Peroxisomal Hydrogen Peroxide Metabolism and Signaling in Health and Disease



Abstract
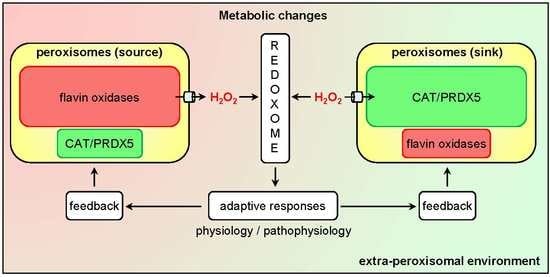
:
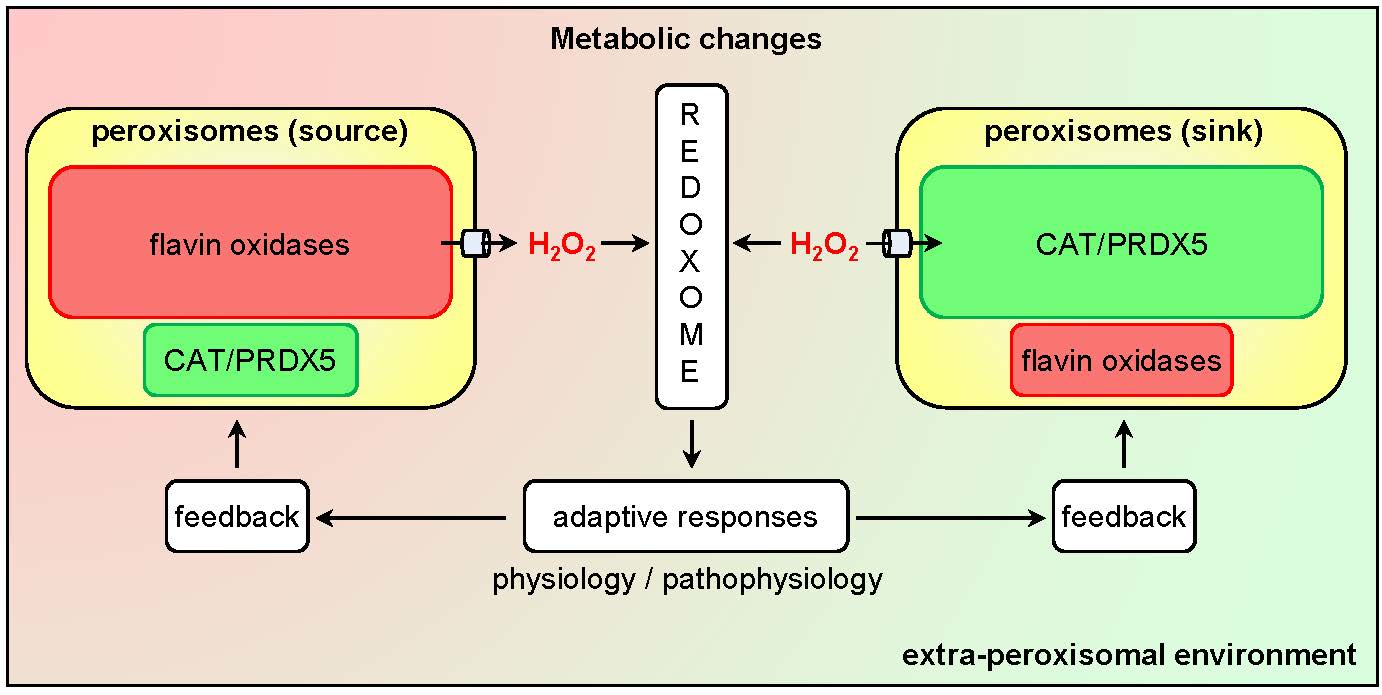{kind=link}
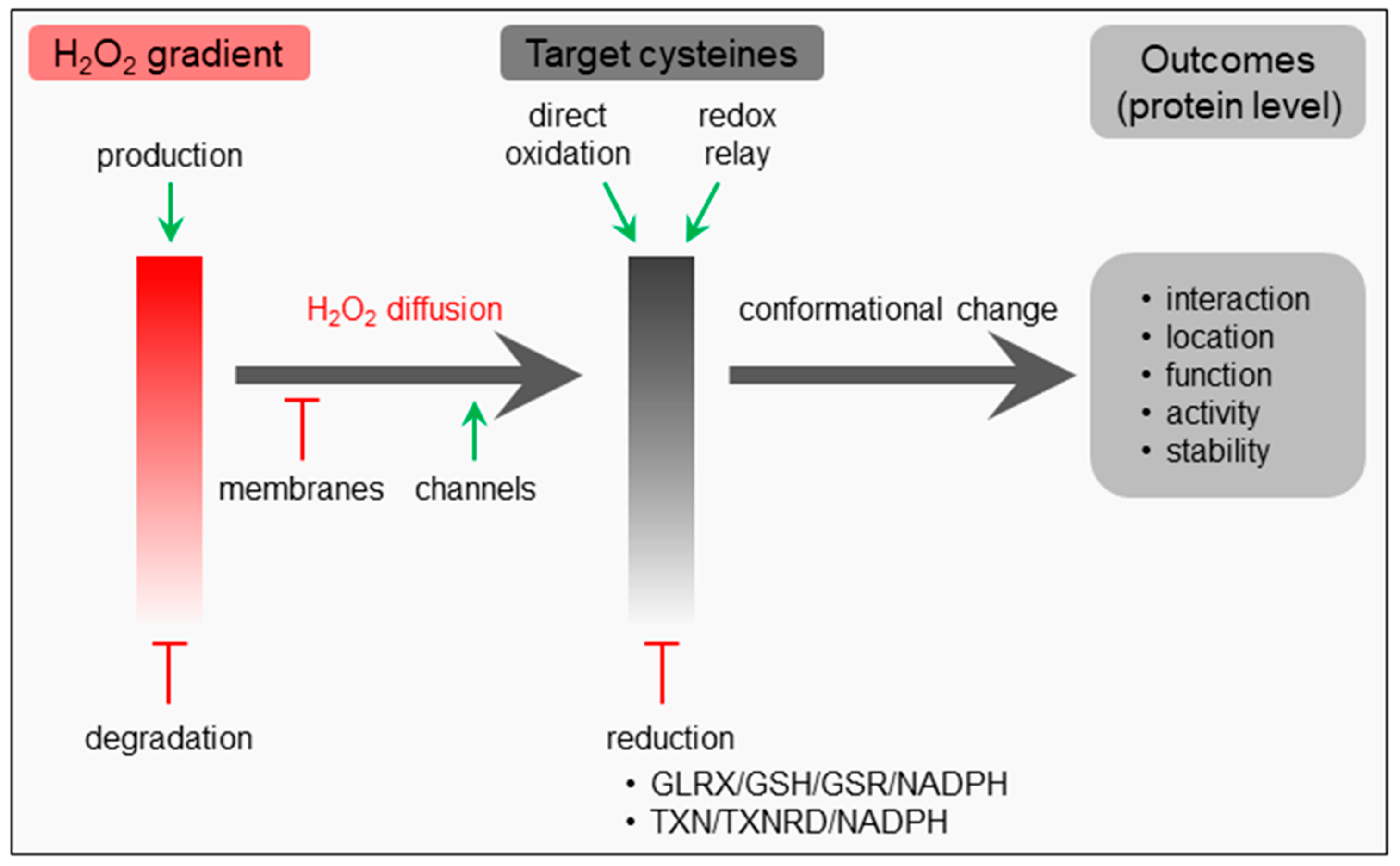{kind=link}
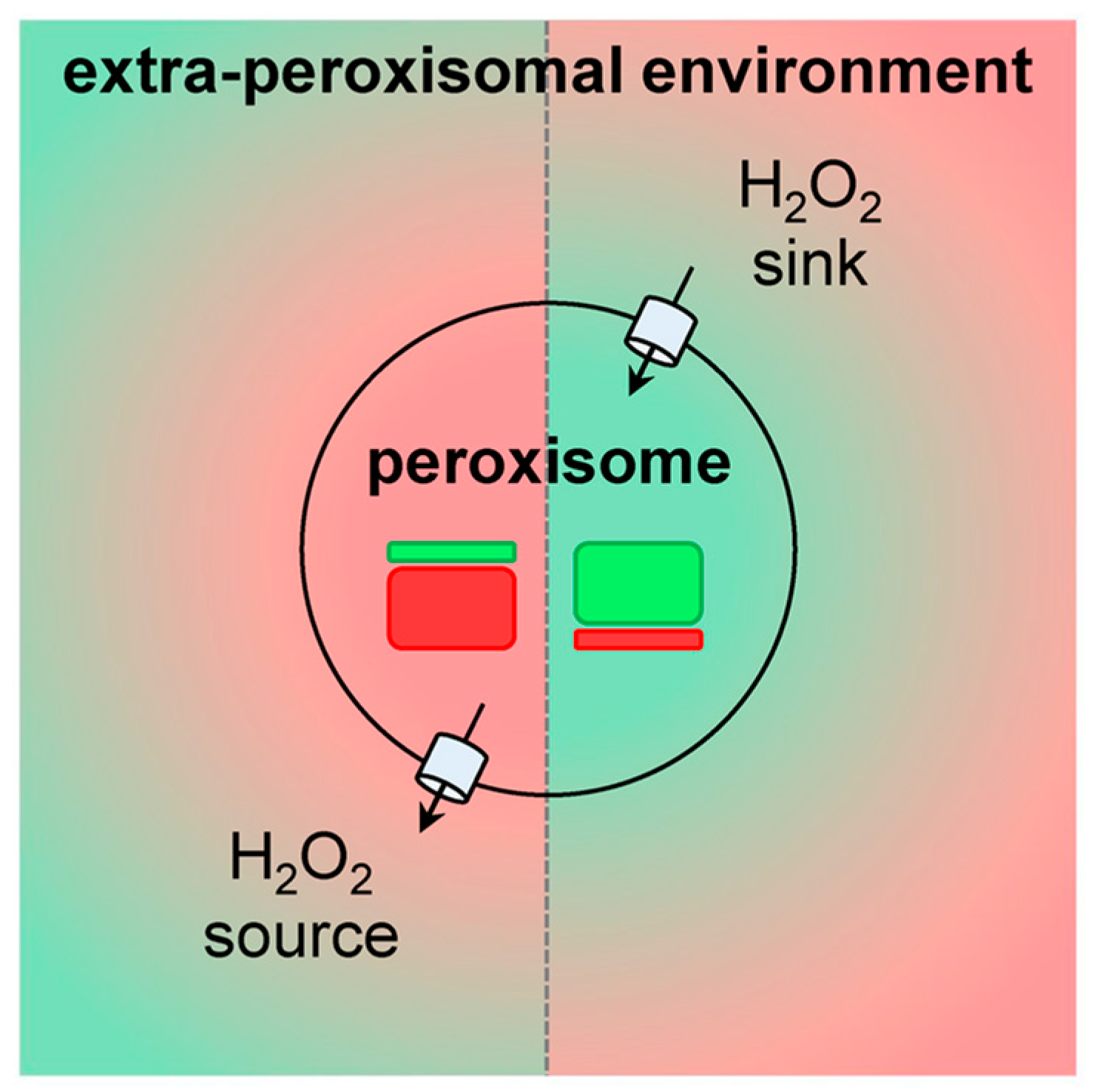{kind=link}
{kind=link}
1. Introduction
2. The Basic Concepts of H2O2 Signaling
3. Players in Peroxisomal H2O2 Metabolism and Transport
3.1. H2O2-Generating Systems
3.2. H2O2-Elimination Systems
3.3. H2O2 Permeation across the Peroxisomal Membrane
4. The Emerging Roles of Peroxisomes in Cellular H2O2 Signaling
4.1. Molecular Targets
4.2. Biological Processes
4.2.1. Gene Expression
4.2.2. Cell Fate Regulation
4.2.3. Mitochondrial Function
4.3. Diseases
4.3.1. Heart Disease
4.3.2. Kidney Disease
4.3.3. Insulin Resistance and Diabetes
4.3.4. Cardiovascular Disease
4.3.5. Cancer
4.3.6. Neurodegenerative Disease
5. Conclusions, Challenges, and Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACOX | Acyl-CoA oxidase |
| Acyl-CoA | Acyl-coenzyme A |
| ABL | Abelson tyrosine-protein kinase |
| CAT | Catalase |
| ERO | Endoplasmic reticulum oxidoreductase |
| FAD | Flavin adenine dinucleotide |
| FMN | Flavin mononucleotide |
| GLRX | Glutaredoxin |
| GPX | Glutathione peroxidase |
| GSH | Glutathione, reduced |
| GSK-3β | Glycogen synthase kinase 3β |
| GSSG | Glutathione, oxidized |
| GSR | Glutathione-disulfide reductase |
| KEAP1 | Kelch-like ECH-associated protein 1 |
| NAD(P)+ | Nicotinamide adenine dinucleotide (phosphate), oxidized |
| NAD(P)H | Nicotinamide adenine dinucleotide (phosphate), reduced |
| NFE2L2 | Nuclear factor erythroid 2-related factor 2 |
| NF-κB | Nuclear factor kappa B |
| NOX | NADPH oxidase |
| PEX | Peroxin |
| PMP | Peroxisomal membrane protein |
| PRDX | Peroxiredoxin |
| RNS | Reactive nitrogen species |
| ROS | Reactive oxygen species |
| SIRT | Sirtuin |
| SOD | Superoxide dismutase |
| TXN | Thioredoxin |
| TXNRD | Thioredoxin reductase |
References
- Sies, H. Role of metabolic H2O2 generation: Redox signaling and oxidative stress. J. Biol. Chem. 2014, 289, 8735–8741. [Google Scholar] [CrossRef] [PubMed]
- Lennicke, C.; Rahn, J.; Lichtenfels, R.; Wessjohann, L.A.; Seliger, B. Hydrogen peroxide—Production, fate and role in redox signaling of tumor cells. Cell Commun. Signal. 2015, 13, 39. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: Oxidative eustress. Redox Biol. 2017, 11, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Auten, R.L.; Davis, J.M. Oxygen toxicity and reactive oxygen species: The devil is in the details. Pediatr. Res. 2009, 66, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B. Reactive species and antioxidants. Redox biology is a fundamental theme of aerobic life. Plant Physiol. 2006, 141, 312–322. [Google Scholar] [CrossRef] [PubMed]
- Veal, E.; Day, A. Hydrogen peroxide as a signaling molecule. Antioxid. Redox Signal. 2011, 15, 147–151. [Google Scholar] [CrossRef] [PubMed]
- Zito, E. ERO1: A protein disulfide oxidase and H2O2 producer. Free Radic. Biol. Med. 2015, 83, 299–304. [Google Scholar] [CrossRef]
- Wang, Y.; Branicky, R.; Noë, A.; Hekimi, S. Superoxide dismutases: Dual roles in controlling ROS damage and regulating ROS signaling. J. Cell Biol. 2018, 217, 1915–1928. [Google Scholar] [CrossRef]
- Wong, H.S.; Dighe, P.A.; Mezera, V.; Monternier, P.A.; Brand, M.D. Production of superoxide and hydrogen peroxide from specific mitochondrial sites under different bioenergetic conditions. J. Biol. Chem. 2017, 292, 16804–16809. [Google Scholar] [CrossRef] [Green Version]
- Parascandolo, A.; Laukkanen, M.O. Carcinogenesis and reactive oxygen species signaling: Interaction of the NADPH oxidase NOX1-5 and superoxide dismutase 1-3 signal transduction pathways. Antioxid. Redox Signal. 2019, 30, 443–486. [Google Scholar] [CrossRef]
- Antunes, F.; Brito, P.M. Quantitative biology of hydrogen peroxide signaling. Redox Biol. 2017, 13, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Benhar, M. Roles of mammalian glutathione peroxidase and thioredoxin reductase enzymes in the cellular response to nitrosative stress. Free Radic. Biol. Med. 2018, 127, 160–164. [Google Scholar] [CrossRef] [PubMed]
- Detienne, G.; De Haes, W.; Mergan, L.; Edwards, S.L.; Temmerman, L.; Van Bael, S. Beyond ROS clearance: Peroxiredoxins in stress signaling and aging. Ageing Res. Rev. 2018, 44, 33–48. [Google Scholar] [CrossRef] [PubMed]
- Wanders, R.J.; Waterham, H.R. Biochemistry of mammalian peroxisomes revisited. Annu. Rev. Biochem. 2006, 75, 295–332. [Google Scholar] [CrossRef] [PubMed]
- Van Veldhoven, P.P. Biochemistry and genetics of inherited disorders of peroxisomal fatty acid metabolism. J. Lipid Res. 2010, 51, 2863–2895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fransen, M.; Nordgren, M.; Wang, B.; Apanasets, O. Role of peroxisomes in ROS/RNS-metabolism: Implications for human disease. Biochim. Biophys. Acta 2012, 1822, 1363–1373. [Google Scholar] [CrossRef] [PubMed]
- Wanders, R.J.A. Peroxisomal disorders: Improved laboratory diagnosis, new defects and the complicated route to treatment. Mol. Cell. Probes 2018, 40, 60–69. [Google Scholar] [CrossRef]
- Wang, B.; Apanasets, O.; Nordgren, M.; Fransen, M. Dissecting peroxisome-mediated signaling pathways: A new and exciting research field. In Molecular Machines Involved in Peroxisome Biogenesis and Maintenance, 1st ed.; Brocard, C., Hartig, A., Eds.; Springer: Wien, Austria, 2014; pp. 255–273. [Google Scholar] [CrossRef]
- Fransen, M. Peroxisome dynamics: Molecular players, mechanisms, and (dys)functions. ISRN Cell Biol. 2012, 2012, 714192. [Google Scholar] [CrossRef]
- Shai, N.; Schuldiner, M.; Zalckvar, E. No peroxisome is an island—Peroxisome contact sites. Biochim. Biophys. Acta 2016, 1863, 1061–1069. [Google Scholar] [CrossRef]
- Distel, B.; Erdmann, R.; Gould, S.J.; Blobel, G.; Crane, D.I.; Cregg, J.M.; Dodt, G.; Fujiki, Y.; Goodman, J.M.; Just, W.W.; et al. A unified nomenclature for peroxisome biogenesis factors. J. Cell Biol. 1996, 135, 1–3. [Google Scholar] [CrossRef]
- Nordgren, M.; Wang, B.; Apanasets, O.; Fransen, M. Peroxisome degradation in mammals: Mechanisms of action, recent advances, and perspectives. Front. Physiol. 2013, 4, 145. [Google Scholar] [CrossRef]
- Waterham, H.R.; Ferdinandusse, S.; Wanders, R.J. Human disorders of peroxisome metabolism and biogenesis. Biochim. Biophys. Acta 2016, 1863, 922–933. [Google Scholar] [CrossRef]
- Wong, C.P.; Xu, Z.; Power, C.; Hobman, T.C. Targeted elimination of peroxisomes during viral infection: Lessons from HIV and other viruses. DNA Cell Biol. 2018, 37, 417–421. [Google Scholar] [CrossRef]
- Fransen, M.; Nordgren, M.; Wang, B.; Apanasets, O.; Van Veldhoven, P.P. Aging, age-related diseases and peroxisomes. Subcell. Biochem. 2013, 69, 45–65. [Google Scholar] [CrossRef]
- Cipolla, C.M.; Lodhi, I.J. Peroxisomal dysfunction in age-related diseases. Trends Endocrinol. Metab. 2017, 28, 297–308. [Google Scholar] [CrossRef]
- Berridge, M.J. Signaling defects and disease. Cell Signal. Biol. 2014, 12.1–12.66. [Google Scholar] [CrossRef]
- D’Autréaux, B.; Toledano, M.B. ROS as signalling molecules: Mechanisms that generate specificity in ROS homeostasis. Nat. Rev. Mol. Cell Biol. 2007, 8, 813–824. [Google Scholar] [CrossRef]
- Bienert, G.P.; Chaumont, F. Aquaporin-facilitated transmembrane diffusion of hydrogen peroxide. Biochim. Biophys. Acta 2014, 1840, 1596–1604. [Google Scholar] [CrossRef]
- Poole, L.B. The basics of thiols and cysteines in redox biology and chemistry. Free Radic. Biol. Med. 2015, 80, 148–157. [Google Scholar] [CrossRef]
- Hanschmann, E.M.; Godoy, J.R.; Berndt, C.; Hudemann, C.; Lillig, C.H. Thioredoxins, glutaredoxins, and peroxiredoxins—Molecular mechanisms and health significance: From cofactors to antioxidants to redox signaling. Antioxid. Redox Signal. 2013, 19, 1539–1605. [Google Scholar] [CrossRef]
- Xiao, Z.; La Fontaine, S.; Bush, A.I.; Wedd, A.G. Molecular mechanisms of glutaredoxin enzymes: Versatile hubs for thiol-disulfide exchange between protein thiols and glutathione. J. Mol. Biol. 2019, 431, 158–177. [Google Scholar] [CrossRef]
- Netto, L.E.; Antunes, F. The roles of peroxiredoxin and thioredoxin in hydrogen peroxide sensing and in signal transduction. Mol. Cells 2016, 39, 65–71. [Google Scholar] [CrossRef]
- Stöcker, S.; Van Laer, K.; Mijuskovic, A.; Dick, T.P. The conundrum of hydrogen peroxide signaling and the emerging role of peroxiredoxins as redox relay hubs. Antioxid. Redox Signal. 2018, 28, 558–573. [Google Scholar] [CrossRef]
- Zou, Y.; Wang, A.; Shi, M.; Chen, X.; Liu, R.; Li, T.; Zhang, C.; Zhang, Z.; Zhu, L.; Ju, Z.; et al. Analysis of redox landscapes and dynamics in living cells and in vivo using genetically encoded fluorescent sensors. Nat. Protoc. 2018, 13, 2362–2386. [Google Scholar] [CrossRef]
- Marinho, H.S.; Real, C.; Cyrne, L.; Soares, H.; Antunes, F. Hydrogen peroxide sensing, signaling and regulation of transcription factors. Redox Biol. 2014, 2, 535–562. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The KEAP1-NRF2 system: A thiol-based sensor-effector apparatus for maintaining redox homeostasis. Physiol. Rev. 2018, 98, 1169–1203. [Google Scholar] [CrossRef]
- Deshmukh, P.; Unni, S.; Krishnappa, G.; Padmanabhan, B. The Keap1-Nrf2 pathway: Promising therapeutic target to counteract ROS-mediated damage in cancers and neurodegenerative diseases. Biophys. Rev. 2017, 9, 41–56. [Google Scholar] [CrossRef]
- Beurel, E.; Grieco, S.F.; Jope, R.S. Glycogen synthase kinase-3 (GSK3): Regulation, actions, and diseases. Pharmacol. Ther. 2015, 148, 114–131. [Google Scholar] [CrossRef]
- Li, W.; Thakor, N.; Xu, E.Y.; Huang, Y.; Chen, C.; Yu, R.; Holcik, M.; Kong, A.N. An internal ribosomal entry site mediates redox-sensitive translation of Nrf2. Nucleic Acids Res. 2010, 38, 778–788. [Google Scholar] [CrossRef]
- Purdom-Dickinson, S.E.; Sheveleva, E.V.; Sun, H.; Chen, Q.M. Translational control of nrf2 protein in activation of antioxidant response by oxidants. Mol. Pharmacol. 2007, 72, 1074–1081. [Google Scholar] [CrossRef]
- Sun, Z.; Chin, Y.E.; Zhang, D.D. Acetylation of Nrf2 by p300/CBP augments promoter-specific DNA binding of Nrf2 during the antioxidant response. Mol Cell. Biol. 2009, 29, 2658–2672. [Google Scholar] [CrossRef]
- Kawai, Y.; Garduño, L.; Theodore, M.; Yang, J.; Arinze, I.J. Acetylation-deacetylation of the transcription factor Nrf2 (nuclear factor erythroid 2-related factor 2) regulates its transcriptional activity and nucleocytoplasmic localization. J. Biol. Chem. 2011, 286, 7629–7640. [Google Scholar] [CrossRef]
- Fransen, M.; Lismont, C. Peroxisomes and cellular oxidant/antioxidant balance: Protein redox modifications and impact on inter-organelle communication. Subcell. Biochem. 2018, 89, 435–461. [Google Scholar] [CrossRef]
- Wanders, R.J. Metabolic functions of peroxisomes in health and disease. Biochimie 2014, 98, 36–44. [Google Scholar] [CrossRef]
- Van Veldhoven, P.P.; Van Rompuy, P.; Fransen, M.; De Béthune, B.; Mannaerts, G.P. Large-scale purification and further characterization of rat pristanoyl-CoA oxidase. Eur. J. Biochem. 1994, 222, 795–801. [Google Scholar] [CrossRef]
- Van Veldhoven, P.P.; Van Rompuy, P.; Vanhooren, J.C.; Mannaerts, G.P. Purification and further characterization of peroxisomal trihydroxycoprostanoyl-CoA oxidase from rat liver. Biochem. J. 1994, 304, 195–200. [Google Scholar] [CrossRef] [Green Version]
- Lismont, C.; Nordgren, M.; Van Veldhoven, P.P.; Fransen, M. Redox interplay between mitochondria and peroxisomes. Front. Cell. Dev. Biol. 2015, 3, 35. [Google Scholar] [CrossRef]
- Pollegioni, L.; Piubelli, L.; Sacchi, S.; Pilone, M.S.; Molla, G. Physiological functions of D-amino acid oxidases: From yeast to humans. Cell. Mol. Life Sci. 2007, 64, 1373–1394. [Google Scholar] [CrossRef]
- Wu, T.; Yankovskaya, V.; McIntire, W.S. Cloning, sequencing, and heterologous expression of the murine peroxisomal flavoprotein, N1-acetylated polyamine oxidase. J. Biol. Chem. 2003, 278, 20514–20525. [Google Scholar] [CrossRef]
- Jones, J.M.; Morrell, J.C.; Gould, S.J. Identification and characterization of HAOX1, HAOX2, and HAOX3, three human peroxisomal 2-hydroxy acid oxidases. J. Biol. Chem. 2000, 275, 12590–12597. [Google Scholar] [CrossRef]
- Peduto, A.; Baumgartner, M.R.; Verhoeven, N.M.; Rabier, D.; Spada, M.; Nassogne, M.C.; Poll-The, B.T.; Bonetti, G.; Jakobs, C.; Saudubray, J.M. Hyperpipecolic acidaemia: A diagnostic tool for peroxisomal disorders. Mol. Genet. Metab. 2004, 82, 224–230. [Google Scholar] [CrossRef]
- Hornbeck, P.V.; Zhang, B.; Murray, B.; Kornhauser, J.M.; Latham, V.; Skrzypek, E. PhosphoSitePlus, 2014: Mutations, PTMs and recalibrations. Nucleic Acids Res. 2015, 43, D512–D520. [Google Scholar] [CrossRef]
- Chen, X.F.; Tian, M.X.; Sun, R.Q.; Zhang, M.L.; Zhou, L.S.; Jin, L.; Chen, L.L.; Zhou, W.J.; Duan, K.L.; Chen, Y.J.; et al. SIRT5 inhibits peroxisomal ACOX1 to prevent oxidative damage and is downregulated in liver cancer. EMBO Rep. 2018, 19, e45124. [Google Scholar] [CrossRef]
- Kirkman, H.N.; Gaetani, G.F. Mammalian catalase: A venerable enzyme with new mysteries. Trends Biochem. Sci. 2007, 32, 44–50. [Google Scholar] [CrossRef]
- Das, K.; Roychoudhury, A. Reactive oxygen species (ROS) and response of antioxidants as ROS-scavengers during environmental stress in plants. Front. Environ. Sci. 2014, 2, 53. [Google Scholar] [CrossRef]
- Yamamoto, K.; Völkl, A.; Fahimi, H.D. Investigation of peroxisomal lipid beta-oxidation enzymes in guinea pig liver peroxisomes by immunoblotting and immunocytochemistry. J. Histochem. Cytochem. 1992, 40, 1909–1918. [Google Scholar] [CrossRef]
- Legakis, J.E.; Koepke, J.I.; Jedeszko, C.; Barlaskar, F.; Terlecky, L.J.; Edwards, H.J.; Walton, P.A.; Terlecky, S.R. Peroxisome senescence in human fibroblasts. Mol. Biol. Cell 2002, 13, 4243–4255. [Google Scholar] [CrossRef]
- Walton, P.A.; Brees, C.; Lismont, C.; Apanasets, O.; Fransen, M. The peroxisomal import receptor PEX5 functions as a stress sensor, retaining catalase in the cytosol in times of oxidative stress. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 1833–1843. [Google Scholar] [CrossRef]
- Rafikov, R.; Kumar, S.; Aggarwal, S.; Hou, Y.; Kangath, A.; Pardo, D.; Fineman, J.R.; Black, S. Endothelin-1 stimulates catalase activity through the PKCδ-mediated phosphorylation of serine 167. Free Radic. Biol. Med. 2014, 67, 255–264. [Google Scholar] [CrossRef]
- Cao, C.; Leng, Y.; Kufe, D. Catalase activity is regulated by c-Abl and Arg in the oxidative stress response. J. Biol. Chem. 2003, 278, 29667–29675. [Google Scholar] [CrossRef]
- Ghosh, S.; Janocha, A.J.; Aronica, M.A.; Swaidani, S.; Comhair, S.A.; Xu, W.; Zheng, L.; Kaveti, S.; Kinter, M.; Hazen, S.L.; et al. Nitrotyrosine proteome survey in asthma identifies oxidative mechanism of catalase inactivation. J. Immunol. 2006, 176, 5587–5597. [Google Scholar] [CrossRef]
- Foye, W.O.; Solis, M.C. Inhibition of catalase and lactate dehydrogenase by radiation-protective thiols and thiol derivatives. J. Pharm. Sci. 1969, 58, 352–355. [Google Scholar] [CrossRef]
- Sun, Y.; Oberley, L.W. The inhibition of catalase by glutathione. Free Radic. Biol. Med. 1989, 7, 595–602. [Google Scholar] [CrossRef]
- Bian, Y.; Song, C.; Cheng, K.; Dong, M.; Wang, F.; Huang, J.; Sun, D.; Wang, L.; Ye, M.; Zou, H. An enzyme assisted RP-RPLC approach for in-depth analysis of human liver phosphoproteome. J. Proteomics 2014, 96, 253–262. [Google Scholar] [CrossRef]
- Rardin, M.J.; Newman, J.C.; Held, J.M.; Cusack, M.P.; Sorensen, D.J.; Li, B.; Schilling, B.; Mooney, S.D.; Kahn, C.R.; Verdin, E.; et al. Label-free quantitative proteomics of the lysine acetylome in mitochondria identifies substrates of SIRT3 in metabolic pathways. Proc. Natl. Acad. Sci. USA 2013, 110, 6601–6606. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Chen, Y.; Tishkoff, D.X.; Peng, C.; Tan, M.; Dai, L.; Xie, Z.; Zhang, Y.; Zwaans, B.M.; Skinner, M.E.; et al. SIRT5-mediated lysine desuccinylation impacts diverse metabolic pathways. Mol. Cell 2013, 50, 919–930. [Google Scholar] [CrossRef]
- Geoghegan, V.; Guo, A.; Trudgian, D.; Thomas, B.; Acuto, O. Comprehensive identification of arginine methylation in primary T cells reveals regulatory roles in cell signalling. Nat. Commun. 2015, 6, 6758. [Google Scholar] [CrossRef]
- Larsen, S.C.; Sylvestersen, K.B.; Mund, A.; Lyon, D.; Mullari, M.; Madsen, M.V.; Daniel, J.A.; Jensen, L.J.; Nielsen, M.L. Proteome-wide analysis of arginine monomethylation reveals widespread occurrence inhuman cells. Sci. Signal. 2016, 9, rs9. [Google Scholar] [CrossRef]
- Kim, W.; Bennett, E.J.; Huttlin, E.L.; Guo, A.; Li, J.; Possemato, A.; Sowa, M.E.; Rad, R.; Rush, J.; Comb, M.J.; et al. Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol. Cell 2011, 44, 325–340. [Google Scholar] [CrossRef]
- Akimov, V.; Barrio-Hernandez, I.; Hansen, S.V.F.; Hallenborg, P.; Pedersen, A.K.; Bekker-Jensen, D.B.; Puglia, M.; Christensen, S.D.K.; Vanselow, J.T.; Nielsen, M.M.; et al. UbiSite approach for comprehensive mapping of lysine and N-terminal ubiquitination sites. Nat. Struct. Mol. Biol. 2018, 25, 631–640. [Google Scholar] [CrossRef]
- Guo, D.; Han, J.; Adam, B.L.; Colburn, N.H.; Wang, M.H.; Dong, Z.; Eizirik, D.L.; She, J.X.; Wang, C.Y. Proteomic analysis of SUMO4 substrates in HEK293 cells under serum starvation-induced stress. Biochem. Biophys. Res. Commun. 2005, 337, 1308–1318. [Google Scholar] [CrossRef]
- Lei, X.G.; Zhu, J.H.; Cheng, W.H.; Bao, Y.; Ho, Y.S.; Reddi, A.R.; Holmgren, A.; Arnér, E.S. Paradoxical roles of antioxidant enzymes: basic mechanisms and health implications. Physiol. Rev. 2016, 96, 307–364. [Google Scholar] [CrossRef]
- Góth, L.; Nagy, T. Inherited catalase deficiency: Is it benign or a factor in various age-related disorders? Mutat. Res. 2013, 753, 147–154. [Google Scholar] [CrossRef]
- Knoops, B.; Goemaere, J.; Van der Eecken, V.; Declercq, J.P. Peroxiredoxin 5: Structure, mechanism, and function of the mammalian atypical 2-Cys peroxiredoxin. Antioxid. Redox Signal. 2011, 15, 817–829. [Google Scholar] [CrossRef]
- Knoops, B.; Argyropoulou, V.; Becker, S.; Ferté, L.; Kuznetsova, O. Multiple Roles of Peroxiredoxins in Inflammation. Mol. Cells 2016, 39, 60–64. [Google Scholar] [CrossRef]
- Huttlin, E.L.; Jedrychowski, M.P.; Elias, J.E.; Goswami, T.; Rad, R.; Beausoleil, S.A.; Villén, J.; Haas, W.; Sowa, M.E.; Gygi, S.P. A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 2010, 143, 1174–1189. [Google Scholar] [CrossRef]
- Lundby, A.; Secher, A.; Lage, K.; Nordsborg, N.B.; Dmytriyev, A.; Lundby, C.; Olsen, J.V. Quantitative maps of protein phosphorylation sites across 14 different rat organs and tissues. Nat. Commun. 2012, 3, 876. [Google Scholar] [CrossRef] [Green Version]
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009, 325, 834–840. [Google Scholar] [CrossRef]
- Fratelli, M.; Demol, H.; Puype, M.; Casagrande, S.; Villa, P.; Eberini, I.; Vandekerckhove, J.; Gianazza, E.; Ghezzi, P. Identification of proteins undergoing glutathionylation in oxidatively stressed hepatocytes and hepatoma cells. Proteomics 2003, 3, 1154–1161. [Google Scholar] [CrossRef]
- Seo, M.S.; Kang, S.W.; Kim, K.; Baines, I.C.; Lee, T.H.; Rhee, S.G. Identification of a new type of mammalian peroxiredoxin that forms an intramolecular disulfide as a reaction intermediate. J. Biol. Chem. 2000, 275, 20346–20354. [Google Scholar] [CrossRef]
- Smeets, A.; Marchand, C.; Linard, D.; Knoops, B.; Declercq, J.P. The crystal structures of oxidized forms of human peroxiredoxin 5 with an intramolecular disulfide bond confirm the proposed enzymatic mechanism for atypical 2-Cys peroxiredoxins. Arch. Biochem. Biophys. 2008, 477, 98–104. [Google Scholar] [CrossRef]
- Wood, Z.A.; Poole, L.B.; Karplus, P.A. Peroxiredoxin evolution and the regulation of hydrogen peroxide signaling. Science 2003, 300, 650–653. [Google Scholar] [CrossRef]
- Zamocky, M.; Furtmüller, P.G.; Obinger, C. Evolution of catalases from bacteria to humans. Antioxid. Redox Signal. 2008, 10, 1527–1548. [Google Scholar] [CrossRef]
- Brigelius-Flohé, R.; Maiorino, M. Glutathione peroxidases. Biochim. Biophys. Acta 2013, 1830, 3289–3303. [Google Scholar] [CrossRef]
- Singh, A.K.; Gupta, M.K.; Orak, J.K. Antioxidant enzymes in peroxisomes: Effect of ischemia. Ann. N. Y. Acad. Sci. 1996, 804, 696–697. [Google Scholar] [CrossRef]
- Antunes, F.; Cadenas, E. Estimation of H2O2 gradients across biomembranes. FEBS Lett. 2000, 475, 121–126. [Google Scholar] [CrossRef]
- Branco, M.R.; Marinho, H.S.; Cyrne, L.; Antunes, F. Decrease of H2O2 plasma membrane permeability during adaptation to H2O2 in Saccharomyces cerevisiae. J. Biol. Chem. 2004, 279, 6501–6506. [Google Scholar] [CrossRef]
- Boveris, A.; Oshino, N.; Chance, B. The cellular production of hydrogen peroxide. Biochem. J. 1972, 128, 617–630. [Google Scholar] [CrossRef]
- Mueller, S.; Weber, A.; Fritz, R.; Mütze, S.; Rost, D.; Walczak, H.; Völkl, A.; Stremmel, W. Sensitive and real-time determination of H2O2 release from intact peroxisomes. Biochem. J. 2002, 363, 483–491. [Google Scholar] [CrossRef]
- Li, Y.; Tharappel, J.C.; Cooper, S.; Glenn, M.; Glauert, H.P.; Spear, B.T. Expression of the hydrogen peroxide-generating enzyme fatty acyl CoA oxidase activates NF-kappaB. DNA Cell Biol. 2000, 19, 113–120. [Google Scholar] [CrossRef]
- Ho, Y.S.; Xiong, Y.; Ma, W.; Spector, A.; Ho, D.S. Mice lacking catalase develop normally but show differential sensitivity to oxidant tissue injury. J. Biol. Chem. 2004, 279, 32804–32812. [Google Scholar] [CrossRef]
- Rokka, A.; Antonenkov, V.D.; Soininen, R.; Immonen, H.L.; Pirilä, P.L.; Bergmann, U.; Sormunen, R.T.; Weckström, M.; Benz, R.; Hiltunen, J.K. Pxmp2 is a channel-forming protein in mammalian peroxisomal membrane. PLoS ONE 2009, 4, e5090. [Google Scholar] [CrossRef]
- Gualdron-López, M.; Vapola, M.H.; Miinalainen, I.J.; Hiltunen, J.K.; Michels, P.A.; Antonenkov, V.D. Channel-forming activities in the glycosomal fraction from the bloodstream form of Trypanosoma brucei. PLoS ONE 2012, 7, e34530. [Google Scholar] [CrossRef]
- Mindthoff, S.; Grunau, S.; Steinfort, L.L.; Girzalsky, W.; Hiltunen, J.K.; Erdmann, R.; Antonenkov, V.D. Peroxisomal Pex11 is a pore-forming protein homologous to TRPM channels. Biochim. Biophys. Acta 2016, 1863, 271–283. [Google Scholar] [CrossRef]
- Lismont, C.; Koster, J.; Provost, S.; Baes, M.; Van Veldhoven, P.P.; Waterham, H.R.; Fransen, M. Deciphering the potential involvement of PXMP2 and PEX11B in hydrogen peroxide permeation across the peroxisomal membrane reveals a role for PEX11B in protein sorting. Biochim. Biophys. Acta Biomembr. 2019. [Google Scholar] [CrossRef]
- Kholodenko, B.N. Cell-signalling dynamics in time and space. Nat. Rev. Mol. Cell Biol. 2006, 7, 165–176. [Google Scholar] [CrossRef]
- Vestergaard, C.L.; Flyvbjerg, H.; Møller, I.M. Intracellular signaling by diffusion: Can waves of hydrogen peroxide transmit intracellular information in plant cells? Front. Plant Sci. 2012, 3, 295. [Google Scholar] [CrossRef]
- Fransen, M.; Lismont, C. Redox signaling from and to peroxisomes: Progress, challenges, and prospects. Antioxid. Redox Signal. 2019, 30, 95–112. [Google Scholar] [CrossRef]
- Fu, L.; Liu, K.; Sun, M.; Tian, C.; Sun, R.; Morales Betanzos, C.; Tallman, K.A.; Porter, N.A.; Yang, Y.; Guo, D.; et al. Systematic and quantitative assessment of hydrogen peroxide reactivity with cysteines across human proteomes. Mol. Cell. Proteom. 2017, 16, 1815–1828. [Google Scholar] [CrossRef]
- Lismont, C.; Nordgren, M.; Brees, C.; Knoops, B.; Van Veldhoven, P.P.; Fransen, M. Peroxisomes as modulators of cellular protein thiol oxidation: A new model system. Antioxid. Redox Signal. 2019, 30, 22–39. [Google Scholar] [CrossRef]
- Zhao, M.X.; Wen, J.L.; Wang, L.; Wang, X.P.; Chen, T.S. Intracellular catalase activity instead of glutathione level dominates the resistance of cells to reactive oxygen species. Cell Stress Chaperones 2019, 24, 609–619. [Google Scholar] [CrossRef]
- Yang, Y.; Song, Y.; Loscalzo, J. Regulation of the protein disulfide proteome by mitochondria in mammalian cells. Proc. Natl. Acad. Sci. USA 2007, 104, 10813–10817. [Google Scholar] [CrossRef] [Green Version]
- Yao, C.; Behring, J.B.; Shao, D.; Sverdlov, A.L.; Whelan, S.A.; Elezaby, A.; Yin, X.; Siwik, D.A.; Seta, F.; Costello, C.E.; et al. Overexpression of catalase diminishes oxidative cysteine modifications of cardiac proteins. PLoS ONE 2015, 10, e0144025. [Google Scholar] [CrossRef]
- Bracalente, C.; Ibañez, I.L.; Berenstein, A.; Notcovich, C.; Cerda, M.B.; Klamt, F.; Chernomoretz, A.; Durán, H. Reprogramming human A375 amelanotic melanoma cells by catalase overexpression: Upregulation of antioxidant genes correlates with regression of melanoma malignancy and with malignant progression when downregulated. Oncotarget 2016, 7, 41154–41171. [Google Scholar] [CrossRef] [Green Version]
- Heit, C.; Marshall, S.; Singh, S.; Yu, X.; Charkoftaki, G.; Zhao, H.; Orlicky, D.J.; Fritz, K.S.; Thompson, D.C.; Vasiliou, V. Catalase deletion promotes prediabetic phenotype in mice. Free Radic. Biol. Med. 2017, 103, 48–56. [Google Scholar] [CrossRef]
- Oliveira-Marques, V.; Marinho, H.S.; Cyrne, L.; Antunes, F. Modulation of NF-kappaB-dependent gene expression by H2O2: A major role for a simple chemical process in a complex biological response. Antioxid. Redox Signal. 2009, 11, 2043–2053. [Google Scholar] [CrossRef]
- Chu, R.; Lin, Y.; Reddy, K.C.; Pan, J.; Rao, M.S.; Reddy, J.K.; Yeldandi, A.V. Transformation of epithelial cells stably transfected with H2O2-generating peroxisomal urate oxidase. Cancer Res. 1996, 56, 4846–4852. [Google Scholar]
- Okamoto, M.; Reddy, J.K.; Oyasu, R. Tumorigenic conversion of a non-tumorigenic rat urothelial cell line by overexpression of H2O2-generating peroxisomal fatty acyl-CoA oxidase. Int. J. Cancer 1997, 70, 716–721. [Google Scholar] [CrossRef]
- Dadras, S.S.; Thorgeirsson, S.S.; Rao, M.S.; Reddy, J.K. Implication of hydrogen peroxide generation and apoptosis in the neoplastic transformation of mouse fibroblasts overexpressing peroxisomal fatty acyl-CoA oxidase. Int. J. Oncol. 1998, 12, 37–44. [Google Scholar] [CrossRef]
- Zmijewski, J.W.; Lorne, E.; Zhao, X.; Tsuruta, Y.; Sha, Y.; Liu, G.; Abraham, E. Antiinflammatory effects of hydrogen peroxide in neutrophil activation and acute lung injury. Am. J. Respir. Crit. Care Med. 2009, 179, 694–704. [Google Scholar] [CrossRef]
- Cong, W.; Ruan, D.; Xuan, Y.; Niu, C.; Tao, Y.; Wang, Y.; Zhan, K.; Cai, L.; Jin, L.; Tan, Y. Cardiac-specific overexpression of catalase prevents diabetes-induced pathological changes by inhibiting NF-κB signaling activation in the heart. J. Mol. Cell. Cardiol. 2015, 89, 314–325. [Google Scholar] [CrossRef]
- Hamanaka, R.B.; Chandel, N.S. Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem. Sci. 2010, 35, 505–513. [Google Scholar] [CrossRef] [Green Version]
- Tatapudy, S.; Aloisio, F.; Barber, D.; Nystul, T. Cell fate decisions: Emerging roles for metabolic signals and cell morphology. EMBO Rep. 2017, 18, 2105–2118. [Google Scholar] [CrossRef]
- Farr, R.L.; Lismont, C.; Terlecky, S.R.; Fransen, M. Peroxisome biogenesis in mammalian cells: The impact of genes and environment. Biochim. Biophys. Acta 2016, 1863, 1049–1060. [Google Scholar] [CrossRef]
- Brown, M.R.; Miller, F.J., Jr.; Li, W.G.; Ellingson, A.N.; Mozena, J.D.; Chatterjee, P.; Engelhardt, J.F.; Zwacka, R.M.; Oberley, L.W.; Fang, X.; et al. Overexpression of human catalase inhibits proliferation and promotes apoptosis in vascular smooth muscle cells. Circ. Res. 1999, 85, 524–533. [Google Scholar] [CrossRef]
- Zanetti, M.; Katusic, Z.S.; O’Brien, T. Adenoviral-mediated overexpression of catalase inhibits endothelial cell proliferation. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H2620–H2626. [Google Scholar] [CrossRef] [Green Version]
- Glorieux, C.; Dejeans, N.; Sid, B.; Beck, R.; Calderon, P.B.; Verrax, J. Catalase overexpression in mammary cancer cells leads to a less aggressive phenotype and an altered response to chemotherapy. Biochem. Pharmacol. 2011, 82, 1384–1390. [Google Scholar] [CrossRef]
- Hachiya, M.; Akashi, M. Catalase regulates cell growth in HL60 human promyelocytic cells: Evidence for growth regulation by H2O2. Radiat. Res. 2005, 163, 271–282. [Google Scholar] [CrossRef]
- Onumah, O.E.; Jules, G.E.; Zhao, Y.; Zhou, L.; Yang, H.; Guo, Z. Overexpression of catalase delays G0/G1- to S-phase transition during cell cycle progression in mouse aortic endothelial cells. Free Radic. Biol. Med. 2009, 46, 1658–1667. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, T.; Sakaguchi, N.; Hachiya, M.; Nakayama, F.; Yamakawa, M.; Akashi, M. Role of catalase in monocytic differentiation of U937 cells by TPA: Hydrogen peroxide as a second messenger. Leukemia 2009, 23, 761–769. [Google Scholar] [CrossRef]
- Koepke, J.I.; Wood, C.S.; Terlecky, L.J.; Walton, P.A.; Terlecky, S.R. Progeric effects of catalase inactivation in human cells. Toxicol. Appl. Pharmacol. 2008, 232, 99–108. [Google Scholar] [CrossRef]
- Rezvani, H.R.; Cario-André, M.; Pain, C.; Ged, C.; deVerneuil, H.; Taïeb, A. Protection of normal human reconstructed epidermis from UV by catalase overexpression. Cancer Gene Ther. 2007, 14, 174–186. [Google Scholar] [CrossRef]
- Xiao, X.; Luo, H.; Vanek, K.N.; LaRue, A.C.; Schulte, B.A.; Wang, G.Y. Catalase inhibits ionizing radiation-induced apoptosis in hematopoietic stem and progenitor cells. Stem Cells Dev. 2015, 24, 1342–1351. [Google Scholar] [CrossRef]
- Song, L.L.; Tu, Y.Y.; Xia, L.; Wang, W.W.; Wei, W.; Ma, C.M.; Wen, D.H.; Lei, H.; Xu, H.Z.; Wu, Y.L. Targeting catalase but not peroxiredoxins enhances arsenic trioxide-induced apoptosis in K562 cells. PLoS ONE 2014, 9, e104985. [Google Scholar] [CrossRef]
- Bai, J.; Cederbaum, A.I. Catalase protects HepG2 cells from apoptosis induced by DNA-damaging agents by accelerating the degradation of p53. J. Biol. Chem. 2003, 278, 4660–4667. [Google Scholar] [CrossRef]
- Walbrecq, G.; Wang, B.; Becker, S.; Hannotiau, A.; Fransen, M.; Knoops, B. Antioxidant cytoprotection by peroxisomal peroxiredoxin-5. Free Radic. Biol. Med. 2015, 84, 215–226. [Google Scholar] [CrossRef]
- Chen, X.; Liang, H.; Van Remmen, H.; Vijg, J.; Richardson, A. Catalase transgenic mice: Characterization and sensitivity to oxidative stress. Arch. Biochem. Biophys. 2004, 422, 197–210. [Google Scholar] [CrossRef]
- Carter, A.B.; Tephly, L.A.; Venkataraman, S.; Oberley, L.W.; Zhang, Y.; Buettner, G.R.; Spitz, D.R.; Hunninghake, G.W. High levels of catalase and glutathione peroxidase activity dampen H2O2 signaling in human alveolar macrophages. Am. J. Respir. Cell. Mol. Biol. 2004, 31, 43–53. [Google Scholar] [CrossRef]
- Li, X.; Chen, H.; Epstein, P.N. Metallothionein and catalase sensitize to diabetes in non-obese diabetic mice: Reactive oxygen species may have a protective role in pancreatic beta-cells. Diabetes 2006, 55, 1592–1604. [Google Scholar] [CrossRef]
- Elsner, M.; Gehrmann, W.; Lenzen, S. Peroxisome-generated hydrogen peroxide as important mediator of lipotoxicity in insulin-producing cells. Diabetes 2011, 60, 200–208. [Google Scholar] [CrossRef]
- Schrader, M.; Kamoshita, M.; Islinger, M. Organelle interplay-peroxisome interactions in health and disease. J. Inherit. Metab. Dis. 2019. [Google Scholar] [CrossRef]
- Peeters, A.; Shinde, A.B.; Dirkx, R.; Smet, J.; De Bock, K.; Espeel, M.; Vanhorebeek, I.; Vanlander, A.; Van Coster, R.; Carmeliet, P.; et al. Mitochondria in peroxisome-deficient hepatocytes exhibit impaired respiration, depleted DNA, and PGC-1α independent proliferation. Biochim. Biophys. Acta 2015, 1853, 285–298. [Google Scholar] [CrossRef]
- Fransen, M.; Lismont, C.; Walton, P. The peroxisome-mitochondria connection: How and why? Int. J. Mol. Sci. 2017, 18, 1126. [Google Scholar] [CrossRef]
- Ivashchenko, O.; Van Veldhoven, P.P.; Brees, C.; Ho, Y.S.; Terlecky, S.R.; Fransen, M. Intraperoxisomal redox balance in mammalian cells: Oxidative stress and interorganellar cross-talk. Mol. Biol. Cell 2011, 22, 1440–1451. [Google Scholar] [CrossRef]
- Hwang, I.; Lee, J.; Huh, J.Y.; Park, J.; Lee, H.B.; Ho, Y.S.; Ha, H. Catalase deficiency accelerates diabetic renal injury through peroxisomal dysfunction. Diabetes 2012, 61, 728–738. [Google Scholar] [CrossRef]
- Walton, P.A.; Pizzitelli, M. Effects of peroxisomal catalase inhibition on mitochondrial function. Front. Physiol. 2012, 3, 108. [Google Scholar] [CrossRef] [Green Version]
- Barbosa, M.R.; Sampaio, I.H.; Teodoro, B.G.; Sousa, T.A.; Zoppi, C.C.; Queiroz, A.L.; Passos, M.A.; Alberici, L.C.; Teixeira, F.R.; Manfiolli, A.O.; et al. Hydrogen peroxide production regulates the mitochondrial function in insulin resistant muscle cells: Effect of catalase overexpression. Biochim. Biophys. Acta 2013, 1832, 1591–1604. [Google Scholar] [CrossRef] [Green Version]
- Qin, F.; Lennon-Edwards, S.; Lancel, S.; Biolo, A.; Siwik, D.A.; Pimentel, D.R.; Dorn, G.W.; Kang, Y.J.; Colucci, W.S. Cardiac-specific overexpression of catalase identifies hydrogen peroxide-dependent and -independent phases of myocardial remodeling and prevents the progression to overt heart failure in G(alpha)q-overexpressing transgenic mice. Circ. Heart Fail. 2010, 3, 306–313. [Google Scholar] [CrossRef]
- Li, G.; Chen, Y.; Saari, J.T.; Kang, Y.J. Catalase-overexpressing transgenic mouse heart is resistant to ischemia-reperfusion injury. Am. J. Physiol. 1997, 273, H1090–H1095. [Google Scholar] [CrossRef]
- Wölkart, G.; Kaber, G.; Kojda, G.; Brunner, F. Role of endogenous hydrogen peroxide in cardiovascular ischaemia/reperfusion function: Studies in mouse hearts with catalase-overexpression in the vascular endothelium. Pharmacol. Res. 2006, 54, 50–56. [Google Scholar] [CrossRef]
- Dong, F.; Fang, C.X.; Yang, X.; Zhang, X.; Lopez, F.L.; Ren, J. Cardiac overexpression of catalase rescues cardiac contractile dysfunction induced by insulin resistance: Role of oxidative stress, protein carbonyl formation and insulin sensitivity. Diabetologia 2006, 49, 1421–1433. [Google Scholar] [CrossRef]
- Turdi, S.; Han, X.; Huff, A.F.; Roe, N.D.; Hu, N.; Gao, F.; Ren, J. Cardiac-specific overexpression of catalase attenuates lipopolysaccharide-induced myocardial contractile dysfunction: Role of autophagy. Free Radic. Biol. Med. 2012, 53, 1327–1338. [Google Scholar] [CrossRef] [Green Version]
- Pendergrass, K.D.; Varghese, S.T.; Maiellaro-Rafferty, K.; Brown, M.E.; Taylor, W.R.; Davis, M.E. Temporal effects of catalase overexpression on healing after myocardial infarction. Circ. Heart Fail. 2011, 4, 98–106. [Google Scholar] [CrossRef]
- Ren, J.; Li, Q.; Wu, S.; Li, S.Y.; Babcock, S.A. Cardiac overexpression of antioxidant catalase attenuates aging-induced cardiomyocyte relaxation dysfunction. Mech. Ageing Dev. 2007, 128, 276–285. [Google Scholar] [CrossRef] [Green Version]
- Godin, N.; Liu, F.; Lau, G.J.; Brezniceanu, M.L.; Chénier, I.; Filep, J.G.; Ingelfinger, J.R.; Zhang, S.L.; Chan, J.S. Catalase overexpression prevents hypertension and tubular apoptosis in angiotensinogen transgenic mice. Kidney Int. 2010, 77, 1086–1097. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Lo, C.S.; Chenier, I.; Maachi, H.; Filep, J.G.; Ingelfinger, J.R.; Zhang, S.L.; Chan, J.S. Overexpression of catalase prevents hypertension and tubulointerstitial fibrosis and normalization of renal angiotensin-converting enzyme-2 expression in Akita mice. Am. J. Physiol. Renal Physiol. 2013, 304, F1335–F1346. [Google Scholar] [CrossRef]
- Abdo, S.; Shi, Y.; Otoukesh, A.; Ghosh, A.; Lo, C.S.; Chenier, I.; Filep, J.G.; Ingelfinger, J.R.; Zhang, S.L.; Chan, J.S. Catalase overexpression prevents nuclear factor erythroid 2-related factor 2 stimulation of renal angiotensinogen gene expression, hypertension, and kidney injury in diabetic mice. Diabetes 2014, 63, 3483–3496. [Google Scholar] [CrossRef]
- Brezniceanu, M.L.; Liu, F.; Wei, C.C.; Chénier, I.; Godin, N.; Zhang, S.L.; Filep, J.G.; Ingelfinger, J.R.; Chan, J.S. Attenuation of interstitial fibrosis and tubular apoptosis in db/db transgenic mice overexpressing catalase in renal proximal tubular cells. Diabetes 2008, 57, 451–459. [Google Scholar] [CrossRef]
- Brezniceanu, M.L.; Liu, F.; Wei, C.C.; Tran, S.; Sachetelli, S.; Zhang, S.L.; Guo, D.F.; Filep, J.G.; Ingelfinger, J.R.; Chan, J.S. Catalase overexpression attenuates angiotensinogen expression and apoptosis in diabetic mice. Kidney Int. 2007, 71, 912–923. [Google Scholar] [CrossRef] [Green Version]
- Rashidi, A.; Kirkwood, T.B.; Shanley, D.P. Metabolic evolution suggests an explanation for the weakness of antioxidant defences in beta-cells. Mech. Ageing Dev. 2009, 130, 216–221. [Google Scholar] [CrossRef]
- Xu, B.; Moritz, J.T.; Epstein, P.N. Overexpression of catalase provides partial protection to transgenic mouse beta cells. Free Radic. Biol. Med. 1999, 27, 830–837. [Google Scholar] [CrossRef]
- Amos, D.L.; Robinson, T.; Massie, M.B.; Cook, C.; Hoffsted, A.; Crain, C.; Santanam, N. Catalase overexpression modulates metabolic parameters in a new ‘stress-less’ leptin-deficient mouse model. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 2293–2306. [Google Scholar] [CrossRef]
- Cervantes Gracia, K.; Llanas-Cornejo, D.; Husi, H. CVD and oxidative stress. J. Clin. Med. 2017, 6, 22. [Google Scholar] [CrossRef]
- Santanam, N.; Aug, N.; Zhou, M.; Keshava, C.; Parthasarathy, S. Overexpression of human catalase gene decreases oxidized lipid-induced cytotoxicity in vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 1912–1917. [Google Scholar] [CrossRef]
- Parastatidis, I.; Weiss, D.; Joseph, G.; Taylor, W.R. Overexpression of catalase in vascular smooth muscle cells prevents the formation of abdominal aortic aneurysms. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2389–2396. [Google Scholar] [CrossRef]
- Maiellaro-Rafferty, K.; Weiss, D.; Joseph, G.; Wan, W.; Gleason, R.L.; Taylor, W.R. Catalase overexpression in aortic smooth muscle prevents pathological mechanical changes underlying abdominal aortic aneurysm formation. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H355–H362. [Google Scholar] [CrossRef] [Green Version]
- Hodara, R.; Weiss, D.; Joseph, G.; Velasquez-Castano, J.C.; Landázuri, N.; Han, J.W.; Yoon, Y.S.; Taylor, W.R. Overexpression of catalase in myeloid cells causes impaired postischemic neovascularization. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2203–2209. [Google Scholar] [CrossRef]
- Moloney, J.N.; Cotter, T.G. ROS signaling in the biology of cancer. Semin. Cell Dev. Biol. 2018, 80, 50–64. [Google Scholar] [CrossRef]
- Syed, D.N.; Mukhtar, H. Gender bias in skin cancer: Role of catalase revealed. J. Investig. Dermatol. 2012, 132, 512–514. [Google Scholar] [CrossRef]
- Chang, D.; Hu, Z.L.; Zhang, L.; Zhao, Y.S.; Meng, Q.H.; Guan, Q.B.; Zhou, J.; Pan, H.Z. Association of catalase genotype with oxidative stress in the predication of colorectal cancer: Modification by epidemiological factors. Biomed. Environ. Sci. 2012, 25, 156–162. [Google Scholar] [CrossRef]
- Sen, S.; Kawahara, B.; Chaudhuri, G. Maintenance of higher H2O2 levels, and its mechanism of action to induce growth in breast cancer cells: Important roles of bioactive catalase and PP2A. Free Radic. Biol. Med. 2012, 53, 1541–1551. [Google Scholar] [CrossRef]
- Castaldo, S.A.; da Silva, A.P.; Matos, A.; Inácio, Â.; Bicho, M.; Medeiros, R.; Alho, I.; Bicho, M.C. The role of CYBA (p22phox) and catalase genetic polymorphisms and their possible epistatic interaction in cervical cancer. Tumour Biol. 2015, 36, 909–914. [Google Scholar] [CrossRef]
- Belotte, J.; Fletcher, N.M.; Saed, M.G.; Abusamaan, M.S.; Dyson, G.; Diamond, M.P.; Saed, G.M. A single nucleotide polymorphism in catalase is strongly associated with ovarian cancer survival. PLoS ONE 2015, 10, e0135739. [Google Scholar] [CrossRef]
- Shen, Y.; Li, D.; Tian, P.; Shen, K.; Zhu, J.; Feng, M.; Wan, C.; Yang, T.; Chen, L.; Wen, F. The catalase C-262T gene polymorphism and cancer risk: A systematic review and meta-analysis. Medicine 2015, 94, e679. [Google Scholar] [CrossRef]
- Hu, J.; Feng, F.; Zhu, S.; Sun, L.; Li, G.; Jiang, N.; Shang, Z.; Niu, Y. Catalase C-262T polymorphism and risk of prostate cancer: Evidence from meta-analysis. Gene 2015, 558, 265–270. [Google Scholar] [CrossRef]
- Liu, K.; Liu, X.; Wang, M.; Wang, X.; Kang, H.; Lin, S.; Yang, P.; Dai, C.; Xu, P.; Li, S.; et al. Two common functional catalase gene polymorphisms (rs1001179 and rs794316) and cancer susceptibility: Evidence from 14,942 cancer cases and 43,285 controls. Oncotarget 2016, 7, 62954–62965. [Google Scholar] [CrossRef]
- Tsai, J.Y.; Lee, M.J.; Dah-Tsyr Chang, M.; Huang, H. The effect of catalase on migration and invasion of lung cancer cells by regulating the activities of cathepsin S, L, and K. Exp. Cell Res. 2014, 323, 28–40. [Google Scholar] [CrossRef]
- Cenini, G.; Lloret, A.; Cascella, R. Oxidative stress in neurodegenerative diseases: From a mitochondrial point of view. Oxid. Med. Cell. Longev. 2019, 2019, 2105607. [Google Scholar] [CrossRef]
- Cobley, J.N.; Fiorello, M.L.; Bailey, D.M. 13 reasons why the brain is susceptible to oxidative stress. Redox Biol. 2018, 15, 490–503. [Google Scholar] [CrossRef]
- Berger, J.; Dorninger, F.; Forss-Petter, S.; Kunze, M. Peroxisomes in brain development and function. Biochim. Biophys. Acta 2016, 1863, 934–955. [Google Scholar] [CrossRef]
- Singhal, A.; Morris, V.B.; Labhasetwar, V.; Ghorpade, A. Nanoparticle-mediated catalase delivery protects human neurons from oxidative stress. Cell Death Dis. 2013, 4, e903. [Google Scholar] [CrossRef]
- Chilumuri, A.; Odell, M.; Milton, N.G. Benzothiazole aniline tetra(ethylene glycol) and 3-amino-1,2,4-triazole inhibit neuroprotection against amyloid peptides by catalase overexpression in vitro. ACS Chem. Neurosci. 2013, 4, 1501–1512. [Google Scholar] [CrossRef]
- Nell, H.J.; Au, J.L.; Giordano, C.R.; Terlecky, S.R.; Walton, P.A.; Whitehead, S.N.; Cechetto, D.F. Targeted antioxidant, catalase-SKL, reduces beta-amyloid toxicity in the rat brain. Brain Pathol. 2017, 27, 86–94. [Google Scholar] [CrossRef]
- Iglesias-González, J.; Sánchez-Iglesias, S.; Méndez-Álvarez, E.; Rose, S.; Hikima, A.; Jenner, P.; Soto-Otero, R. Differential toxicity of 6-hydroxydopamine in SH-SY5Y human neuroblastoma cells and rat brain mitochondria: Protective role of catalase and superoxide dismutase. Neurochem. Res. 2012, 37, 2150–2160. [Google Scholar] [CrossRef]
- Habib, L.K.; Lee, M.T.; Yang, J. Inhibitors of catalase-amyloid interactions protect cells from beta-amyloid-induced oxidative stress and toxicity. J. Biol. Chem. 2010, 285, 38933–38943. [Google Scholar] [CrossRef]
- Gsell, W.; Conrad, R.; Hickethier, M.; Sofic, E.; Frölich, L.; Wichart, I.; Jellinger, K.; Moll, G.; Ransmayr, G.; Beckmann, H.; et al. Decreased catalase activity but unchanged superoxide dismutase activity in brains of patients with dementia of Alzheimer type. J. Neurochem. 1995, 64, 1216–1223. [Google Scholar] [CrossRef]
- Capurso, C.; Solfrizzi, V.; D’Introno, A.; Colacicco, A.M.; Capurso, S.A.; Bifaro, L.; Menga, R.; Santamato, A.; Seripa, D.; Pilotto, A.; et al. Short arm of chromosome 11 and sporadic Alzheimer’s disease: Catalase and cathepsin D gene polymorphisms. Neurosci. Lett. 2008, 432, 237–242. [Google Scholar] [CrossRef]
- Gasser, T.; Wszolek, Z.K.; Trofatter, J.; Ozelius, L.; Uitti, R.J.; Lee, C.S.; Gusella, J.; Pfeiffer, R.F.; Calne, D.B.; Breakefield, X.O. Genetic linkage studies in autosomal dominant parkinsonism: Evaluation of seven candidate genes. Ann. Neurol. 1994, 36, 387–396. [Google Scholar] [CrossRef]
- Parboosingh, J.S.; Rousseau, M.; Rogan, F.; Amit, Z.; Chertkow, H.; Johnson, W.G.; Manganaro, F.; Schipper, H.N.; Curran, T.J.; Stoessl, J.; et al. Absence of mutations in superoxide dismutase and catalase genes in patients with Parkinson’s disease. Arch. Neurol. 1995, 52, 1160–1163. [Google Scholar] [CrossRef]
- Przedborski, S.; Donaldson, D.M.; Murphy, P.L.; Hirsch, O.; Lange, D.; Naini, A.B.; McKenna-Yasek, D.; Brown, R.H., Jr. Blood superoxide dismutase, catalase and glutathione peroxidase activities in familial and sporadic amyotrophic lateral sclerosis. Neurodegeneration 1996, 5, 57–64. [Google Scholar] [CrossRef]
- Waszczak, C.; Akter, S.; Eeckhout, D.; Persiau, G.; Wahni, K.; Bodra, N.; Van Molle, I.; De Smet, B.; Vertommen, D.; Gevaert, K.; et al. Sulfenome mining in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2014, 111, 11545–11550. [Google Scholar] [CrossRef]
- Jones, D.P.; Sies, H. The redox code. Antioxid. Redox Signal. 2015, 23, 734–746. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lismont, C.; Revenco, I.; Fransen, M. Peroxisomal Hydrogen Peroxide Metabolism and Signaling in Health and Disease. Int. J. Mol. Sci. 2019, 20, 3673. https://doi.org/10.3390/ijms20153673
Lismont C, Revenco I, Fransen M. Peroxisomal Hydrogen Peroxide Metabolism and Signaling in Health and Disease. International Journal of Molecular Sciences. 2019; 20(15):3673. https://doi.org/10.3390/ijms20153673
Chicago/Turabian StyleLismont, Celien, Iulia Revenco, and Marc Fransen. 2019. "Peroxisomal Hydrogen Peroxide Metabolism and Signaling in Health and Disease" International Journal of Molecular Sciences 20, no. 15: 3673. https://doi.org/10.3390/ijms20153673
APA StyleLismont, C., Revenco, I., & Fransen, M. (2019). Peroxisomal Hydrogen Peroxide Metabolism and Signaling in Health and Disease. International Journal of Molecular Sciences, 20(15), 3673. https://doi.org/10.3390/ijms20153673