Glutathione Metabolism in Renal Cell Carcinoma Progression and Implications for Therapies



Abstract
:1. Introduction
2. Renal Cell Carcinoma: An Overview
2.1. Clear Cell Renal Cell Carcinoma
2.2. Papillary Renal Cell Carcinoma
2.3. Chromophobe Renal Cell Carcinoma
3. Rewired Glutathione Metabolism in RCC Is a Key Metabolic Alteration Involved in Tumor Progression
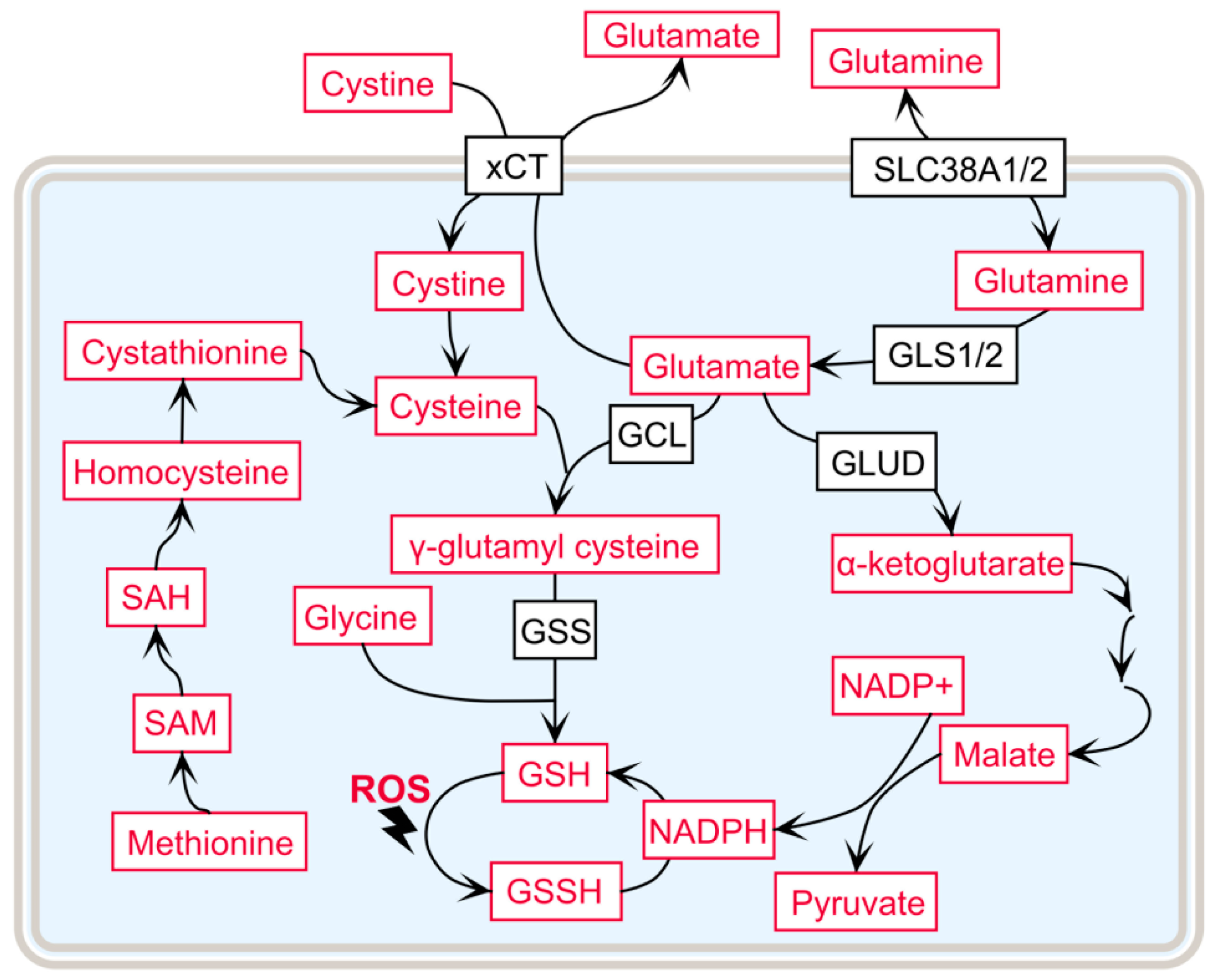
3.1. γ-Glutamyl Cycle and ccRCC Progression
3.2. Precursor Amino Acid Availability for GSH de novo Synthesis
3.3. Increased Flux of the Pentose Phosphate Pathway in ccRCC to Support GSH Synthesis
3.4. Fumarate Hydratase Mutations and GSH in Type II pRCC
3.5. Glutathione Salvage Pathway in chRCC
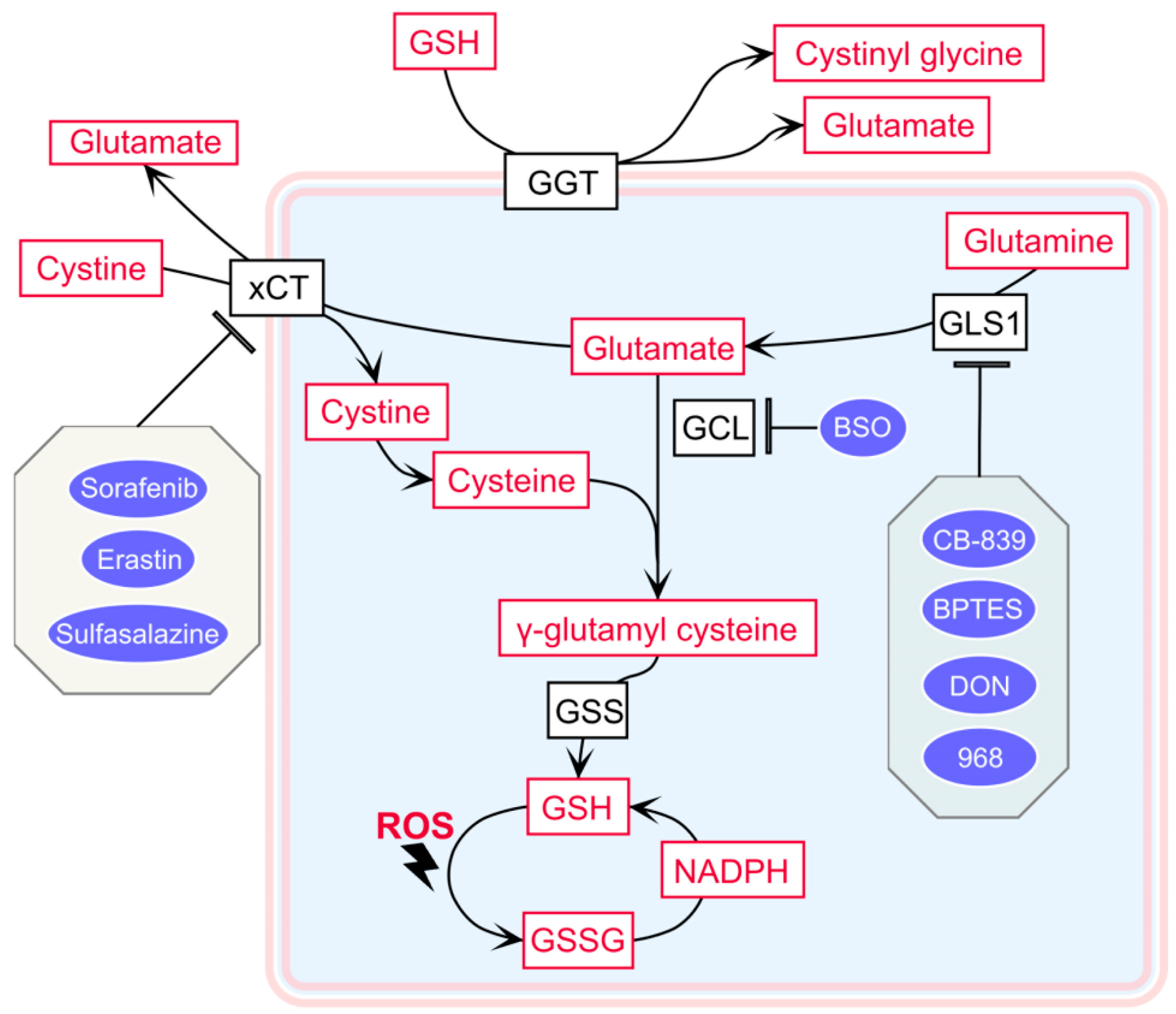
4. Therapeutic Strategies to Exploit Increased GSH Levels in RCC
4.1. The Cystine–Glutamate Shuttle Inhibitor
4.2. Glutaminase 1 Inhibitor
4.3. The Glutamate–Cysteine Ligase Inhibitor Buthionine Sulfoximine
4.4. Inhibition of Deubiquitinating Enzymes Initiates Proteotoxicity
4.5. The Role of GSH Metabolism in the Immune Microenvironment of the Tumor
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 968 | 5-[3-bromo-4-(dimethylamino)phenyl]-2,3,5,6-tetrahydro-2,2-dimethyl-benzo[A]phenanth-ridin-4(1H)-one |
| ALDOB | Aldolase B |
| ARE | Antioxidant response element |
| BPTES | Bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-Yl)ethyl sulfide |
| BRAF | Proto-oncogene B-RAF |
| BSO | Buthionine sulfoximine |
| ccRCC | Clear cell renal cell carcinoma |
| CDKN2A | Cyclin-dependent kinase inhibitor 2A |
| CDKN2B | Cyclin-dependent kinase inhibitor 2B |
| chRCC | Chromophobe renal cell carcinoma |
| CRAF | Proto-oncogene C-RAF |
| CTLA-4 | Cytotoxic T lymphocyte-associated protein 4 |
| CUL3 | Cullin 3 |
| DCR | Disease control rate |
| DON | 6-diazo-5-oxo-l-norleucine |
| DUB | Deubiquitinating enzyme |
| EGFR | Epidermal growth factor receptor |
| EGLN | Egl-nine homolog |
| FAK | Focal adhesion kinase |
| FBP | Fructose 1,6-bisphosphate |
| FBP1 | Fructose 1,6-bisphosphatase 1 |
| FH | Fumarate hydratase |
| FLT3 | Fms-like tyrosine kinase 3 |
| G6PD | Glucose-6-phosphate dehydrogenase |
| GCL | Glutamate cysteine ligase |
| GCLC | Glutamate cysteine ligase catalytic subunit |
| GCLM | Glutamate cysteine ligase modulatory subunit |
| GGT | γ-glutamyl transferase |
| GGT1 | γ-glutamyl transferase 1 |
| GLS1 | Glutaminase 1 |
| GLS2 | Glutaminase 2 |
| GLUD | Glutamate dehydrogenase |
| GLUT1 | Glucose transporter type 1 |
| GPX | Glutathione peroxidase |
| GR | Glutathione reductase |
| GSH | Glutathione reduced form |
| GSS | Glutathione synthetase |
| GSSG | Glutathione oxidized form |
| GST | Glutathione S-transferase |
| HGF | Hepatocyte growth factor |
| HIF | Hypoxia inducible factor |
| KEAP1 | Kelch-like ECH-associated protein 1 |
| KIT | Tyrosine-protein kinase kit |
| MET | Proto-oncogene c-Met |
| MYC | MYC proto-oncogene |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| NOX4 | NADPH oxidase 4 |
| NRAS | NRAS proto-oncogene, GTPase |
| NRF2 | Nuclear factor erythroid 2-related factor 2 |
| ODD | Oxygen-dependent degradation |
| PC | Pyruvate carboxylase |
| PD-1 | Programmed cell death 1 |
| PD-L1 | Programmed cell death-ligand 1 |
| PDGFR | Platelet-derived growth factor receptor |
| PPP | Pentose phosphate pathway |
| pRCC | Papillary renal cell carcinoma |
| R5P | Ribose 5-phosphate |
| RCC | Renal cell carcinoma |
| RET | RET proto-oncogene |
| ROS | Reactive oxygen species |
| RTK | Receptor tyrosine kinase |
| SAH | S-adenosyl homocysteine |
| SAM | S-adenosyl methionine |
| SETD2 | Set domain-containing 2, histone lysine methyltransferase |
| SLC38A1 | Solute carrier family 38 member 1 |
| SLC7A11 | Solute carrier family 7 member 11 |
| TERT | Telomerase reverse transcriptase |
| TFE3 | Transcription factor E3 |
| TSC1 | Tuberous sclerosis 1 protein |
| TSC2 | Tuberous sclerosis 2 protein |
| VDAC1 | Voltage-dependent anion channel 1 |
| VDAC2 | Voltage-dependent anion channel 2 |
| VDAC3 | Voltage-dependent anion channel 3 |
| VEGF | Vascular endothelial growth factor |
| VEGFR | Vascular endothelial growth factor receptor |
| VHL | von Hippel-Lindau |
References
- Kim, J.; Kim, J.; Bae, J.S. ROS homeostasis and metabolism: A critical liaison for cancer therapy. Exp. Mol. Med. 2016, 48, e269. [Google Scholar] [CrossRef] [PubMed]
- Meister, A. On the discovery of glutathione. Trends. Biochem. Sci. 1988, 13, 185–188. [Google Scholar] [CrossRef]
- Kaplowitz, N.; Aw, T.Y.; Ookhtens, M. The regulation of hepatic glutathione. Annu. Rev. Pharmacol. Toxicol. 1985, 25, 715–744. [Google Scholar] [CrossRef] [PubMed]
- Meister, A.; Anderson, M.E. Glutathione. Annu. Rev. Biochem. 1983, 52, 711–760. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.C. Regulation of glutathione synthesis. Mol. Asp. Med. 2009, 30, 42–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, E.S.; Johnson, N.; Snider, B.C. Tissue glutathione as a cyst(e)ine reservoir during cystine depletion in growing rats. J. Nutr. 1984, 114, 1853–1862. [Google Scholar] [CrossRef]
- Sipos, K.; Lange, H.; Fekete, Z.; Ullmann, P.; Lill, R.; Kispal, G. Maturation of cytosolic iron-sulfur proteins requires glutathione. J. Biol. Chem. 2002, 277, 26944–26949. [Google Scholar] [CrossRef]
- Awasthi, Y.C.; Misra, G.; Rassin, D.K.; Srivastava, S.K. Detoxification of xenobiotics by glutathione S-transferases in erythrocytes: The transport of the conjugate of glutathione and 1-chloro-2,4-dinitrobenzene. Br. J. Haematol. 1983, 55, 419–425. [Google Scholar] [CrossRef]
- Duan, J.; Kodali, V.K.; Gaffrey, M.J.; Guo, J.; Chu, R.K.; Camp, D.G.; Smith, R.D.; Thrall, B.D.; Qian, W.J. Quantitative Profiling of Protein S-Glutathionylation Reveals Redox-Dependent Regulation of Macrophage Function during Nanoparticle-Induced Oxidative Stress. ACS Nano 2016, 10, 524–538. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, P.; Zhang, C.; Chiewchengchol, D.; Zhao, F.; Yu, H.; Li, J.; Kambara, H.; Luo, K.Y.; Venkataraman, A.; et al. Positive Regulation of Interleukin-1beta Bioactivity by Physiological ROS-Mediated Cysteine S-Glutathionylation. Cell. Rep. 2017, 20, 224–235. [Google Scholar] [CrossRef]
- Ren, X.; Zou, L.; Zhang, X.; Branco, V.; Wang, J.; Carvalho, C.; Holmgren, A.; Lu, J. Redox Signaling Mediated by Thioredoxin and Glutathione Systems in the Central Nervous System. Antioxid. Redox Signal. 2017, 27, 989–1010. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.P.; Hofseth, L.J.; Harris, C.C. Radical causes of cancer. Nat. Rev. Cancer 2003, 3, 276–285. [Google Scholar] [CrossRef] [PubMed]
- Hatem, E.; El Banna, N.; Huang, M.E. Multifaceted Roles of Glutathione and Glutathione-Based Systems in Carcinogenesis and Anticancer Drug Resistance. Antioxid. Redox Signal. 2017, 27, 1217–1234. [Google Scholar] [CrossRef] [PubMed]
- Thyagarajan, A.; Sahu, R.P. Potential Contributions of Antioxidants to Cancer Therapy: Immunomodulation and Radiosensitization. Integr. Cancer 2018, 17, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA. Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, Y.; Yoshizato, T.; Shiraishi, Y.; Maekawa, S.; Okuno, Y.; Kamura, T.; Shimamura, T.; Sato-Otsubo, A.; Nagae, G.; Suzuki, H.; et al. Integrated molecular analysis of clear-cell renal cell carcinoma. Nat. Genet. 2013, 45, 860–867. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network; Linehan, W.M.; Spellman, P.T.; Ricketts, C.J.; Creighton, C.J.; Fei, S.S.; Davis, C.; Wheeler, D.A.; Murray, B.A.; Schmidt, L.; et al. Comprehensive Molecular Characterization of Papillary Renal-Cell Carcinoma. N. Engl. J. Med. 2016, 374, 135–145. [Google Scholar] [CrossRef]
- Davis, C.F.; Ricketts, C.J.; Wang, M.; Yang, L.; Cherniack, A.D.; Shen, H.; Buhay, C.; Kang, H.; Kim, S.C.; Fahey, C.C.; et al. The somatic genomic landscape of chromophobe renal cell carcinoma. Cancer Cell 2014, 26, 319–330. [Google Scholar] [CrossRef]
- Ricketts, C.J.; De Cubas, A.A.; Fan, H.; Smith, C.C.; Lang, M.; Reznik, E.; Bowlby, R.; Gibb, E.A.; Akbani, R.; Beroukhim, R.; et al. The Cancer Genome Atlas Comprehensive Molecular Characterization of Renal Cell Carcinoma. Cell. Rep. 2018, 23, 313–326 e5. [Google Scholar] [CrossRef]
- Hoadley, K.A.; Yau, C.; Hinoue, T.; Wolf, D.M.; Lazar, A.J.; Drill, E.; Shen, R.; Taylor, A.M.; Cherniack, A.D.; Thorsson, V.; et al. Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer. Cell 2018, 173, 291–304 e6. [Google Scholar] [CrossRef] [PubMed]
- Moch, H.; Cubilla, A.L.; Humphrey, P.A.; Reuter, V.E.; Ulbright, T.M. The 2016 WHO Classification of Tumours of the Urinary System and Male Genital Organs-Part A: Renal, Penile, and Testicular Tumours. Eur. Urol. 2016, 70, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Chin, A.I.; Lam, J.S.; Figlin, R.A.; Belldegrun, A.S. Surveillance strategies for renal cell carcinoma patients following nephrectomy. Rev. Urol. 2006, 8, 1–7. [Google Scholar] [PubMed]
- Motzer, R.J.; Bacik, J.; Mazumdar, M. Prognostic factors for survival of patients with stage IV renal cell carcinoma: Memorial sloan-kettering cancer center experience. Clin. Cancer Res. 2004, 10, 6302S–6303S. [Google Scholar] [CrossRef] [PubMed]
- Delahunt, B.; Srigley, J.R.; Montironi, R.; Egevad, L. Advances in renal neoplasia: Recommendations from the 2012 International Society of Urological Pathology Consensus Conference. Urology 2014, 83, 969–974. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 2013, 499, 43–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latif, F.; Tory, K.; Gnarra, J.; Yao, M.; Duh, F.M.; Orcutt, M.L.; Stackhouse, T.; Kuzmin, I.; Modi, W.; Geil, L.; et al. Identification of the von Hippel-Lindau disease tumor suppressor gene. Science 1993, 260, 1317–1320. [Google Scholar] [CrossRef]
- Jaakkola, P.; Mole, D.R.; Tian, Y.M.; Wilson, M.I.; Gielbert, J.; Gaskell, S.J.; von Kriegsheim, A.; Hebestreit, H.F.; Mukherji, M.; Schofield, C.J.; et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 2001, 292, 468–472. [Google Scholar] [CrossRef]
- Ivan, M.; Kondo, K.; Yang, H.; Kim, W.; Valiando, J.; Ohh, M.; Salic, A.; Asara, J.M.; Lane, W.S.; Kaelin, W.G., Jr. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: Implications for O2 sensing. Science 2001, 292, 464–468. [Google Scholar] [CrossRef]
- Gossage, L.; Eisen, T.; Maher, E.R. VHL, the story of a tumour suppressor gene. Nat. Rev. Cancer 2015, 15, 55–64. [Google Scholar] [CrossRef]
- Riazalhosseini, Y.; Lathrop, M. Precision medicine from the renal cancer genome. Nat. Rev. Nephrol. 2016, 12, 655–666. [Google Scholar] [CrossRef] [PubMed]
- Wettersten, H.I.; Aboud, O.A.; Lara, P.N., Jr.; Weiss, R.H. Metabolic reprogramming in clear cell renal cell carcinoma. Nat. Rev. Nephrol. 2017, 13, 410–419. [Google Scholar] [CrossRef] [PubMed]
- Posadas, E.M.; Limvorasak, S.; Figlin, R.A. Targeted therapies for renal cell carcinoma. Nat. Rev. Nephrol. 2017, 13, 496–511. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Qiu, B.; Lee, D.S.; Walton, Z.E.; Ochocki, J.D.; Mathew, L.K.; Mancuso, A.; Gade, T.P.; Keith, B.; Nissim, I.; et al. Fructose-1,6-bisphosphatase opposes renal carcinoma progression. Nature 2014, 513, 251–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, T.; Kouvonen, P.; Koh, C.C.; Gillet, L.C.; Wolski, W.E.; Rost, H.L.; Rosenberger, G.; Collins, B.C.; Blum, L.C.; Gillessen, S.; et al. Rapid mass spectrometric conversion of tissue biopsy samples into permanent quantitative digital proteome maps. Nat. Med. 2015, 21, 407–413. [Google Scholar] [CrossRef] [Green Version]
- Hakimi, A.A.; Reznik, E.; Lee, C.H.; Creighton, C.J.; Brannon, A.R.; Luna, A.; Aksoy, B.A.; Liu, E.M.; Shen, R.; Lee, W.; et al. An Integrated Metabolic Atlas of Clear Cell Renal Cell Carcinoma. Cancer Cell 2016, 29, 104–116. [Google Scholar] [CrossRef] [Green Version]
- Steffens, S.; Janssen, M.; Roos, F.C.; Becker, F.; Schumacher, S.; Seidel, C.; Wegener, G.; Thüroff, J.W.; Hofmann, R.; Stöckle, M.; et al. Incidence and long-term prognosis of papillary compared to clear cell renal cell carcinoma—A multicentre study. Eur. J. Cancer 2012, 48, 2347–2352. [Google Scholar] [CrossRef]
- Delahunt, B.; Eble, J.N. Papillary renal cell carcinoma: A clinicopathologic and immunohistochemical study of 105 tumors. Mod. Pathol. 1997, 10, 537–544. [Google Scholar]
- Pignot, G.; Elie, C.; Conquy, S.; Vieillefond, A.; Flam, T.; Zerbib, M.; Debre, B.; Amsellem-Ouazana, D. Survival analysis of 130 patients with papillary renal cell carcinoma: Prognostic utility of type 1 and type 2 subclassification. Urology 2007, 69, 230–235. [Google Scholar] [CrossRef]
- Pal, S.K.; Ali, S.M.; Yakirevich, E.; Geynisman, D.M.; Karam, J.A.; Elvin, J.A.; Frampton, G.M.; Huang, X.; Lin, D.I.; Rosenzweig, M.; et al. Characterization of Clinical Cases of Advanced Papillary Renal Cell Carcinoma via Comprehensive Genomic Profiling. Eur. Urol. 2018, 73, 71–78. [Google Scholar] [CrossRef]
- Fay, A.P.; Signoretti, S.; Choueiri, T.K. MET as a target in papillary renal cell carcinoma. Clin. Cancer Res. 2014, 20, 3361–3363. [Google Scholar] [CrossRef]
- Li, S.; Shuch, B.M.; Gerstein, M.B. Whole-genome analysis of papillary kidney cancer finds significant noncoding alterations. PLoS Genet. 2017, 13, e1006685. [Google Scholar] [CrossRef]
- Tomlinson, I.; Alam, N.; Rowan, A.; Barclay, E.; Jaeger, E.; Kelsell, D.; Leigh, I.; Gorman, P.; Lamlum, H.; Rahman, S. Multiple Leiomyoma Consortium: Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat. Genet. 2002, 30, 406–410. [Google Scholar]
- Pollard, P.J.; Briere, J.J.; Alam, N.A.; Barwell, J.; Barclay, E.; Wortham, N.C.; Hunt, T.; Mitchell, M.; Olpin, S.; Moat, S.J.; et al. Accumulation of Krebs cycle intermediates and over-expression of HIF1alpha in tumours which result from germline FH and SDH mutations. Hum. Mol. Genet. 2005, 14, 2231–2239. [Google Scholar] [CrossRef]
- Sullivan, L.B.; Martinez-Garcia, E.; Nguyen, H.; Mullen, A.R.; Dufour, E.; Sudarshan, S.; Licht, J.D.; Deberardinis, R.J.; Chandel, N.S. The proto-oncometabolite fumarate binds glutathione to amplify ROS-dependent signaling. Mol. Cell 2013, 51, 236–248. [Google Scholar] [CrossRef]
- Alahmad, A.; Paffrath, V.; Clima, R.; Busch, J.F.; Rabien, A.; Kilic, E.; Villegas, S.; Timmermann, B.; Attimonelli, M.; Jung, K.; et al. Papillary renal cell carcinomas rewire glutathione metabolism and are deficient in anabolic glucose synthesis. bioRxiv 2019, 651265. [Google Scholar] [Green Version]
- Aretz, I.; Hardt, C.; Wittig, I.; Meierhofer, D. An Impaired Respiratory Electron Chain Triggers Down-regulation of the Energy Metabolism and De-ubiquitination of Solute Carrier Amino Acid Transporters. Mol. Cell. Proteom. MCP 2016, 15, 1526–1538. [Google Scholar] [CrossRef] [Green Version]
- Hu, H.; Nan, J.; Sun, Y.; Zhu, D.; Xiao, C.; Wang, Y.; Zhu, L.; Wu, Y.; Zhao, J.; Wu, R.; et al. Electron leak from NDUFA13 within mitochondrial complex I attenuates ischemia-reperfusion injury via dimerized STAT3. Proc. Natl. Acad. Sci. USA 2017, 114, 11908–11913. [Google Scholar] [CrossRef] [Green Version]
- Jastroch, M.; Divakaruni, A.S.; Mookerjee, S.; Treberg, J.R.; Brand, M.D. Mitochondrial proton and electron leaks. Essays Biochem. 2010, 47, 53–67. [Google Scholar] [CrossRef] [Green Version]
- Hirst, J.; Roessler, M.M. Energy conversion, redox catalysis and generation of reactive oxygen species by respiratory complex I. Biochim. Biophys. Acta 2016, 1857, 872–883. [Google Scholar] [CrossRef]
- Reichart, G.; Mayer, J.; Zehm, C.; Kirschstein, T.; Tokay, T.; Lange, F.; Baltrusch, S.; Tiedge, M.; Fuellen, G.; Ibrahim, S.; et al. Mitochondrial complex IV mutation increases reactive oxygen species production and reduces lifespan in aged mice. Acta Physiol. (Oxf.) 2019, 225, e13214. [Google Scholar] [CrossRef]
- Thoenes, W.; Storkel, S.; Rumpelt, H.J. Human chromophobe cell renal carcinoma. Virchows Arch. B Cell Pathol. Incl. Mol. Pathol. 1985, 48, 207–217. [Google Scholar] [CrossRef]
- Yusenko, M.V. Molecular pathology of chromophobe renal cell carcinoma: A review. Int. J. Urol. 2010, 17, 592–600. [Google Scholar] [CrossRef]
- Haake, S.M.; Weyandt, J.D.; Rathmell, W.K. Insights into the Genetic Basis of the Renal Cell Carcinomas from The Cancer Genome Atlas. Mol. Cancer Res. 2016, 14, 589–598. [Google Scholar] [CrossRef]
- Brunelli, M.; Eble, J.N.; Zhang, S.; Martignoni, G.; Delahunt, B.; Cheng, L. Eosinophilic and classic chromophobe renal cell carcinomas have similar frequent losses of multiple chromosomes from among chromosomes 1, 2, 6, 10, and 17, and this pattern of genetic abnormality is not present in renal oncocytoma. Mod. Pathol. Off. J. United States Can. Acad. Pathol. Inc. 2005, 18, 161–169. [Google Scholar] [CrossRef]
- Casuscelli, J.; Weinhold, N.; Gundem, G.; Wang, L.; Zabor, E.C.; Drill, E.; Wang, P.I.; Nanjangud, G.J.; Redzematovic, A.; Nargund, A.M.; et al. Genomic landscape and evolution of metastatic chromophobe renal cell carcinoma. JCI Insight 2017, 2. [Google Scholar] [CrossRef]
- Xiao, Y.; Clima, R.; Busch, J.F.; Rabien, A.; Kilic, E.; Villegas, S.; Türkmen, S.; Timmermann, B.; Attimonelli, M.; Jung, K.; et al. Metabolic reprogramming and elevation of glutathione in chromophobe renal cell carcinomas. bioRxiv. 2019, 649046. [Google Scholar]
- Jinzaki, M.; Tanimoto, A.; Mukai, M.; Ikeda, E.; Kobayashi, S.; Yuasa, Y.; Narimatsu, Y.; Murai, M. Double-phase helical CT of small renal parenchymal neoplasms: Correlation with pathologic findings and tumor angiogenesis. J. Comput. Assist. Tomogr. 2000, 24, 835–842. [Google Scholar] [CrossRef]
- Nakajima, R.; Nozaki, S.; Kondo, T.; Nagashima, Y.; Abe, K.; Sakai, S. Evaluation of renal cell carcinoma histological subtype and fuhrman grade using (18)F-fluorodeoxyglucose-positron emission tomography/computed tomography. Eur. Radiol. 2017, 27, 4866–4873. [Google Scholar] [CrossRef]
- Priolo, C.; Khabibullin, D.; Reznik, E.; Filippakis, H.; Ogorek, B.; Kavanagh, T.R.; Nijmeh, J.; Herbert, Z.T.; Asara, J.M.; Kwiatkowski, D.J.; et al. Impairment of gamma-glutamyl transferase 1 activity in the metabolic pathogenesis of chromophobe renal cell carcinoma. Proc. Natl. Acad. Sci. USA 2018, 115, E6274–E6282. [Google Scholar] [CrossRef] [Green Version]
- Kurschner, G.; Zhang, Q.; Clima, R.; Xiao, Y.; Busch, J.F.; Kilic, E.; Jung, K.; Berndt, N.; Bulik, S.; Holzhutter, H.G.; et al. Renal oncocytoma characterized by the defective complex I of the respiratory chain boosts the synthesis of the ROS scavenger glutathione. Oncotarget 2017, 8, 105882–105904. [Google Scholar] [CrossRef]
- Gopal, R.K.; Calvo, S.E.; Shih, A.R.; Chaves, F.L.; McGuone, D.; Mick, E.; Pierce, K.A.; Li, Y.; Garofalo, A.; Van Allen, E.M.; et al. Early loss of mitochondrial complex I and rewiring of glutathione metabolism in renal oncocytoma. Proc. Natl. Acad. Sci. USA 2018, 115, E6283–E6290. [Google Scholar] [CrossRef] [Green Version]
- Orlowski, M.; Meister, A. The gamma-glutamyl cycle: A possible transport system for amino acids. Proc. Natl. Acad. Sci. USA 1970, 67, 1248–1255. [Google Scholar] [CrossRef]
- Franklin, C.C.; Backos, D.S.; Mohar, I.; White, C.C.; Forman, H.J.; Kavanagh, T.J. Structure, function, and post-translational regulation of the catalytic and modifier subunits of glutamate cysteine ligase. Mol. Asp. Med. 2009, 30, 86–98. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Zhang, Z.; Yuan, J.; Zhang, Y.; Jin, X. Altered glutamate cysteine ligase expression and activity in renal cell carcinoma. Biomed. Rep. 2014, 2, 831–834. [Google Scholar] [CrossRef] [Green Version]
- Miess, H.; Dankworth, B.; Gouw, A.M.; Rosenfeldt, M.; Schmitz, W.; Jiang, M.; Saunders, B.; Howell, M.; Downward, J.; Felsher, D.W.; et al. The glutathione redox system is essential to prevent ferroptosis caused by impaired lipid metabolism in clear cell renal cell carcinoma. Oncogene 2018, 37, 5435–5450. [Google Scholar] [CrossRef]
- Terzyan, S.S.; Burgett, A.W.; Heroux, A.; Smith, C.A.; Mooers, B.H.; Hanigan, M.H. Human Gamma-Glutamyl Transpeptidase 1: Structures Of The Free Enzyme, Inhibitor-Bound Tetrahedral Transition States, And Glutamate-Bound Enzyme Reveal Novel Movement Within The Active Site During Catalysis. J. Biol. Chem. 2015, 290, 17576–17586. [Google Scholar] [CrossRef]
- Simic, T.; Dragicevic, D.; Savic-Radojevic, A.; Cimbaljevic, S.; Tulic, C.; Mimic-Oka, J. Serum gamma glutamyl-transferase is a sensitive but unspecific marker of metastatic renal cell carcinoma. Int. J. Urol. 2007, 14, 289–293. [Google Scholar] [CrossRef]
- Hofbauer, S.L.; Stangl, K.I.; de Martino, M.; Lucca, I.; Haitel, A.; Shariat, S.F.; Klatte, T. Pretherapeutic gamma-glutamyltransferase is an independent prognostic factor for patients with renal cell carcinoma. Br. J. Cancer 2014, 111, 1526–1531. [Google Scholar] [CrossRef] [Green Version]
- Fu, Q.; Xu, L.; Wang, Y.; Jiang, Q.; Liu, Z.; Zhang, J.; Zhou, Q.; Zeng, H.; Tong, S.; Wang, T.; et al. Tumor-associated Macrophage-derived Interleukin-23 Interlinks Kidney Cancer Glutamine Addiction with Immune Evasion. Eur. Urol. 2019, 75, 752–763. [Google Scholar] [CrossRef]
- Abu Aboud, O.; Habib, S.L.; Trott, J.; Stewart, B.; Liang, S.; Chaudhari, A.J.; Sutcliffe, J.; Weiss, R.H. Glutamine Addiction in Kidney Cancer Suppresses Oxidative Stress and Can Be Exploited for Real-Time Imaging. Cancer Res. 2017, 77, 6746–6758. [Google Scholar] [CrossRef]
- Hoerner, C.R.; Chen, V.J.; Fan, A.C. The ‘Achilles Heel’ of Metabolism in Renal Cell Carcinoma: Glutaminase Inhibition as a Rational Treatment Strategy. Kidney Cancer 2019, 3, 15–29. [Google Scholar] [CrossRef]
- Broer, A.; Rahimi, F.; Broer, S. Deletion of Amino Acid Transporter ASCT2 (SLC1A5) Reveals an Essential Role for Transporters SNAT1 (SLC38A1) and SNAT2 (SLC38A2) to Sustain Glutaminolysis in Cancer Cells. J. Biol. Chem. 2016, 291, 13194–131205. [Google Scholar] [CrossRef]
- Bailey, S.T.; Smith, A.M.; Kardos, J.; Wobker, S.E.; Wilson, H.L.; Krishnan, B.; Saito, R.; Lee, H.J.; Zhang, J.; Eaton, S.C.; et al. MYC activation cooperates with Vhl and Ink4a/Arf loss to induce clear cell renal cell carcinoma. Nat. Commun. 2017, 8, 15770. [Google Scholar] [CrossRef] [Green Version]
- Tang, S.W.; Chang, W.H.; Su, Y.C.; Chen, Y.C.; Lai, Y.H.; Wu, P.T.; Hsu, C.I.; Lin, W.C.; Lai, M.K.; Lin, J.Y. MYC pathway is activated in clear cell renal cell carcinoma and essential for proliferation of clear cell renal cell carcinoma cells. Cancer Lett. 2009, 273, 35–43. [Google Scholar] [CrossRef]
- Son, J.; Lyssiotis, C.A.; Ying, H.; Wang, X.; Hua, S.; Ligorio, M.; Perera, R.M.; Ferrone, C.R.; Mullarky, E.; Shyh-Chang, N.; et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 2013, 496, 101. [Google Scholar] [CrossRef]
- Kredich, N.M. Biosynthesis of Cysteine. EcoSal Plus 2008, 3. [Google Scholar] [CrossRef]
- Bansal, A.; Simon, M.C. Glutathione metabolism in cancer progression and treatment resistance. J. Cell Biol. 2018, 217, 2291–2298. [Google Scholar] [CrossRef] [Green Version]
- Lucarelli, G.; Galleggiante, V.; Rutigliano, M.; Sanguedolce, F.; Cagiano, S.; Bufo, P.; Lastilla, G.; Maiorano, E.; Ribatti, D.; Giglio, A.; et al. Metabolomic profile of glycolysis and the pentose phosphate pathway identifies the central role of glucose-6-phosphate dehydrogenase in clear cell-renal cell carcinoma. Oncotarget 2015, 6, 13371–13386. [Google Scholar] [CrossRef]
- Wang, J.; Wu, Q.; Qiu, J. Accumulation of fructose 1,6-bisphosphate protects clear cell renal cell carcinoma from oxidative stress. Lab. Investig. 2019. [Google Scholar] [CrossRef]
- Mullen, A.R.; Wheaton, W.W.; Jin, E.S.; Chen, P.H.; Sullivan, L.B.; Cheng, T.; Yang, Y.; Linehan, W.M.; Chandel, N.S.; DeBerardinis, R.J. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature 2011, 481, 385–388. [Google Scholar] [CrossRef] [Green Version]
- Ooi, A.; Wong, J.C.; Petillo, D.; Roossien, D.; Perrier-Trudova, V.; Whitten, D.; Min, B.W.; Tan, M.H.; Zhang, Z.; Yang, X.J.; et al. An antioxidant response phenotype shared between hereditary and sporadic type 2 papillary renal cell carcinoma. Cancer Cell 2011, 20, 511–523. [Google Scholar] [CrossRef]
- Adam, J.; Hatipoglu, E.; O’Flaherty, L.; Ternette, N.; Sahgal, N.; Lockstone, H.; Baban, D.; Nye, E.; Stamp, G.W.; Wolhuter, K.; et al. Renal cyst formation in Fh1-deficient mice is independent of the Hif/Phd pathway: Roles for fumarate in KEAP1 succination and Nrf2 signaling. Cancer Cell 2011, 20, 524–537. [Google Scholar] [CrossRef]
- Jaramillo, M.C.; Zhang, D.D. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev. 2013, 27, 2179–2191. [Google Scholar] [CrossRef]
- Kansanen, E.; Kuosmanen, S.M.; Leinonen, H.; Levonen, A.L. The Keap1-Nrf2 pathway: Mechanisms of activation and dysregulation in cancer. Redox Biol. 2013, 1, 45–49. [Google Scholar] [CrossRef] [Green Version]
- Tong, K.I.; Katoh, Y.; Kusunoki, H.; Itoh, K.; Tanaka, T.; Yamamoto, M. Keap1 recruits Neh2 through binding to ETGE and DLG motifs: Characterization of the two-site molecular recognition model. Mol. Cell Biol. 2006, 26, 2887–2900. [Google Scholar] [CrossRef]
- Taguchi, K.; Yamamoto, M. The KEAP1-NRF2 System in Cancer. Front. Oncol. 2017, 7, 85. [Google Scholar] [CrossRef]
- Jin, L.; Li, D.; Alesi, G.N.; Fan, J.; Kang, H.B.; Lu, Z.; Boggon, T.J.; Jin, P.; Yi, H.; Wright, E.R.; et al. Glutamate dehydrogenase 1 signals through antioxidant glutathione peroxidase 1 to regulate redox homeostasis and tumor growth. Cancer Cell 2015, 27, 257–270. [Google Scholar] [CrossRef]
- Cairns, P. Renal cell carcinoma. Cancer Biomark 2010, 9, 461–473. [Google Scholar] [CrossRef]
- Shojaei, F.; Lee, J.H.; Simmons, B.H.; Wong, A.; Esparza, C.O.; Plumlee, P.A.; Feng, J.; Stewart, A.E.; Hu-Lowe, D.D.; Christensen, J.G. HGF/c-Met acts as an alternative angiogenic pathway in sunitinib-resistant tumors. Cancer Res. 2010, 70, 10090–10100. [Google Scholar] [CrossRef]
- Bielecka, Z.F.; Czarnecka, A.M.; Solarek, W.; Kornakiewicz, A.; Szczylik, C. Mechanisms of Acquired Resistance to Tyrosine Kinase Inhibitors in Clear - Cell Renal Cell Carcinoma (ccRCC). Curr. Signal. Transduct. 2014, 8, 218–228. [Google Scholar] [CrossRef]
- Harris, I.S.; Endress, J.E.; Coloff, J.L.; Selfors, L.M.; McBrayer, S.K.; Rosenbluth, J.M.; Takahashi, N.; Dhakal, S.; Koduri, V.; Oser, M.G.; et al. Deubiquitinases Maintain Protein Homeostasis and Survival of Cancer Cells upon Glutathione Depletion. Cell Metab. 2019, 29, 1166–1181. [Google Scholar] [CrossRef]
- Lewerenz, J.; Hewett, S.J.; Huang, Y.; Lambros, M.; Gout, P.W.; Kalivas, P.W.; Massie, A.; Smolders, I.; Methner, A.; Pergande, M. The cystine/glutamate antiporter system xc− in health and disease: From molecular mechanisms to novel therapeutic opportunities. Antioxid. Redox Signal. 2013, 18, 522–555. [Google Scholar] [CrossRef]
- Dixon, S.J.; Patel, D.N.; Welsch, M.; Skouta, R.; Lee, E.D.; Hayano, M.; Thomas, A.G.; Gleason, C.E.; Tatonetti, N.P.; Slusher, B.S.; et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. eLife 2014, 3, e02523. [Google Scholar] [CrossRef]
- Keating, G.M. Sorafenib: A Review in Hepatocellular Carcinoma. Target. Oncol. 2017, 12, 243–253. [Google Scholar] [CrossRef]
- Sehm, T.; Rauh, M.; Wiendieck, K.; Buchfelder, M.; Eyupoglu, I.Y.; Savaskan, N.E. Temozolomide toxicity operates in a xCT/SLC7a11 dependent manner and is fostered by ferroptosis. Oncotarget 2016, 7, 74630–74647. [Google Scholar] [CrossRef]
- Roh, J.L.; Kim, E.H.; Jang, H.; Shin, D. Aspirin plus sorafenib potentiates cisplatin cytotoxicity in resistant head and neck cancer cells through xCT inhibition. Free Radic. Biol. Med. 2017, 104, 1–9. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Yagoda, N.; von Rechenberg, M.; Zaganjor, E.; Bauer, A.J.; Yang, W.S.; Fridman, D.J.; Wolpaw, A.J.; Smukste, I.; Peltier, J.M.; Boniface, J.J.; et al. RAS–RAF–MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 2007, 447, 865. [Google Scholar] [CrossRef]
- Tang, X.; Wu, J.; Ding, C.K.; Lu, M.; Keenan, M.M.; Lin, C.C.; Lin, C.A.; Wang, C.C.; George, D.; Hsu, D.S.; et al. Cystine Deprivation Triggers Programmed Necrosis in VHL-Deficient Renal Cell Carcinomas. Cancer Res. 2016, 76, 1892–1903. [Google Scholar] [CrossRef]
- Momcilovic, M.; Bailey, S.T.; Lee, J.T.; Fishbein, M.C.; Magyar, C.; Braas, D.; Graeber, T.; Jackson, N.J.; Czernin, J.; Emberley, E.; et al. Targeted Inhibition of EGFR and Glutaminase Induces Metabolic Crisis in EGFR Mutant Lung Cancer. Cell Rep. 2017, 18, 601–610. [Google Scholar] [CrossRef]
- Emberley, E.; Bennett, M.; Chen, J.; Gross, M.; Huang, T.; Li, W.; Mackinnon, A.; Pan, A.; Rodriguez, M.; Steggerda, S.; et al. CB-839, a selective glutaminase inhibitor, has anti-tumor activity in renal cell carcinoma and synergizes with everolimus and receptor tyrosine kinase inhibitors. Eur. J. Cancer 2016, 69, S124. [Google Scholar] [CrossRef]
- MacKinnon, A.L.; Bennett, M.; Gross, M.; Janes, J.; Li, W.Q.; Rodriquez, M.; Wang, T.; Zhang, W.; Parlati, F. Metabolomic, Proteomic and Genomic Profiling Identifies Biomarakers of Sensitivity to Glutaminase Inhibitor CB-839 in Multiple Myeloma. Blood 2015, 126, 1802. [Google Scholar]
- Meric-Bernstam, F.; Tannir, N.M.; Mier, J.W.; DeMichele, A.; Telli, M.L.; Fan, A.C.; Munster, P.N.; Carvajal, R.D.; Orford, K.W.; Bennett, M.K.; et al. Phase 1 study of CB-839, a small molecule inhibitor of glutaminase (GLS), alone and in combination with everolimus (E) in patients (pts) with renal cell cancer (RCC). J. Clin. Oncol. 2016, 34, 4568. [Google Scholar] [CrossRef]
- Okazaki, A.; Gameiro, P.A.; Christodoulou, D.; Laviollette, L.; Schneider, M.; Chaves, F.; Stemmer-Rachamimov, A.; Yazinski, S.A.; Lee, R.; Stephanopoulos, G.; et al. Glutaminase and poly(ADP-ribose) polymerase inhibitors suppress pyrimidine synthesis and VHL-deficient renal cancers. J. Clin. Investig. 2017, 127, 1631–1645. [Google Scholar] [CrossRef] [Green Version]
- Song, M.; Kim, S.H.; Im, C.Y.; Hwang, H.J. Recent Development of Small Molecule Glutaminase Inhibitors. Curr. Top. Med. Chem. 2018, 18, 432–443. [Google Scholar] [CrossRef]
- Bailey, H.H. l-S,R-buthionine sulfoximine: Historical development and clinical issues. Chem. Biol. Interact. 1998, 111–112, 239–254. [Google Scholar] [CrossRef]
- Griffith, O.W.; Meister, A. Potent and specific inhibition of glutathione synthesis by buthionine sulfoximine (Sn-butyl homocysteine sulfoximine). J. Biol. Chem. 1979, 254, 7558–7560. [Google Scholar]
- Tagde, A.; Singh, H.; Kang, M.H.; Reynolds, C.P. The glutathione synthesis inhibitor buthionine sulfoximine synergistically enhanced melphalan activity against preclinical models of multiple myeloma. Blood Cancer J. 2014, 4, e229. [Google Scholar] [CrossRef]
- Anderson, C.P.; Matthay, K.K.; Perentesis, J.P.; Neglia, J.P.; Bailey, H.H.; Villablanca, J.G.; Groshen, S.; Hasenauer, B.; Maris, J.M.; Seeger, R.C.; et al. Pilot study of intravenous melphalan combined with continuous infusion l-S,R-buthionine sulfoximine for children with recurrent neuroblastoma. Pediatr. Blood Cancer 2015, 62, 1739–1746. [Google Scholar] [CrossRef]
- Villablanca, J.G.; Volchenboum, S.L.; Cho, H.; Kang, M.H.; Cohn, S.L.; Anderson, C.P.; Marachelian, A.; Groshen, S.; Tsao-Wei, D.; Matthay, K.K.; et al. A Phase I New Approaches to Neuroblastoma Therapy Study of Buthionine Sulfoximine and Melphalan With Autologous Stem Cells for Recurrent/Refractory High-Risk Neuroblastoma. Pediatr. Blood Cancer 2016, 63, 1349–1356. [Google Scholar] [CrossRef]
- Zhang, L.; Bhasin, M.; Schor-Bardach, R.; Wang, X.; Collins, M.P.; Panka, D.; Putheti, P.; Signoretti, S.; Alsop, D.C.; Libermann, T.; et al. Resistance of renal cell carcinoma to sorafenib is mediated by potentially reversible gene expression. PLoS ONE 2011, 6, e19144. [Google Scholar] [CrossRef]
- Panka, D.; Kumar, M.; Schor-Bardach, R.; Zhang, L.; Atkins, M.; Libermann, T.; Goldberg, N.; Bhatt, R.; Mier, J. Mechanism of acquired resistance to sorafenib in RCC. Cancer Res. 2008, 68, 2500. [Google Scholar]
- Nikinmaa, M.; Pursiheimo, S.; Soitamo, A.J. Redox state regulates HIF-1alpha and its DNA binding and phosphorylation in salmonid cells. J. Cell Sci. 2004, 117, 3201–3206. [Google Scholar] [CrossRef]
- Chen, H.; Shi, H. A reducing environment stabilizes HIF-2alpha in SH-SY5Y cells under hypoxic conditions. FEBS Lett. 2008, 582, 3899–3902. [Google Scholar] [CrossRef]
- Jin, W.-s.; Kong, Z.-l.; Shen, Z.-f.; Jin, Y.-z.; Zhang, W.-k.; Chen, G.-f. Regulation of hypoxia inducible factor-1α expression by the alteration of redox status in HepG2 cells. J. Exp. Clin. Cancer Res. CR 2011, 30, 61. [Google Scholar] [CrossRef]
- Fyfe, G.; Fisher, R.I.; Rosenberg, S.A.; Sznol, M.; Parkinson, D.R.; Louie, A.C. Results of treatment of 255 patients with metastatic renal cell carcinoma who received high-dose recombinant interleukin-2 therapy. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 1995, 13, 688–696. [Google Scholar] [CrossRef]
- Negrier, S.; Escudier, B.; Lasset, C.; Douillard, J.Y.; Savary, J.; Chevreau, C.; Ravaud, A.; Mercatello, A.; Peny, J.; Mousseau, M.; et al. Recombinant human interleukin-2, recombinant human interferon alfa-2a, or both in metastatic renal-cell carcinoma. Groupe Francais d’Immunotherapie. N. Engl. J. Med. 1998, 338, 1272–1278. [Google Scholar] [CrossRef]
- Alsharedi, M.; Katz, H. Check point inhibitors a new era in renal cell carcinoma treatment. Med. Oncol. 2018, 35, 85. [Google Scholar] [CrossRef]
- Motzer, R.J.; Escudier, B.; McDermott, D.F.; George, S.; Hammers, H.J.; Srinivas, S.; Tykodi, S.S.; Sosman, J.A.; Procopio, G.; Plimack, E.R.; et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2015, 373, 1803–1813. [Google Scholar] [CrossRef]
- McDermott, D.F.; Atkins, M.B. PD-1 as a potential target in cancer therapy. Cancer Med. 2013, 2, 662–673. [Google Scholar] [CrossRef]
- Siska, P.J.; Kim, B.; Ji, X.; Hoeksema, M.D.; Massion, P.P.; Beckermann, K.E.; Wu, J.; Chi, J.T.; Hong, J.; Rathmell, J.C. Fluorescence-based measurement of cystine uptake through xCT shows requirement for ROS detoxification in activated lymphocytes. J. Immunol. Methods 2016, 438, 51–58. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Zhu, B. Editorial: Metabolism of Cancer Cells and Immune Cells in the Tumor Microenvironment. Front. Immunol. 2018, 9, 3080. [Google Scholar] [CrossRef] [Green Version]
- Nakaya, M.; Xiao, Y.; Zhou, X.; Chang, J.H.; Chang, M.; Cheng, X.; Blonska, M.; Lin, X.; Sun, S.C. Inflammatory T cell responses rely on amino acid transporter ASCT2 facilitation of glutamine uptake and mTORC1 kinase activation. Immunity 2014, 40, 692–705. [Google Scholar] [CrossRef]
- Klysz, D.; Tai, X.; Robert, P.A.; Craveiro, M.; Cretenet, G.; Oburoglu, L.; Mongellaz, C.; Floess, S.; Fritz, V.; Matias, M.I. Glutamine-dependent α-ketoglutarate production regulates the balance between T helper 1 cell and regulatory T cell generation. Sci. Signal. 2015, 8, ra97. [Google Scholar] [CrossRef]
- Crawford, J.; Cohen, H.J. The essential role of L-glutamine in lymphocyte differentiation in vitro. J. Cell. Physiol. 1985, 124, 275–282. [Google Scholar] [CrossRef]
- Fraternale, A.; Brundu, S.; Magnani, M. Glutathione and glutathione derivatives in immunotherapy. Biol. Chem. 2017, 398, 261–275. [Google Scholar] [CrossRef]
- Gamcsik, M.P.; Kasibhatla, M.S.; Teeter, S.D.; Colvin, O.M. Glutathione levels in human tumors. Biomarkers 2012, 17, 671–691. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| RCC Subtypes | Clear Cell | Papillary | Chromophobe |
|---|---|---|---|
| Incidence | 75% | 15% | 5% |
| Main mutations | VHL | MET, FH | TP53, PTEN |
| Metabolites | GSH, GSSG increased | GSH, GSSG increased | GSH, GSSG increased |
| GSH regulation | 1. GCL protein abundance increases; 2. Increased serum GGT as a marker for metastatic ccRCC; 3. GLS1, glutamine importers, and cysteine antiporter xCT enhance to favor GSH synthesis; 4. Increased PPP flux to produce NADPH for GSH conversion. | 1. FH mutation causes HIF stabilization; 2. FH mutation activates NRF2–ARE pathway, leading to increased GSH synthesis and enhanced expression of antioxidant proteins. | Loss of GGT1 increases sensitivity to oxidative stress in chRCC cells. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiao, Y.; Meierhofer, D. Glutathione Metabolism in Renal Cell Carcinoma Progression and Implications for Therapies. Int. J. Mol. Sci. 2019, 20, 3672. https://doi.org/10.3390/ijms20153672
Xiao Y, Meierhofer D. Glutathione Metabolism in Renal Cell Carcinoma Progression and Implications for Therapies. International Journal of Molecular Sciences. 2019; 20(15):3672. https://doi.org/10.3390/ijms20153672
Chicago/Turabian StyleXiao, Yi, and David Meierhofer. 2019. "Glutathione Metabolism in Renal Cell Carcinoma Progression and Implications for Therapies" International Journal of Molecular Sciences 20, no. 15: 3672. https://doi.org/10.3390/ijms20153672
APA StyleXiao, Y., & Meierhofer, D. (2019). Glutathione Metabolism in Renal Cell Carcinoma Progression and Implications for Therapies. International Journal of Molecular Sciences, 20(15), 3672. https://doi.org/10.3390/ijms20153672