Structural Mapping of Missense Mutations in the Pex1/Pex6 Complex

Abstract
:1. Introduction
2. Results
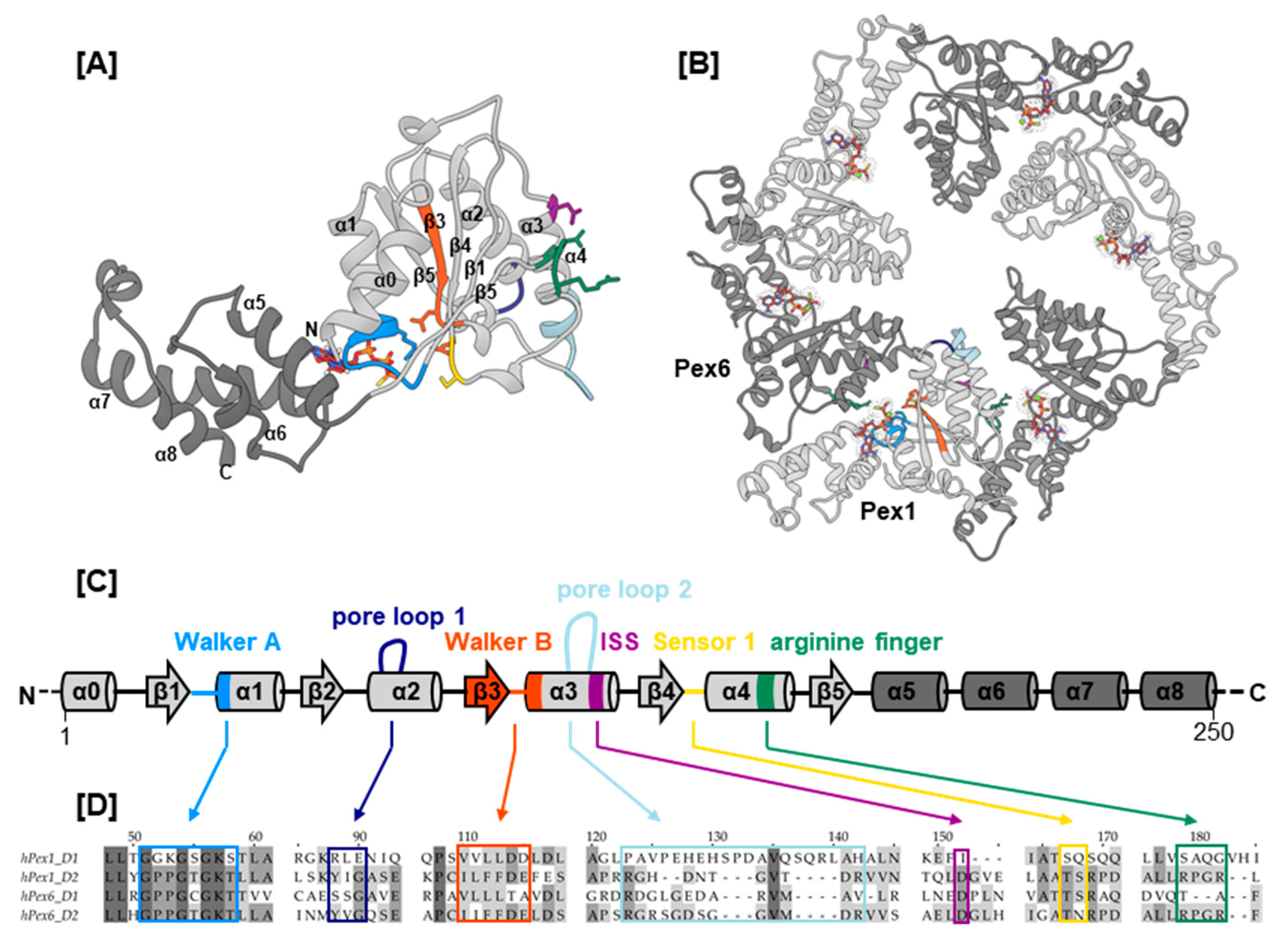
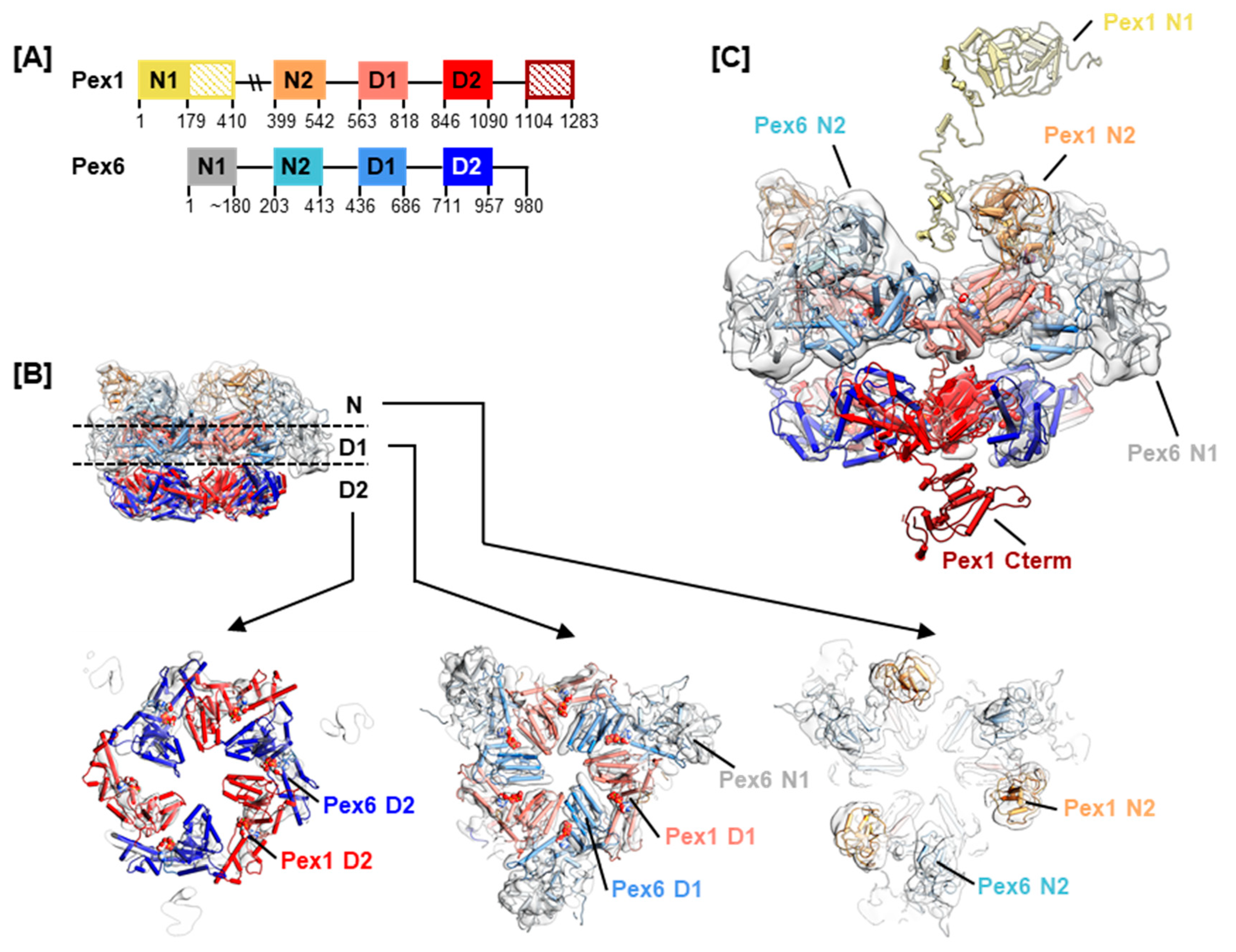
2.1. Selection of Missense Mutations and Generation of Pex1/Pex6 Homolgy Model
2.2. Mutations Concerning ATP Binding and Hydrolysis
2.3. Mutations Concerning Substrate Interaction
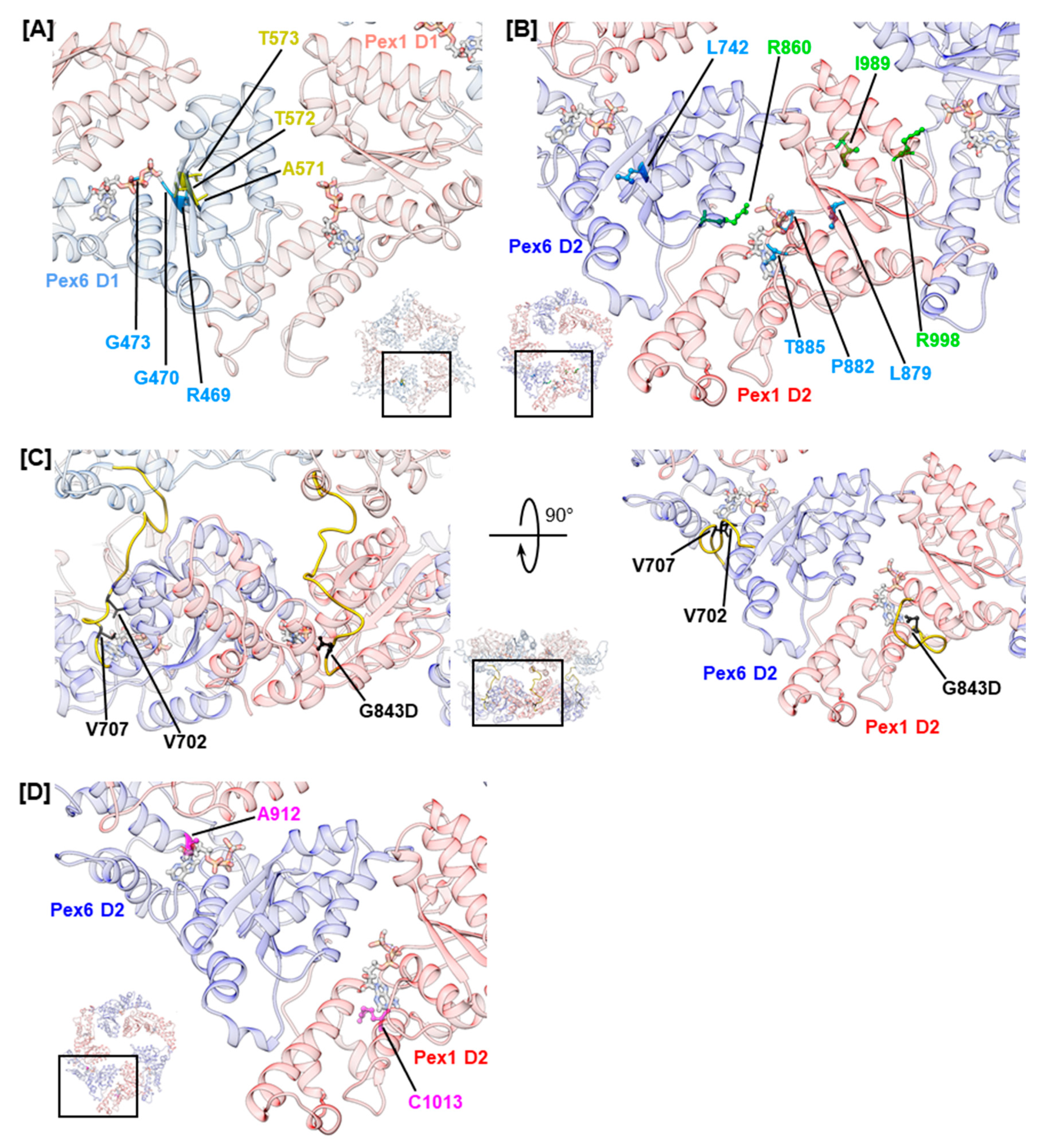
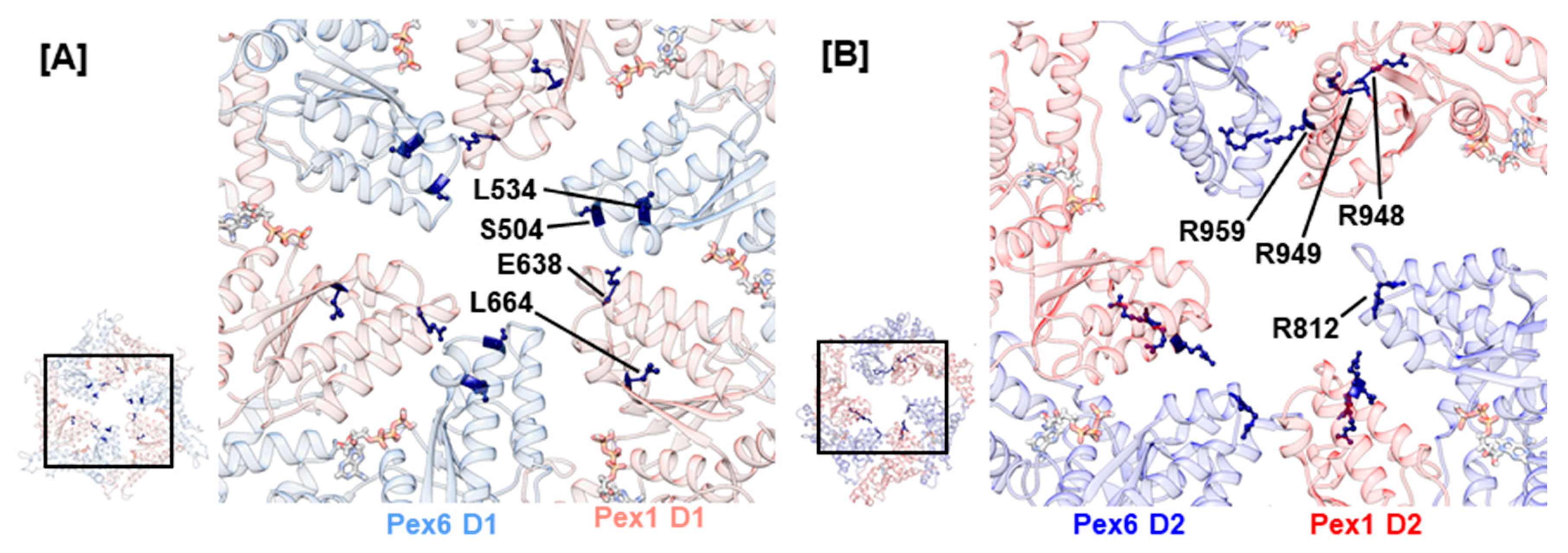
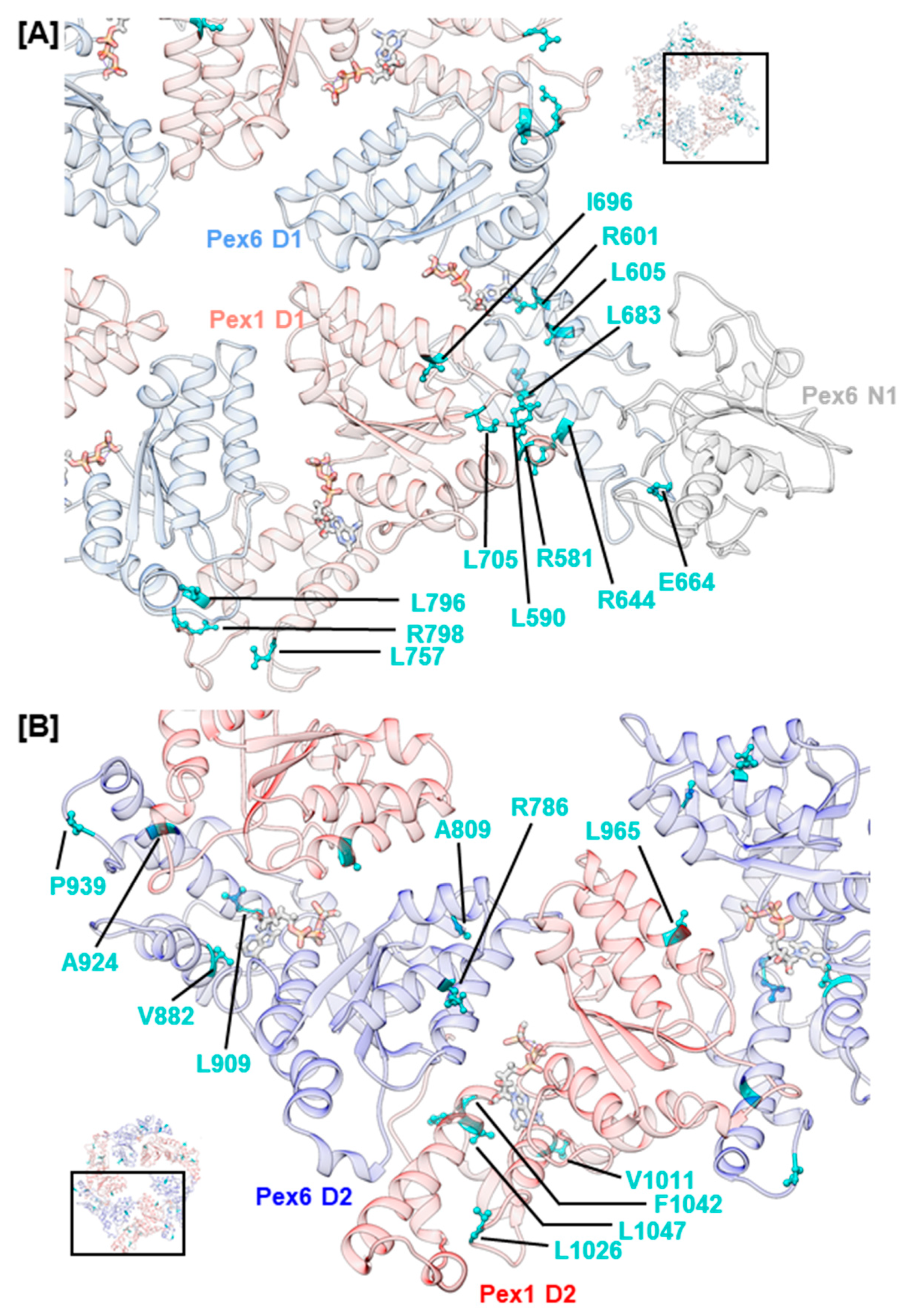
2.4. Mutations Concerning the Interaction between Pex1 and Pex6
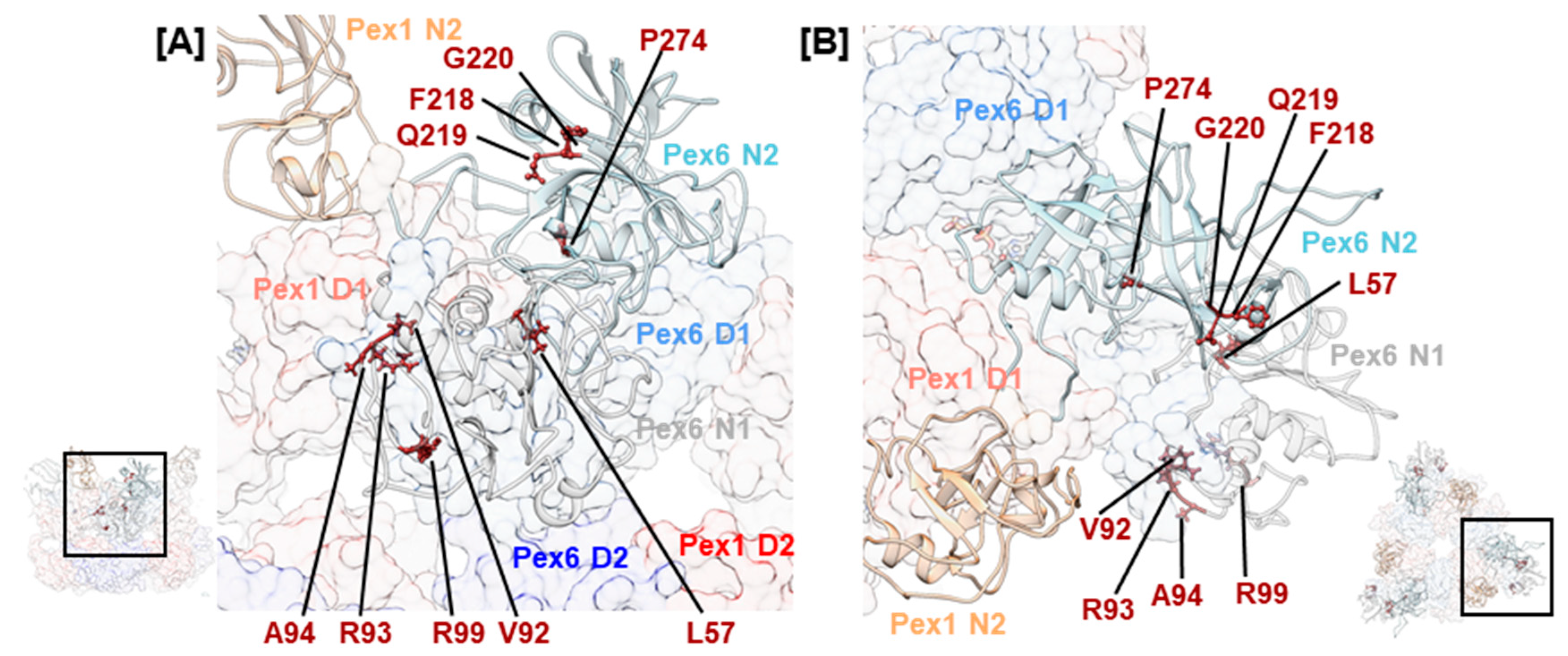
2.5. Mutations Concerning Interactions with Cofactors
3. Discussion
4. Materials and Methods
4.1. Homology Modeling
4.2. Sequence Alignments and Conservation Score
- sequences whose name indicated a Pex1 or Pex6 sequence were retrieved;
- sequences whose name indicated a putative annotation or a low quality were excluded;
- doubled or contained sequences were excluded; and
- one sequence per species was kept, generally, the sequence referring to isoform 1 or a RefSeq sequence.
4.3. Analysis of Mutations Described in Peroxisome Biogenesis Disorders (PBDs) Patients
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| NTD | N-terminal domain |
| ZSSD | Zellweger syndrome spectrum disease |
| ZS | Zellweger syndrome |
| HS | Heimler syndrome |
| EM | Electron microscopy |
| PBD | Peroxisome biogenesis disorder |
| NSF | N-ethylmaleimide sensitive factor |
References
- Waterham, H.R.; Ebberink, M.S. Genetics and molecular basis of human peroxisome biogenesis disorders. Biochim. Et Biophys. Acta 2012, 1822, 1430–1441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klouwer, F.C.C.; Berendse, K.; Ferdinandusse, S.; Wanders, R.J.A.; Engelen, M.; Poll-The, B.T. Zellweger spectrum disorders: Clinical overview and management approach. Orphanet J. Rare Dis. 2015, 10, 151. [Google Scholar] [CrossRef] [PubMed]
- Ratbi, I.; Falkenberg, K.D.; Sommen, M.; Al-Sheqaih, N.; Guaoua, S.; Vandeweyer, G.; Urquhart, J.E.; Chandler, K.E.; Williams, S.G.; Roberts, N.A.; et al. Heimler Syndrome Is Caused by Hypomorphic Mutations in the Peroxisome-Biogenesis Genes PEX1 and PEX6. Am. J. Hum. Genet. 2015, 97, 535–545. [Google Scholar] [CrossRef] [PubMed]
- Gould, S.J.; Raymond, G.V.; Valle, D. The peroxisome biogenesis disorders. In The Metabolic & Molecular Bases of Inherited Disease, 8th ed.; Scriver, C.R., Ed.; McGraw-Hill: New York, NY, USA, 2001; Volume 4. [Google Scholar]
- Waterham, H.R.; Ferdinandusse, S.; Wanders, R.J.A. Human disorders of peroxisome metabolism and biogenesis. Biochim. Et Biophys. Acta Mol. Cell Res. 2016, 1863, 922–933. [Google Scholar] [CrossRef] [PubMed]
- Motley, A.M.; Galvin, P.C.; Ekal, L.; Nuttall, J.M.; Hettema, E.H. Reevaluation of the role of Pex1 and dynamin-related proteins in peroxisome membrane biogenesis. J. Cell Biol. 2015, 211, 1041–1056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knoops, K.; de Boer, R.; Kram, A.; van der Klei, I.J. Yeast pex1 cells contain peroxisomal ghosts that import matrix proteins upon reintroduction of Pex1. J. Cell Biol. 2015, 211, 955–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, M.J.; Hoefler, S.; Moser, A.B.; Moser, H.W.; Lazarow, P.B. Peroxisome assembly mutations in humans: Structural heterogeneity in Zellweger syndrome. J. Cell. Physiol. 1992, 151, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Soliman, K.; Göttfert, F.; Rosewich, H.; Thoms, S.; Gärtner, J. Super-resolution imaging reveals the sub-diffraction phenotype of Zellweger Syndrome ghosts and wild-type peroxisomes. Sci. Rep. 2018, 8, 7809. [Google Scholar] [CrossRef] [PubMed]
- Preuss, N.; Brosius, U.; Biermanns, M.; Muntau, A.C.; Conzelmann, E.; Gärtner, J. PEX1 mutations in complementation group 1 of Zellweger spectrum patients correlate with severity of disease. Pediatric Res. 2002, 51, 706–714. [Google Scholar] [CrossRef] [PubMed]
- Rosewich, H.; Ohlenbusch, A.; Gärtner, J. Genetic and clinical aspects of Zellweger spectrum patients with PEX1 mutations. J. Med. Genet. 2005, 42, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Walter, C.; Gootjes, J.; Mooijer, P.A.; Portsteffen, H.; Klein, C.; Waterham, H.R.; Barth, P.G.; Epplen, J.T.; Kunau, W.-H.; Wanders, R.J.A.; et al. Disorders of Peroxisome Biogenesis Due to Mutations in PEX1: Phenotypes and PEX1 Protein Levels. Am. J. Hum. Genet. 2002, 69, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Ebberink, M.S.; Mooijer, P.A.W.; Gootjes, J.; Koster, J.; Wanders, R.J.A.; Waterham, H.R. Genetic classification and mutational spectrum of more than 600 patients with a Zellweger syndrome spectrum disorder. Hum. Mutat. 2011, 32, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, M.A.; Allen, T.; Solly, P.B.; Svingen, T.; Paton, B.C.; Crane, D.I. Novel PEX1 mutations and genotype-phenotype correlations in Australasian peroxisome biogenesis disorder patients. Hum. Mutat. 2002, 20, 342–351. [Google Scholar] [CrossRef] [PubMed]
- Berendse, K.; Ebberink, M.S.; Ijlst, L.; Poll-The, B.T.; Wanders, R.J.A.; Waterham, H.R. Arginine improves peroxisome functioning in cells from patients with a mild peroxisome biogenesis disorder. Orphanet J. Rare Dis. 2013, 8, 1–8. [Google Scholar] [CrossRef] [PubMed]
- MacLean, G.E.; Argyriou, C.; Di Pietro, E.; Sun, X.; Birjandian, S.; Saberian, P.; Hacia, J.G.; Braverman, N.E. Zellweger spectrum disorder patient–derived fibroblasts with the PEX1-Gly843Asp allele recover peroxisome functions in response to flavonoids. J. Cell. Biochem. 2019, 120, 3243–3258. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Chen, L.; Jiralerspong, S.; Snowden, A.; Steinberg, S.; Braverman, N. Recovery of PEX1-Gly843Asp peroxisome dysfunction by small-molecule compounds. Proc. Natl. Acad. Sci. USA 2010, 107, 5569–5574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klouwer, F.C.C.; Meester-Delver, A.; Vaz, F.M.; Waterham, H.R.; Hennekam, R.C.M.; Poll-The, B.T. Development and validation of a severity scoring system for Zellweger spectrum disorders. Clin. Genet. 2018, 93, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Berendse, K.; Engelen, M.; Ferdinandusse, S.; Majoie, C.B.L.M.; Waterham, H.R.; Vaz, F.M.; Koelman, J.H.T.M.; Barth, P.G.; Wanders, R.J.A.; Poll-The, B.T. Zellweger spectrum disorders: Clinical manifestations in patients surviving into adulthood. J. Inherit. Metab. Dis. 2016, 39, 93–106. [Google Scholar] [CrossRef] [PubMed]
- Iyer, L.M.; Leipe, D.D.; Koonin, E.V.; Aravind, L. Evolutionary history and higher order classification of AAA+ ATPases. J. Struct. Biol. 2004, 146, 11–31. [Google Scholar] [CrossRef]
- Miller, J.M.; Enemark, E.J. Fundamental Characteristics of AAA+ Protein Family Structure and Function. Archaea 2016, 2016, 12. [Google Scholar] [CrossRef]
- Wendler, P.; Ciniawsky, S.; Kock, M.; Kube, S. Structure and function of the AAA+ nucleotide binding pocket. Biochim. Et Biophys. Acta 2012, 1823, 2–14. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Bartesaghi, A.; Merk, A.; Rao, P.; Bulfer, S.L.; Yan, Y.; Green, N.; Mroczkowski, B.; Neitz, R.J.; Wipf, P.; et al. 2.3 Å resolution cryo-EM structure of human p97 and mechanism of allosteric inhibition. Science 2016, 351, 871–875. [Google Scholar] [CrossRef] [PubMed]
- Ciniawsky, S.; Grimm, I.; Saffian, D.; Girzalsky, W.; Erdmann, R.; Wendler, P. Molecular snapshots of the Pex1/6 AAA+ complex in action. Nat. Commun. 2015, 6, 7331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooney, I.; Han, H.; Stewart, M.G.; Carson, R.H.; Hansen, D.T.; Iwasa, J.H.; Price, J.C.; Hill, C.P.; Shen, P.S. Structure of the Cdc48 segregase in the act of unfolding an authentic substrate. Science 2019. [Google Scholar] [CrossRef] [PubMed]
- Monroe, N.; Han, H.; Shen, P.S.; Sundquist, W.I.; Hill, C.P. Structural basis of protein translocation by the Vps4-Vta1 AAA ATPase. eLife 2017, 6, e24487. [Google Scholar] [CrossRef] [PubMed]
- Ripstein, Z.A.; Huang, R.; Augustyniak, R.; Kay, L.E.; Rubinstein, J.L. Structure of a AAA+ unfoldase in the process of unfolding substrate. eLife 2017, 6, e25754. [Google Scholar] [CrossRef] [PubMed]
- White, K.I.; Zhao, M.; Choi, U.B.; Pfuetzner, R.A.; Brunger, A.T. Structural principles of SNARE complex recognition by the AAA+ protein NSF. eLife 2018, 7, e38888. [Google Scholar] [CrossRef] [PubMed]
- Rizo, A.N.; Lin, J.; Gates, S.N.; Tse, E.; Bart, S.M.; Castellano, L.M.; DiMaio, F.; Shorter, J.; Southworth, D.R. Structural basis for substrate gripping and translocation by the ClpB AAA+ disaggregase. Nat. Commun. 2019, 10, 2393. [Google Scholar] [CrossRef] [PubMed]
- Puchades, C.; Rampello, A.J.; Shin, M.; Giuliano, C.J.; Wiseman, R.L.; Glynn, S.E.; Lander, G.C. Structure of the mitochondrial inner membrane AAA+ protease YME1 gives insight into substrate processing. Science 2017, 358, eaao0464. [Google Scholar] [CrossRef] [Green Version]
- Blok, N.B.; Tan, D.; Wang, R.Y.-R.; Penczek, P.A.; Baker, D.; DiMaio, F.; Rapoport, T.A.; Walz, T. Unique double-ring structure of the peroxisomal Pex1/Pex6 ATPase complex revealed by cryo-electron microscopy. Proc. Natl. Acad. Sci. USA 2015, 112, E4017–E4025. [Google Scholar] [CrossRef] [Green Version]
- Gardner, B.M.; Chowdhury, S.; Lander, G.C.; Martin, A. The Pex1/Pex6 complex is a heterohexameric AAA+ motor with alternating and highly coordinated subunits. J. Mol. Biol. 2015, 427, 1375–1388. [Google Scholar] [CrossRef] [PubMed]
- Gardner, B.M.; Castanzo, D.T.; Chowdhury, S.; Stjepanovic, G.; Stefely, M.S.; Hurley, J.H.; Lander, G.C.; Martin, A. The peroxisomal AAA-ATPase Pex1/Pex6 unfolds substrates by processive threading. Nat. Commun. 2018, 9, 135. [Google Scholar] [CrossRef] [PubMed]
- Pedrosa, A.G.; Francisco, T.; Bicho, D.; Dias, A.F.; Barros-Barbosa, A.; Hagmann, V.; Dodt, G.; Rodrigues, T.A.; Azevedo, J.E. Peroxisomal monoubiquitinated PEX5 interacts with the AAA ATPases PEX1 and PEX6 and is unfolded during its dislocation into the cytosol. J. Biol. Chem. 2018, 293, 11553–11563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamura, S.; Yasutake, S.; Matsumoto, N.; Fujiki, Y. Dynamic and Functional Assembly of the AAA Peroxins, Pex1p and Pex6p, and Their Membrane Receptor Pex26p. J. Biol. Chem. 2006, 281, 27693–27704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyata, N.; Okumoto, K.; Mukai, S.; Noguchi, M.; Fujiki, Y. AWP1/ZFAND6 Functions in Pex5 Export by Interacting with Cys-Monoubiquitinated Pex5 and Pex6 AAA ATPase. Traffic 2012, 13, 168–183. [Google Scholar] [CrossRef]
- Shiozawa, K.; Maita, N.; Tomii, K.; Seto, A.; Goda, N.; Akiyama, Y.; Shimizu, T.; Shirakawa, M.; Hiroaki, H. Structure of the N-terminal Domain of PEX1 AAA-ATPase: CHARACTERIZATION OF A PUTATIVE ADAPTOR-BINDING DOMAIN. J. Biol. Chem. 2004, 279, 50060–50068. [Google Scholar] [CrossRef]
- Hänzelmann, P.; Schindelin, H. The Interplay of Cofactor Interactions and Post-translational Modifications in the Regulation of the AAA+ ATPase p97. Front. Mol. Biosci. 2017, 4, 21. [Google Scholar] [CrossRef]
- Twomey, E.C.; Ji, Z.; Wales, T.E.; Bodnar, N.O.; Ficarro, S.B.; Marto, J.A.; Engen, J.R.; Rapoport, T.A. Substrate processing by the Cdc48 ATPase complex is initiated by ubiquitin unfolding. Science 2019, eaax1033. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2017, 46, D1062–D1067. [Google Scholar] [CrossRef]
- Stenson, P.D.; Ball, E.V.; Mort, M.; Phillips, A.D.; Shiel, J.A.; Thomas, N.S.T.; Abeysinghe, S.; Krawczak, M.; Cooper, D.N. Human Gene Mutation Database (HGMD®): 2003 update. Hum. Mutat. 2003, 21, 577–581. [Google Scholar] [CrossRef]
- Fokkema, I.F.A.C.; Taschner, P.E.M.; Schaafsma, G.C.P.; Celli, J.; Laros, J.F.J.; den Dunnen, J.T. LOVD v.2.0: The next generation in gene variant databases. Hum. Mutat. 2011, 32, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Šali, A.; Blundell, T.L. Comparative Protein Modelling by Satisfaction of Spatial Restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein structure and function prediction. Nat. Methods 2014, 12, 7. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Zhang, Y. Ab initio protein structure assembly using continuous structure fragments and optimized knowledge-based force field. Proteins Struct. Funct. Bioinform. 2012, 80, 1715–1735. [Google Scholar] [CrossRef] [PubMed]
- Oates, M.E.; Romero, P.; Ishida, T.; Ghalwash, M.; Mizianty, M.J.; Xue, B.; Dosztányi, Z.; Uversky, V.N.; Obradovic, Z.; Kurgan, L.; et al. D2P2: Database of disordered protein predictions. Nucleic Acids Res. 2012, 41, D508–D516. [Google Scholar] [CrossRef] [PubMed]
- Reuber, B.E.; Germain-Lee, E.; Collins, C.S.; Morrell, J.C.; Ameritunga, R.; Moser, H.W.; Valle, D.; Gould, S.J. Mutations in PEX1 are the most common cause of peroxisome biogenesis disorders. Nat. Genet. 1997, 17, 445–448. [Google Scholar] [CrossRef] [PubMed]
- Geisbrecht, B.V.; Collins, C.S.; Reuber, B.E.; Gould, S.J. Disruption of a PEX1-PEX6 interaction is the most common cause of the neurologic disorders Zellweger syndrome, neonatal adrenoleukodystrophy, and infantile Refsum disease. Proc. Natl. Acad. Sci. USA 1998, 95, 8630–8635. [Google Scholar] [CrossRef]
- Collins, C.S.; Gould, S.J. Identification of a common PEX1 mutation in Zellweger syndrome. Hum. Mutat. 1999, 14, 45–53. [Google Scholar] [CrossRef]
- TAMURA, S.; MATSUMOTO, N.; IMAMURA, A.; SHIMOZAWA, N.; SUZUKI, Y.; KONDO, N.; FUJIKI, Y. Phenotype–genotype relationships in peroxisome biogenesis disorders of PEX1-defective complementation group 1 are defined by Pex1p–Pex6p interaction. Biochem. J. 2001, 357, 417–426. [Google Scholar]
- Steinberg, S.; Chen, L.; Wei, L.; Moser, A.; Moser, H.; Cutting, G.; Braverman, N. The PEX Gene Screen: Molecular diagnosis of peroxisome biogenesis disorders in the Zellweger syndrome spectrum. Mol. Genet. Metab. 2004, 83, 252–263. [Google Scholar] [CrossRef]
- Westberry, D.; Pugh, L. Zellweger syndrome: An older child with progressive foot deformity. J. Pediatric Genet. 2013, 2, 203–207. [Google Scholar]
- Maxwell, M.A.; Leane, P.B.; Paton, B.C.; Crane, D.I. Novel PEX1 coding mutations and 5′ UTR regulatory polymorphisms. Hum. Mutat. 2005, 26, 279. [Google Scholar] [CrossRef] [PubMed]
- Thoms, S.; Grønborg, S.; Rabenau, J.; Ohlenbusch, A.; Rosewich, H.; Gärtner, J. Characterization of two common 5′ polymorphisms in PEX1 and correlation to survival in PEX1 peroxisome biogenesis disorder patients. BMC Med. Genet. 2011, 12, 109. [Google Scholar] [CrossRef] [PubMed]
- Alshenaifi, J.; Ewida, N.; Anazi, S.; Shamseldin, H.E.; Patel, N.; Maddirevula, S.; Al-Sheddi, T.; Alomar, R.; Alobeid, E.; Ibrahim, N.; et al. The many faces of peroxisomal disorders: Lessons from a large Arab cohort. Clin. Genet. 2019, 95, 310–319. [Google Scholar] [CrossRef] [PubMed]
- Ebberink, M.S.; Koster, J.; Wanders, R.J.A.; Waterham, H.R. Spectrum of PEX6 mutations in Zellweger syndrome spectrum patients. Hum. Mutat. 2010, 31, 1058–1070. [Google Scholar] [CrossRef] [PubMed]
- Raas-Rothschild, A.; Wanders, R.J.A.; Mooijer, P.A.W.; Gootjes, J.; Waterham, H.R.; Gutman, A.; Suzuki, Y.; Shimozawa, N.; Kondo, N.; Eshel, G.; et al. A PEX6-Defective Peroxisomal Biogenesis Disorder with Severe Phenotype in an Infant, versus Mild Phenotype Resembling Usher Syndrome in the Affected Parents. Am. J. Hum. Genet. 2002, 70, 1062–1068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, C.E.L.; Poulter, J.A.; Levin, A.V.; Capasso, J.E.; Price, S.; Ben-Yosef, T.; Sharony, R.; Newman, W.G.; Shore, R.C.; Brookes, S.J.; et al. Spectrum of PEX1 and PEX6 variants in Heimler syndrome. Eur. J. Hum. Genet. 2016, 24, 1565–1571. [Google Scholar] [CrossRef] [PubMed]
- Grunert, M.; Dorn, C.; Schueler, M.; Dunkel, I.; Schlesinger, J.; Mebus, S.; Alexi-Meskishvili, V.; Perrot, A.; Wassilew, K.; Timmermann, B.; et al. Rare and private variations in neural crest, apoptosis and sarcomere genes define the polygenic background of isolated Tetralogy of Fallot. Hum. Mol. Genet. 2014, 23, 3115–3128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krause, C.; Rosewich, H.; Gärtner, J. Rational diagnostic strategy for Zellweger syndrome spectrum patients. Eur. J. Hum. Genet. 2009, 17, 741–748. [Google Scholar] [CrossRef] [PubMed]
- Falkenberg, K.D.; Braverman, N.E.; Moser, A.B.; Steinberg, S.J.; Klouwer, F.C.C.; Schlüter, A.; Ruiz, M.; Pujol, A.; Engvall, M.; Naess, K.; et al. Allelic Expression Imbalance Promoting a Mutant PEX6 Allele Causes Zellweger Spectrum Disorder. Am. J. Hum. Genet. 2017, 101, 965–976. [Google Scholar] [CrossRef] [Green Version]
- Yik, W.Y.; Steinberg, S.J.; Moser, A.B.; Moser, H.W.; Hacia, J.G. Identification of novel mutations and sequence variation in the Zellweger syndrome spectrum of peroxisome biogenesis disorders. Hum. Mutat. 2009, 30, E467–E480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamura, S.; Okumoto, K.; Toyama, R.; Shimozawa, N.; Tsukamoto, T.; Suzuki, Y.; Osumi, T.; Kondo, N.; Fujiki, Y. Human PEX1 cloned by functional complementation on a CHO cell mutant is responsible for peroxisome-deficient Zellweger syndrome of complementation group I. Proc. Natl. Acad. Sci. USA 2002, 95, 4350–4355. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.Y.; Chang, Y.P.; Park, J.Y.; Park, H.-D.; Sohn, Y.B.; Park, S.W.; Kim, S.J.S.H.; Ji, S.; Kim, S.J.S.H.; Choi, E.W.; et al. Zellweger spectrum disorder patient–derived fibroblasts with the PEX1-Gly843Asp allele recover peroxisome functions in response to flavonoids. Hum. Mutat. 2011, 120, 109. [Google Scholar]
- Najmabadi, H.; Hu, H.; Garshasbi, M.; Zemojtel, T.; Abedini, S.S.; Chen, W.; Hosseini, M.; Behjati, F.; Haas, S.; Jamali, P.; et al. Deep sequencing reveals 50 novel genes for recessive cognitive disorders. Nature 2011, 478, 57. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Suzuki, Y.; Shimozawa, N.; Fukuda, S.; Imamura, A.; Tsukamoto, T.; Osumi, T.; Fujiki, Y.; Orii, T.; Wanders, R.J.A.; et al. Genomic structure and identification of 11 novel mutations of the PEX6 (peroxisome assembly factor-2) gene in patients with peroxisome biogenesis disorders. Hum. Mutat. 1999, 13, 487–496. [Google Scholar] [CrossRef]
- Bousfiha, A.; Bakhchane, A.; Charoute, H.; Riahi, Z.; Snoussi, K.; Rouba, H.; Bonnet, C.; Petit, C.; Barakat, A. A novel PEX1 mutation in a Moroccan family with Zellweger spectrum disorders. Hum. Genome Var. 2017, 4, 17009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ratbi, I.; Jaouad, I.C.; Elorch, H.; Al-Sheqaih, N.; Elalloussi, M.; Lyahyai, J.; Berraho, A.; Newman, W.G.; Sefiani, A. Severe early onset retinitis pigmentosa in a Moroccan patient with Heimler syndrome due to novel homozygous mutation of PEX1 gene. Eur. J. Med. Genet. 2016, 59, 507–511. [Google Scholar] [CrossRef] [Green Version]
- Imamura, A.; Shimozawa, N.; Suzuki, Y.; Zhang, Z.; Tsukamoto, T.; Fujiki, Y.; Orii, T.; Osumi, T.; Wanders, R.J.A.; Kondo, N. Temperature-Sensitive Mutation of PEX6 in Peroxisome Biogenesis Disorders in Complementation Group C (CG-C): Comparative Study of PEX6 and PEX1. Pediatric Res. 2000, 48, 541–545. [Google Scholar] [CrossRef] [Green Version]
- Rydzanicz, M.; Stradomska, T.J.; Jurkiewicz, E.; Jamroz, E.; Gasperowicz, P.; Kostrzewa, G.; Płoski, R.; Tylki-Szymańska, A. Mild Zellweger syndrome due to a novel PEX6 mutation: Correlation between clinical phenotype and in silico prediction of variant pathogenicity. J. Appl. Genet. 2017, 58, 475–480. [Google Scholar] [CrossRef]
- Ogura, T.; Whiteheart, S.W.; Wilkinson, A.J. Conserved arginine residues implicated in ATP hydrolysis, nucleotide-sensing, and inter-subunit interactions in AAA and AAA+ ATPases. J. Struct. Biol. 2004, 146, 106–112. [Google Scholar] [CrossRef]
- Hänzelmann, P.; Schindelin, H. Structural Basis of ATP Hydrolysis and Intersubunit Signaling in the AAA+ ATPase p97. Structure 2016, 24, 127–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birschmann, I.; Rosenkranz, K.; Erdmann, R.; Kunau, W.-H. Structural and functional analysis of the interaction of the AAA-peroxins Pex1p and Pex6p. FEBS J. 2005, 272, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Augustin, S.; Gerdes, F.; Lee, S.; Tsai, F.T.F.; Langer, T.; Tatsuta, T. An Intersubunit Signaling Network Coordinates ATP Hydrolysis by m-AAA Proteases. Mol. Cell 2009, 35, 574–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, W.K.; Xia, D. Role of the D1-D2 Linker of Human VCP/p97 in the Asymmetry and ATPase Activity of the D1-domain. Sci. Rep. 2016, 6, 20037. [Google Scholar] [CrossRef] [PubMed]
- Johnston, D.M.; Miot, M.; Hoskins, J.R.; Wickner, S.; Doyle, S.M. Substrate Discrimination by ClpB and Hsp104. Front. Mol. Biosci. 2017, 4, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlieker, C.; Weibezahn, J.; Patzelt, H.; Tessarz, P.; Strub, C.; Zeth, K.; Erbse, A.; Schneider-Mergener, J.; Chin, J.W.; Schultz, P.G.; et al. Substrate recognition by the AAA+ chaperone ClpB. Nat. Struct. Mol. Biol. 2004, 11, 607–615. [Google Scholar] [CrossRef] [PubMed]
- DeLaBarre, B.; Christianson, J.C.; Kopito, R.R.; Brunger, A.T. Central Pore Residues Mediate the p97/VCP Activity Required for ERAD. Mol. Cell 2006, 22, 451–462. [Google Scholar] [CrossRef]
- Worth, C.L.; Blundell, T.L. On the evolutionary conservation of hydrogen bonds made by buried polar amino acids: The hidden joists, braces and trusses of protein architecture. BMC Evol. Biol. 2010, 10, 161. [Google Scholar] [CrossRef]
- Hänzelmann, P.; Schindelin, H. The Structural and Functional Basis of the p97/Valosin-containing Protein (VCP)-interacting Motif (VIM): MUTUALLY EXCLUSIVE BINDING OF COFACTORS TO THE N-TERMINAL DOMAIN OF p97. J. Biol. Chem. 2011, 286, 38679–38690. [Google Scholar] [CrossRef]
- Buentzel, J.; Vilardi, F.; Lotz-Havla, A.; Gärtner, J.; Thoms, S. Conserved targeting information in mammalian and fungal peroxisomal tail-anchored proteins. Sci. Rep. 2015, 5, 17420. [Google Scholar] [CrossRef] [Green Version]
- Lindquist, S.L.; Kelly, J.W. Chemical and Biological Approaches for Adapting Proteostasis to Ameliorate Protein Misfolding and Aggregation Diseases–Progress and Prognosis. Cold Spring Harb. Perspect. Biol. 2011, 3. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, F.W.; Sergeev, Y.V. Global computational mutagenesis of domain structures associated with inherited eye disease. Sci. Rep. 2019, 9, 3676. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.K.; Xia, D. Mutations in the Human AAA+ Chaperone p97 and Related Diseases. Front. Mol. Biosci. 2016, 3, 79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, J.M.M.; Deinhardt, K.; Rosewell, I.; Warren, G.; Shima, D.T. Targeted deletion of p97 (VCP/CDC48) in mouse results in early embryonic lethality. Biochem. Biophys. Res. Commun. 2007, 354, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Nazarko, T.Y. Pexophagy is responsible for 65% of cases of peroxisome biogenesis disorders. Autophagy 2017, 13, 991–994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Law, K.B.; Bronte-Tinkew, D.; Di Pietro, E.; Snowden, A.; Jones, R.O.; Moser, A.; Brumell, J.H.; Braverman, N.; Kim, P.K. The peroxisomal AAA ATPase complex prevents pexophagy and development of peroxisome biogenesis disorders. Autophagy 2017, 13, 868–884. [Google Scholar] [CrossRef] [PubMed]
- Nuttall, J.M.; Motley, A.M.; Hettema, E.H. Deficiency of the exportomer components Pex1, Pex6, and Pex15 causes enhanced pexophagy in Saccharomyces cerevisiae. Autophagy 2014, 10, 835–845. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, L.; Stephens, A.; Nam, S.-Z.; Rau, D.; Kübler, J.; Lozajic, M.; Gabler, F.; Söding, J.; Lupas, A.N.; Alva, V. A Completely Reimplemented MPI Bioinformatics Toolkit with a New HHpred Server at its Core. J. Mol. Biol. 2018, 430, 2237–2243. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Joseph, A.P.; Malhotra, S.; Burnley, T.; Wood, C.; Clare, D.K.; Winn, M.; Topf, M. Refinement of atomic models in high resolution EM reconstructions using Flex-EM and local assessment. Methods 2016, 100, 42–49. [Google Scholar] [CrossRef] [Green Version]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Soding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.A.; Clamp, M.; Barton, G.J. Jalview Version 2—A multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| I—Mutations Concerning ATP Binding and Hydrolysis | ||||||||||
| Domain | Putative SSE a | Concerned Motif b | Exon | cDNA Level | Protein Level | Clinical Significance c | Condition | Patient d | Database | Publication |
| Pex1 D1–D2 | 15 | c.2528G>A | p.G843D | p [40] | p.G843D/p.G843D, 2 years 9 months (d)/9 years (d), n/a [12]; p.G843D/p.G843D, 8–45 years (a), n/a [12] | [40,41,42] | [10,12,14,18,47,48,49,50,51,52] | |||
| Pex1 D2 | β1 | (Walker A) | 16 | c.2636T>C | p.L879S | u | ZSSD [13] | [41] | [13,18] | |
| β1–α1 | Walker A | 16 | c.2645C>T | p.P882L | u | n/a | [40] | |||
| α1 | Walker A | 16 | c.2654C>G | p.T885R | u | ZSSD [13] | [41] | [13] | ||
| α4 | ISS | 19 | c.2993G>A | p.R998Q | p(−) [53] | ZSSD [53] | p.I989T/p.R998Q, 127 months (d), ZS [53] | [41,42] | [53] | |
| α5 | 20 | c.3038G>A | p.R1013H | u | ZSSD | p.R1013H/p.S1096X, 4 months (d), NALD [11] | [40,41] | [11] | ||
| α5 | 20 | c.3037C>G | p.R1013G | u | ZSSD [54] | p.R1013G/p.I700Yfs42X, <10 months (d), n/a [54] | [41] | [54] | ||
| α5 | 20 | c.3037C>T | p.R1013C | p(−) [55] | ZSSD [55] | p.R1013C/p.R1013C, 1 month (d)/20–36 months (a), n/a (5 patients) [55] | [41] | [55] | ||
| Pex6 D1 | β1 | (Walker A) | 6 | c.1405C>T | p.R469W | u | n/a | [40] | ||
| β1–α1 | Walker A | 6 | c.1409G>C | p.G470A | u | ZSSD | [40] | |||
| β1–α1 | Walker A | 6 | c.1417G>A | p.G473S | u | n/a | [40] | |||
| β4 | (sensor 1) | 8 | c.1711G>A | p.A571T | u | ZSSD [56] | [41,42] | [56] | ||
| β4–α4 | (sensor 1) | 8 | c.1715C>T | p.T572I | p [40] | ZSSD [57], HS [58] | p.T572I/p.T572I, adult (a), IRD [57]; p.T572I/splice variant, 17 months (d), ZS–NALD [57]; p.T572I/p.T572I, 22 years (a), HS [58]; p.T572I/p.T572I, 35 years (a), HS [58] | [40,41,42] | [57,58] | |
| β4–α4 | sensor 1 | 8 | c.1718C>T | p.T573I | u | ZSSD | [40,41] | [59] | ||
| Pex6 D1–D2 | 11 | c.2104G>A | p.V702M | u | n/a | [40] | ||||
| 11 | c.2120T>G | p.V707G | u | ZSSD [60] | [41,42] | [60] | ||||
| Pex6 D2 | β1 | (Walker A) | 11 | c.2225T>C | p.L742P | u | ZSSD | [40,41,42] | [56] | |
| α4 | arginine finger | 14 | c.2578C>T | p.R860W | p [61] | ZSSD | p.R860W/p.P285A, n/a, IRD [42]; p.R860W/WT, n/a, n/a [62]; p.R860W/WT, 8–20 years (d), ZSSD [61] | [41,42] | [61,62] | |
| α4 | arginine finger | 14 | c.2579G>A | p.R860Q | u | ZSSD [62] | p.R860Q/p.R601Q, n/a, IRD [42,62] | [41,42] | [62] | |
| α7 | 16 | c.2735C>T | p.A912V | p(−) [42] | ZSSD [42] | p.A912V/p.A912V, n/a, NALD [42] | [42] | [18] | ||
| II—Mutations Concerning Substrate Processing | ||||||||||
| Domain | Putative SSE a | Concerned Motif b | Exon | cDNA Level | Protein Level | Clinical Significance c | Condition | Patient d | Data Base | Publication |
| Pex1 D1 | α2 | pore loop 1 | 12 | c.1913A>C | p.E638A | u | n/a | [40] | ||
| α3 | (pore loop 2) | 12 | c.1991T>C | p.L664P | p(−) [63] | ZSSD | p.L664P/p.634del690, 2 months (d), ZS [63] | [40,42] | [11,63] | |
| Pex1 D2 | α3 | (pore loop 2) | 18 | c.2843G>A | p.R948Q | u | ZSSD [62] | Pex1:p.R948Q/WT, Pex26:p.R98W/p.R98W, n/a, n/a [62] | [40,41] | [62] |
| α3 | (pore loop 2) | 18 | c.2842C>T | p.R948W | u | ZSSD | [40] | |||
| α3 | pore loop 2 | 18 | c.2846G>A | p.R949Q | p(−) [12] | ZSSD | p.G843D/p. R949Q, 3 months (d), ZS [12]; p.R949Q/?, n/a, ZS [11] | [40,41,42] | [11,12] | |
| α3 | pore loop 2 | 18 | c.2845C>T | p.R949W | u | ZSSD | p.R949W/p.V336A/p.S555P, n/a, n/a [13,18]; p.R494W/p.H678fsX3, 2 months (a), ZS [64] | [40,41] | [13,18,64] | |
| α3 | pore loop 2 | 18 | c.2876G>C | p.R959P | u | ZSSD | [40] | |||
| α3 | pore loop 2 | 18 | c.2876G>A | p.R959Q | u | n/a | [40] | |||
| Pex6 D1 | α2 | pore loop 1 | 7 | c.1511G>A | p.S504N | u | n/a | [40] | ||
| α3 | (pore loop 2) | 7 | c.1601T>C | p.L534P | p [40] | ZSSD | [40] | [65] | ||
| Pex6 D2 | β3 | pore loop 2 | 13 | c.2434C>T | p.R812W | p [66] | ZSSD [66] | p.R812W/p.R601Q, 3 years 6 months (a), nonclassical ZS [42,66] | [41,42] | [66] |
| β3 | pore loop 2 | 13 | c.2345G>A | p.R812Q | p(−) [40] | ZSSD [66] | p.R812Q/splice variant, n/a, ZS [66] | [40,41,42] | [66] | |
| III—Mutations Concerning the Interaction Between Pex1 and Pex6 | ||||||||||
| Domain | Putative SSE a | Exon | cDNA Level | Protein Level | Clinical Significance c | Condition | Patient d | Data Base | Publication | |
| Pex1 D1 | α0 | 10 | c.1742G>C | p.R581P | p [40] | HS [3] | p.R581P/splice variant, 19 years (a), HS [3]; p.R581P/p.I700YfsX42, 24 years (a), HS [3] | [40] | [3] | |
| α0 | 10 | c.1769T>G | p.L590R | p(−) [62] | ZSSD [62] | p.L590R/p.L590R, n/a, n/a [62] | [41] | [62] | ||
| α3 | 13 | c.2088A>G | p.I696M | b [40] | n/a | [40,42] | [51] | |||
| α3–β4 | 13 | c.2114T>G | p.L705W | u | HS [3] | p.L705W/p.I700YfsX42, 29 and 31 years (a), HS [3] | [40] | [3] | ||
| α5–α6 | 14 | c.2271G>C | p.L757F | b [40] | n/a | [40] | ||||
| α7 | 14 | c.2387T>C | p.L796P | u | p.L796P/p.S304CfsX4, 5 months (a), ZS [11] | [41] | [11] | |||
| α7 | 14 | c.2392C>G | p.R798G | u | p.R798G/p.G843D, 15 months (d), ZS [14] | [41,42] | [14,53] | |||
| Pex1 D2 | α3 | 18 | c.2894T>C | p.L965P | p(−) [40] | n/a | [40] | |||
| β4–α4 | 19 | c.2966T>C | p.I989T | c [40] | ZSSD | p.I989T/p.R998Q, 127 months (d), ZS [53]; p.I989T/p.p.Q598TfsX11, 45 years (a), HS [58] | [40,41,42] | [53,58] | ||
| α5 | 20 | c.3031G>A | p.V1011M | c [40] | ZSSD | [40] | ||||
| α5–α6 | 20 | c.3077T>C | p.L1026P | p(−) [67] | ZSSD | p.L1026P/p.L1026P, 6 years (a)/12.5 years (d), NALD [67] | [40] | [67] | ||
| Pex1 C-term | 23 | c.3691_3694delCAGT | p.Q1231HfsX3 | p(−) [11] | ZSSD [11] | p.Q1231HfsX3/p.Q1231HfsX3, 2 months (d), ZS [11] | [11] | |||
| 23 | c.3750G>A | p.W1250X | p(−) [68] | HS | p.W1250X/p.W1250X, 12 and 16 years (a), HS [68] | [68] | ||||
| Pex6 D1 | α5 | 8 | c.1802G>A | p.R601Q | c [40] | ZSSD, HS | p.R601Q/p.L614RfsX5, 21 years (a), HS [3]; p.R601Q/p.R860Q, n/a, IRD [62]; p.R601Q/p.R812W, 3 years 6 months (a), nonclassical ZS [42,66] | [40,41,42] | [3,56,58,62,66] | |
| α5 | 8 | c.1814T>G | p.L605R | u | ZSSD [56] | [41,42] | [56] | |||
| α7 | 9 | c.1930C>T | p.R644W | p [40] | HS | p.P274L/p.R644W, 21 years (a), HS [3]; |p.P274L/p.R644W, 16 years (a), HS [3] | [40] | [3] | ||
| α7–α8 | 10 | c.1992G>C | p.E664D | u | ZSSD [56] | [41,42] | [13,18] | |||
| α8 | 10 | c.2048T>C | p.L683P | u | ZSSD [60] | [41,42] | [60] | |||
| Pex6 D2 | α2 | 12 | c.2356C>T | p.R786W | u | ZSSD [56] | [40,41,42] | [56] | ||
| α3 | 13 | c.2426C>T | p.A809V | b [40,56] | ZSSD [56] | splice variant/p.A809V/p.I845T, adult (a), n/a [57] | [40,41,42] | [56,57,60] | ||
| α5 | 15 | c.2644G>A | p.V882I | b [40] | n/a | [40,42] | ||||
| α6–α7 | 16 | c.2726T>A | p.L909Q | u | ZSSD [56] | [41,42] | [56] | |||
| α7 | 16 | c.2770G>T | p.A924S | b(−) [40] | n/a | [40,42] | ||||
| α8 | 17 | c.2816C>A | p.P939Q | b [56,62] | n/a | [40,42] | [56,62] | |||
| IV—Mutations Concerning the Interaction with Cofactors | ||||||||||
| Domain | Exon | cDNA Level | Protein Level | Clinical Significance c | Condition | Patient d | Data Base | Publication | ||
| Pex1 N-term | 3 | c.274G>C | p.V92L | p(−) [11] | ZSSD [11] | p.V92L/p.V92L, 1 year 11 months (d), nonclassical ZS [11] | [41] | [11] | ||
| Pex6 N1 | 1 | c.170T>C | p.L57P | p(−) [69] | ZSSD [69] | p.L57P/p.L57P, n/a, NALD [69] | [41,42] | [69] | ||
| 1 | c.275T>G | p.V92G | p [40] | ZSSD | p.R92L/p.R601Q, 12 years (a), HS [58] | [40] | [58] | |||
| 1 | c.277C>G | p.R93G | u | ZSSD [56] | [41,42] | [56] | ||||
| 1 | c280G>C | p.A94P | p(−) [70] | ZSSD [70] | p.A94P/p.A94P, 6 years (d), mild ZS [70] | [70] | ||||
| 1 | c.281C>T | p.A94E | u | ZSSD [51] | [41] | [51] | ||||
| 1 | c.281C>A | p.A94K | u | ZSSD [51] | [42] | [51] | ||||
| 1 | c.296G>T | p.R99L | p [40] | ZSSD | p.R99L/R601Q, 7 years (a), HS [58] | [40] | [58] | |||
| Pex6 N2 | 1 | c.654C>G | p.F218L | p(−) [58] | ZSSD | p.F218L/p.R601Q, n/a, HS [58] | [40] | [58] | ||
| 1 | c.656A>C | p.Q219P | u | ZSSD | [41,42] | [56] | ||||
| 1 | c.659G>T | p.G220V | u | ZSSD | [40,41,42] | [56] | ||||
| 1 | c.821C>T | p.P274L | p [40] | ZSSD [62], HS [3] | P274L/R644W, 21 years (a), HS [3]; P274L/R644W, 16 years (a), HS [3]; P274L/E439fsX3, n/a, n/a [15]; P274L/splice variant, n/a, n/a [62] | [40,41,42] | [3,15,51,62] | |||
| Domain | Residues b | Model a | Resource a | Range a | Templates a |
|---|---|---|---|---|---|
| Pex1 N1 | 1–179 | 1 | iTASSER | 1–399 | 1WLF (88%), 3HU3 (13%), 3JC8 (9%), 4KO8 (12%), 1TZL (8%) |
| 178–399 | |||||
| Pex1 N2 | 410–542 | 2 | MODELLER | 1–1238 | 1WLF (89%), 5E7P (32.9%), 5VC7 (38.5%), 5G4F (32.9%), 5KWA (36.3%), 6MAT (30.6%), 6MCK (36.1%) |
| Pex1 D1 | 563–818 | ||||
| Pex1 D2 | 846–1090 | 3 | iTASSER | 838–1238 | 5G4F (49%), 6MAT (23%), 6AZ0 (23%), 5VC7 (48%) |
| Pex1 C-terminus | 1104–1283 | ||||
| Pex6 N1 | 1 to about 180 | 4 | QUARK | 1–200 | |
| Pex6 N2 | 191–413 | 5 | MODELLER | 200–400 | 4RV0 (18.9%), 5B6C (15.8%), 1CZ4 (15.5%), 5G4F (13.6%), 1WLF (17.2%), 1ZC1 (13.8%), 2YUJ (12.3%), 5E7P (16.5%), 1QCS (10.1%), 5FTJ (15.1%) |
| Pex6 D1 | 440–687 | 6 | MODELLER | 1–980 | 5FTJ (27.6%), 5G4F (28.2%), 5E7P (25.3%), 6MCK (33.1%), 6MAT (30.8%) |
| Pex6 D2 | 709–958 | ||||
| Pex6 C-terminus | 963–980 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schieferdecker, A.; Wendler, P. Structural Mapping of Missense Mutations in the Pex1/Pex6 Complex. Int. J. Mol. Sci. 2019, 20, 3756. https://doi.org/10.3390/ijms20153756
Schieferdecker A, Wendler P. Structural Mapping of Missense Mutations in the Pex1/Pex6 Complex. International Journal of Molecular Sciences. 2019; 20(15):3756. https://doi.org/10.3390/ijms20153756
Chicago/Turabian StyleSchieferdecker, Anne, and Petra Wendler. 2019. "Structural Mapping of Missense Mutations in the Pex1/Pex6 Complex" International Journal of Molecular Sciences 20, no. 15: 3756. https://doi.org/10.3390/ijms20153756
APA StyleSchieferdecker, A., & Wendler, P. (2019). Structural Mapping of Missense Mutations in the Pex1/Pex6 Complex. International Journal of Molecular Sciences, 20(15), 3756. https://doi.org/10.3390/ijms20153756