. Aldo-keto reductase family 1 member B10 promotes cell survival by regulating lipid synthesis and eliminating carbonyls. J. Biol. Chem. 2009, 284, 26742–26748. [Google Scholar] [CrossRef]
- Beretta, G.L.; Benedetti, V.; Cossa, G.; Assaraf, Y.G.; Bram, E.; Gatti, L.; Zunino, F. Increased levels and defective glycosylation of MRPs in ovarian carcinoma cells resistant to oxaliplatin. Biochem. Pharmacol. 2010, 79, 1108–1117. [Google Scholar] [CrossRef] [Green Version]
- Myint, K.; Li, Y.; Paxton, J.; McKeage, M. Multidrug Resistance-Associated Protein 2 (MRP2) Mediated Transport of Oxaliplatin-Derived Platinum in Membrane Vesicles. PLoS ONE 2015, 10, e0130727. [Google Scholar] [CrossRef] [PubMed]
- Soulie, P.; Poupon, M.F.; Remvikos, Y.; Dutrillaux, B.; Muleris, M. Distinct chromosomal alterations associated with TP53 status of LoVo cells under PALA selective pressure: A parallel with cytogenetic pathways of colorectal cancers. Oncogene 1999, 18, 775–781. [Google Scholar] [CrossRef] [PubMed]
- Stordal, B.; Peters, G.; Davey, R. Similar chromosomal changes in cisplatin and oxaliplatin-resistant sublines of the H69 SCLC cell line are not associated with platinum resistance. Genes Chromosomes Cancer 2006, 45, 1094–1105. [Google Scholar] [CrossRef] [PubMed]
- Pocard, M.; Chevillard, S.; Villaudy, J.; Poupon, M.F.; Dutrillaux, B.; Remvikos, Y. Different p53 mutations produce distinct effects on the ability of colon carcinoma cells to become blocked at the G1/S boundary after irradiation. Oncogene 1996, 12, 875–882. [Google Scholar] [PubMed]
- Giovannetti, E.; Backus, H.H.; Wouters, D.; Ferreira, C.G.; Van Houten, V.M.M.; Brakenhoff, R.H.; Peters, G.J. Changes in the status of p53 affect drug sensitivity to thymidylate synthase (TS) inhibitors by altering TS levels. Br. J. Cancer 2007, 96, 769–775. [Google Scholar] [CrossRef] [Green Version]
- Keepers, Y.P.; Pizao, P.E.; Peters, G.J.; van Ark-Otte, J.; Winograd, B.; Pinedo, H.M. Comparison of the sulforhodamine B protein and tetrazolium (MTT) assays for in vitro chemosensitivity testing. Eur. J. Cancer 1991, 27, 897–900. [Google Scholar] [CrossRef] [Green Version]
- Bergman, A.M.; Ruiz van Haperen, V.W.; Veerman, G.; Kuiper, C.M.; Peters, G.J. Synergistic interaction between cisplatin and gemcitabine in vitro. Clin. Cancer Res. 1996, 2, 521–530. [Google Scholar]
- Van Moorsel, C.J.; Pinedo, H.M.; Veerman, G.; Bergman, A.M.; Kuiper, C.M.; Vermorken, J.B.; Peters, G.J. Mechanisms of synergism between cisplatin and gemcitabine in ovarian and non-small-cell lung cancer cell lines. Br. J. Cancer 1999, 80, 981–990. [Google Scholar] [CrossRef] [Green Version]
- Rots, M.G.; Willey, J.C.; Jansen, G.; Van Zantwijk, C.H.; Noordhuis, P.; DeMuth, J.P.; Peters, G.J. mRNA expression levels of methotrexate resistance-related proteins in childhood leukemia as determined by a standardized competitive template-based RT-PCR method. Leukemia 2000, 14, 2166–2175. [Google Scholar] [CrossRef] [Green Version]
- Buffart, T.E.; Israeli, D.; Tijssen, M.; Vosse, S.J.; Mršić, A.; Meijer, G.A.; Ylstra, B. Across array comparative genomic hybridization: A strategy to reduce reference channel hybridizations. Genes Chromosomes Cancer 2008, 47, 994–1004. [Google Scholar] [CrossRef]
- Van de Wiel, M.A.; Kim, K.I.; Vosse, S.J.; Van Wieringen, W.N.; Wilting, S.M.; Ylstra, B. CGHcall: Calling aberrations for array CGH tumor profiles. Bioinformatics 2007, 23, 892–894. [Google Scholar] [CrossRef] [PubMed]
- Van Wieringen, W.N.; Van de Wiel, M.A. Nonparametric testing for DNA copy number induced differential mRNA gene expression. Biometrics 2009, 65, 19–29. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
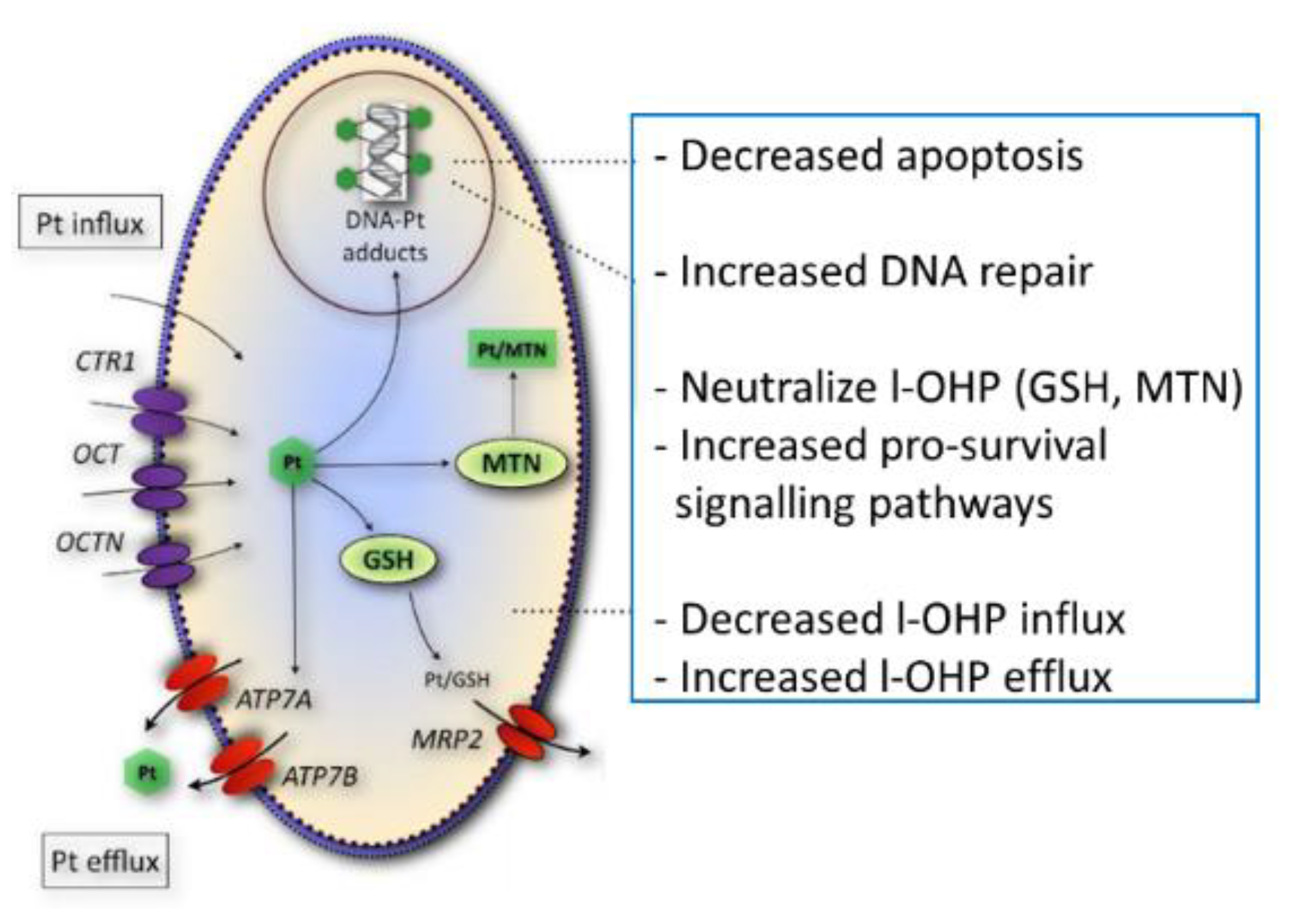{kind=link}
| Cell Line | l-OHP (µM) | CDDP (µM) | RF (l-OHP) | RF (CDDP) |
|---|---|---|---|---|
| LoVo-92 | 0.21 ± 0.04 | 1.50 ± 0.40 | ||
| LoVo-92/4OHP | 1.69 ± 0.25 | 2.50 ± 1.31 | 7.9 | 1.7 |
| LoVo-92/cOHP | 2.51 ± 0.25 | 4.03 ± 1.64 | 11.8 | 2.7 |
| LoVo-Li | 0.75 ± 0.08 | 4.23 ± 1.65 | ||
| LoVo-Li/4OHP | 3.03 ± 0.64 | 4.65 ± 0.35 | 4.0 | 1.1 |
| LoVo-Li/cOHP | 4.23 ± 0.92 | 7.57 ± 1.23 | 5.6 | 1.8 |
| A2780 | 0.32 ± 0.03 | 0.50 ± 0.14 | ||
| A2780/4OHP | 1.73 ± 0.40 | 3.50 ± 2.25 | 5.3 | 7.0 |
| A2780/cOHP | 3.61 ± 0.33 | 4.60 ± 0.70 | 11.1 | 9.3 |
| ADDP | 2.43 ± 0.35 | 20.3 ± 2.7 | 7.5 | 40.9 |
| Pearson Correlation | Spearman’s Rho | ||||||
|---|---|---|---|---|---|---|---|
| Total Pt | Pt-DNA | IC50 | Total Pt | Pt-DNA | IC50 | ||
| Total Pt | Correlation | 0.760 * | −0.667 * | 0.533 | −0.700 * | ||
| p-value | 0.018 | 0.050 | 0.139 | 0.036 | |||
| Pt-DNA | Correlation | 0.760 * | −0.398 | 0.533 | −0.283 | ||
| p-value | 0.018 | 0.289 | 0.139 | 0.460 | |||
| OCT1 | Correlation | 0.882 * | 0.841 * | −0.557 | 0.867 * | 0.733 * | −0.467 |
| p-value | 0.007 | 0.005 | 0.119 | 0.002 | 0.025 | 0.205 | |
| OCT2 | Correlation | 0.802 * | 0.152 | 0.638 | 0.714 | −0.214 | −0.821 * |
| p-value | 0.030 | 0.745 | 0.123 | 0.071 | 0.645 | 0.023 | |
| OCT3 | Correlation | 0.949 * | 0.688 | −0.675 | 0.771 | 0.029 | −0.829 * |
| p-value | 0.004 | 0.131 | 0.141 | 0.072 | 0.975 | 0.042 | |
| CTR1 | Correlation | 0.475 | 0.145 | −0.413 | 0.762 * | −0.071 | −0.619 |
| p-value | 0.234 | 0.732 | 0.269 | 0.028 | 0.867 | 0.102 | |
| ATP7A | Correlation | −0.489 | −0.494 | −0.107 | −0.644 | −0.745 * | 0.259 |
| p-value | 0.182 | 0.177 | 0.784 | 0.061 | 0.021 | 0.500 | |
| ATP7B | Correlation | 0.351 | −0.054 | −0.588 | 0.517 | −0.217 | −0.617 |
| p-value | 0.354 | 0.890 | 0.096 | 0.154 | 0.576 | 0.077 | |
| ERCC1 | Correlation | 0.572 | 0.672 * | −0.457 | 0.500 | 0.650 | −0.367 |
| p-value | 0.108 | 0.048 | 0.216 | 0.170 | 0.058 | 0.332 | |
| LoVo-92 | LoVo-Li | A2780 | ||||
|---|---|---|---|---|---|---|
| IPA Pathway | 4OHP | cOHP | 4OHP | cOHP | 4OHP | cOHP |
| Axonal guidance signaling | 3.16 | 2.35 | 3.56 | 1.92 | ||
| Aryl hydrocarbon receptor signaling | 2.81 | 3.95 | 2.32 | 3.06 | ||
| p53 Signaling | 1.71 | 2.90 | 3.84 | |||
| Colorectal cancer metastasis signaling | 2.72 | 4.10 | 2.97 | |||
| ILK signaling | 1.99 | 3.44 | 2.57 | |||
| RAR activation | 2.17 | 2.42 | 2.11 | 2.66 | ||
| Role of macrophages, fibroblasts and endothelial cells in rheumatoid arthritis | 2.00 | 2.98 | 3.93 | 2.04 | ||
| Virus entry via endocytic pathways | 1.78 | 3.08 | 3.28 | |||
| Agrin interactions at neuromuscular junction | 1.84 | 2.71 | ||||
| Cardiac hypertrophy signaling | 2.00 | 2.02 | ||||
| Caveolar-mediated endocytosis signaling | 1.71 | 2.89 | ||||
| Cysteine metabolism | 1.42 | 2.32 | 1.89 | |||
| Glycine, serine and threonine metabolism | 1.48 | 3.38 | 2.96 | |||
| Hepatic fibrosis/hepatic stellate cell activation | 2.94 | 2.32 | ||||
| Molecular mechanisms of cancer | 3.51 | 2.43 | ||||
| p38 MAPK signaling | 1.52 | 3.01 | 1.95 | |||
| Semaphorin signaling in neurons | 2.45 | 2.84 | ||||
| Sphingosine-1-phosphate signaling | 3.11 | 2.06 | ||||
| Starch and sucrose metabolism | 1.77 | 1.40 | 4.36 | |||
| Tyrosine metabolism | 1.83 | 1.75 | 2.37 | |||
| Role of BCRA1 in DNA damage response | 2.30 | 5.76 | ||||
| CXCR4 Signaling | 2.42 | |||||
| Xenobiotic metabolism by cytochrome P450 | 9.41 | |||||
| Cell cycle: G1/S checkpoint regulation | 4.63 | |||||
| Xenobiotic metabolism signaling | 5.57 | |||||
| Decreased Proapoptotic Genes | Increased “Inhibition of Apoptosis” Genes |
|---|---|
| Bax | PCNA (replication) |
| PUMA | DNA damage response |
| Apaf1 | IAP |
| Tumor suppressor serpin B5 | Survivin |
| BRCA1 in DNA damage |
| Chr | Chr. Band | Probe Position | Aberration | Cell Lines |
|---|---|---|---|---|
| 2 | q37.1 | 232396451–233411853 | Focal gain | LoVo-Li/4OHP, LoVo-Li/cOHP |
| 4 | p16.3–16.1 | 6503780–880103 | Gain | A2780/4OHP, A2780/cOHP |
| 7 | q31.1 | 110361335–110795919 | Focal loss | LoVo-Li/4OHP, LoVo-Li/cOHP |
| 10 | q21.3 | 69418458–69551488 | Focal loss | A2780/4OHP, A2780/cOHP |
| 12 | q24.23–q24.31 | 120316650–120358330 | Focal gain | LoVo-Li/4OHP, LoVo-Li/cOHP |
| 15 | q22–q26.2 | 66499644–98087372 | Loss | LoVo-92/4OHP, LoVo-92/cOHP |
| 16 | p13.3 | 3427264–4180609 | Focal gain | LoVo-Li/4OHP, LoVo-Li/cOHP, A2780/4OHP |
| 17 | q21.2 | 39782285–39993938 | Focal gain | LoVo-Li/4OHP, LoVo-Li/cOHP, A2780/cOHP |
| 19 | p13.3 | 232080–637653 | Focal gain | A2780/4OHP, A2780/cOHP |
| 19 | p13.11 | 17268193–17536526 | Focal gain | LoVo-Li/4OHP, LoVo-Li/cOHP |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Noordhuis, P.; Laan, A.C.; van de Born, K.; Honeywell, R.J.; Peters, G.J. Coexisting Molecular Determinants of Acquired Oxaliplatin Resistance in Human Colorectal and Ovarian Cancer Cell Lines. Int. J. Mol. Sci. 2019, 20, 3619. https://doi.org/10.3390/ijms20153619
Noordhuis P, Laan AC, van de Born K, Honeywell RJ, Peters GJ. Coexisting Molecular Determinants of Acquired Oxaliplatin Resistance in Human Colorectal and Ovarian Cancer Cell Lines. International Journal of Molecular Sciences. 2019; 20(15):3619. https://doi.org/10.3390/ijms20153619
Chicago/Turabian StyleNoordhuis, Paul, Adrianus C. Laan, Kasper van de Born, Richard J. Honeywell, and Godefridus J. Peters. 2019. "Coexisting Molecular Determinants of Acquired Oxaliplatin Resistance in Human Colorectal and Ovarian Cancer Cell Lines" International Journal of Molecular Sciences 20, no. 15: 3619. https://doi.org/10.3390/ijms20153619
APA StyleNoordhuis, P., Laan, A. C., van de Born, K., Honeywell, R. J., & Peters, G. J. (2019). Coexisting Molecular Determinants of Acquired Oxaliplatin Resistance in Human Colorectal and Ovarian Cancer Cell Lines. International Journal of Molecular Sciences, 20(15), 3619. https://doi.org/10.3390/ijms20153619