Lipid and Lipid Raft Alteration in Aging and Neurodegenerative Diseases: A Window for the Development of New Biomarkers



 and
and 
Abstract
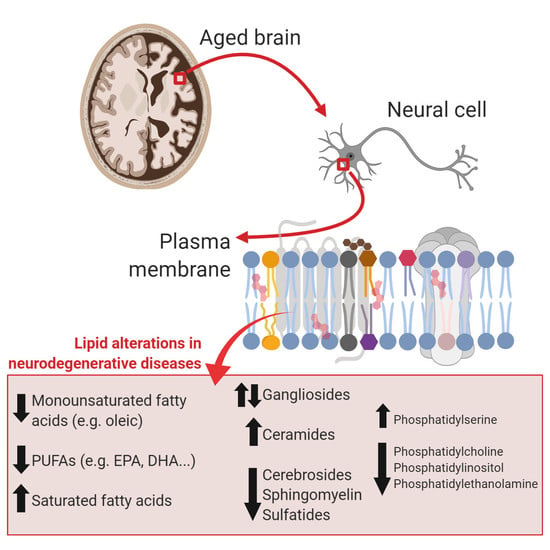
:
1. Introduction
2. Brain Lipids in Aging
2.1. Lipid and Lipid Raft Profile Alterations in Alzheimer’s Disease
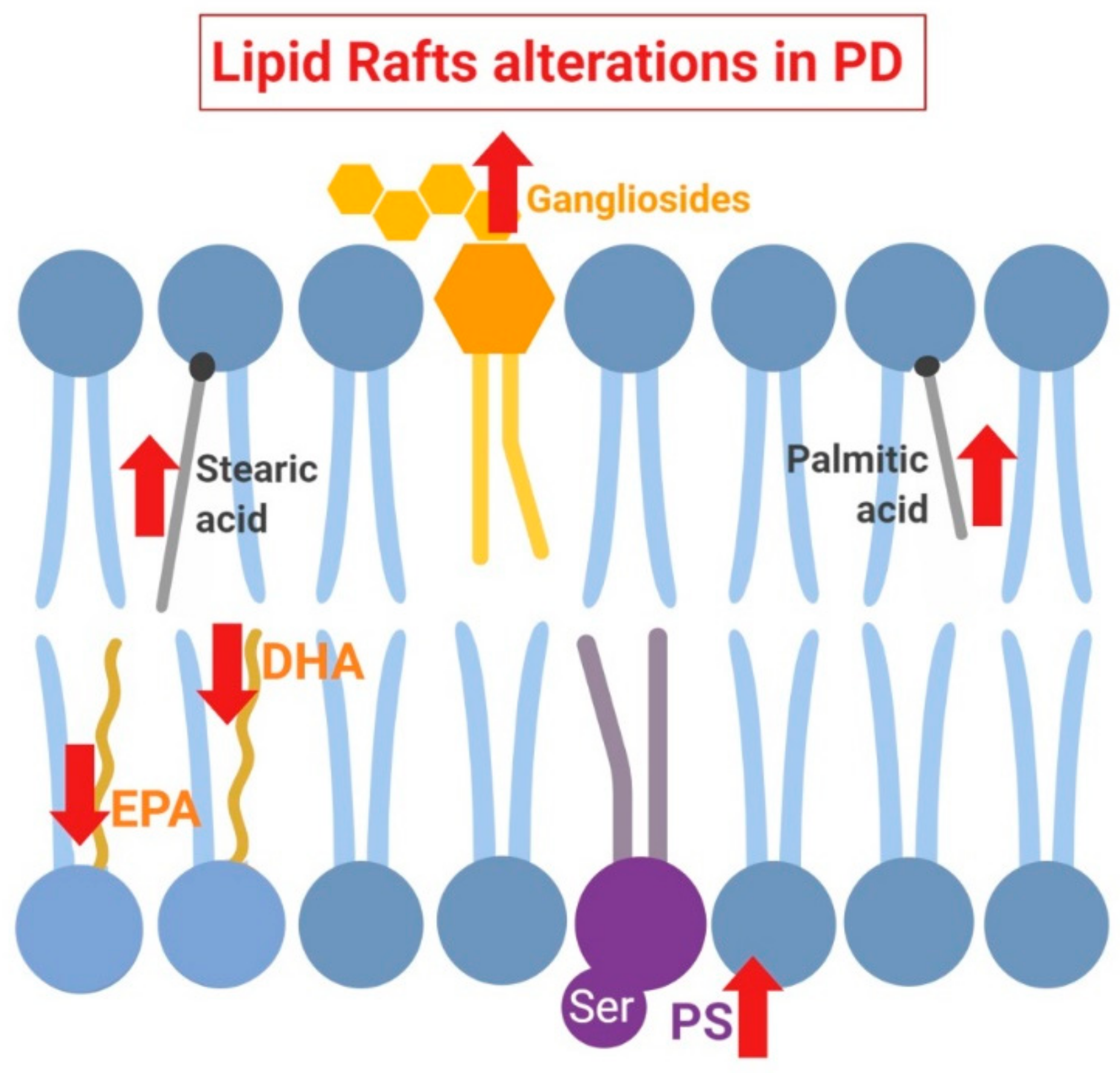
2.2. Lipids and Lipid Raft Profile Alterations in Parkinson’s Disease
3. Lipids Alterations in Lipid Rafts as Potential Biomarkers for Neurodegenerative Diseases
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 24-OH Cho | 24-hydroxycholesterol |
| 27-OH Cho | 27- hydroxycholesterol |
| 8-OHdG | 8-hydroxyguanosine oxized |
| 4-HNE | 4-hydroxynonenal |
| 8-OHG | 8-hydroxyguanine |
| AA | Arachidonic acid |
| AD | Alzheimer’s Disease |
| ALA | Linolenic acid |
| Aβ | β-amyloid peptide |
| B4GALNT1 | beta-1,4 N-acetylgalactosaminyltransferase 1 |
| CNS | Central nervous system |
| cPLA2 | Cytosolic phospholipase A2 |
| CSF | Cerebrospinal fluid |
| DHA | Docosahexaenoic acid |
| DHEA | Dehydroepiandrosterone |
| DLBD | Dementia with Lewy bodies disease |
| EPA | Eicosapentaenoic acid |
| FABP | Fatty acid-binding protein |
| GM1 | Ganglioside 1 |
| GM2 | Ganglioside 2 |
| GPI | Glycosylphosphatidylinositol |
| LA | Linoleic acid |
| LB | Lewy Body |
| MDA | Malondialdehyde |
| MCI | Mild cognitive impairment |
| NFTs | Neurofibrillary tangles |
| PC | phosphatidylcholine |
| PD | Parkinson’s Disease |
| PE | Phosphatidylethanolamine |
| PG | Phosphatidylglycerol |
| PI | Phosphatidylinositol |
| PREG | Pregnenolone |
| PS | Phosphatidylserine |
| PUFA | Polyunsaturated fatty acid |
| ROS | Reactive oxygen species |
| TBARS | Thiobarbituric acid-reactive substances |
| sAPPβ | Soluble amyloid precursor protein |
| SMase | Sphingomyelinase |
| SNpc | substantia nigra pars compacta |
| Sphk1 | Sphingosine Kinase |
| PSP | Progressive supranuclear palsy |
| MSA | Multiple system atrophy |
| HDL | High density lipoprotein |
References
- Kalvodova, L.; Kahya, N.; Schwille, P.; Ehehalt, R.; Verkade, P.; Drechsel, D.; Simons, K. Lipids as Modulators of Proteolytic Activity of BACE: Involvement of Cholesterol, Glycosphingolipids, and Anionic Phospholipids in Vitro. J. Biol. Chem. 2005, 280, 36815–36823. [Google Scholar] [CrossRef] [PubMed]
- Vetrivel, K.S.; Thinakaran, G. Membrane Rafts in Alzheimer’s Disease Beta-Amyloid Production. Biochim. Biophys. Acta—Mol. Cell Biol. Lipids 2010, 1801, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Spener, F.; Dennis, E.A.; Raetz, C.R.H.; Nishijima, M.; Subramaniam, S.; van Meer, G.; Shimizu, T.; Wakelam, M.J.O.; Murphy, R.C.; Fahy, E. Update of the LIPID MAPS Comprehensive Classification System for Lipids. J. Lipid Res. 2008, 50, S9–S14. [Google Scholar] [CrossRef]
- Cermenati, G.; Mitro, N.; Audano, M.; Melcangi, R.C.; Crestani, M.; De Fabiani, E.; Caruso, D. Lipids in the Nervous System: From Biochemistry and Molecular Biology to Patho-Physiology. Biochim. Biophys. Acta—Mol. Cell Biol. Lipids 2015, 1851, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Saeed, A.A.; Genové, G.; Li, T.; Lütjohann, D.; Olin, M.; Mast, N.; Pikuleva, I.A.; Crick, P.; Wang, Y.; Griffiths, W.; et al. Effects of a Disrupted Blood-Brain Barrier on Cholesterol Homeostasis in the Brain. J. Biol. Chem. 2014, 289, 23712–23722. [Google Scholar] [CrossRef] [Green Version]
- Di Scala, C.; Troadec, J.D.; Lelièvre, C.; Garmy, N.; Fantini, J.; Chahinian, H. Mechanism of Cholesterol-Assisted Oligomeric Channel Formation by a Short Alzheimer β-Amyloid Peptide. J. Neurochem. 2014, 128, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Varilly, P.; Chandler, D. APOE and Cholesterol in Aging and Disease in the Brain. J. Stat. Phys. 2012, 145, 265–275. [Google Scholar] [CrossRef]
- Wong, M.W.; Braidy, N.; Poljak, A.; Pickford, R.; Thambisetty, M.; Sachdev, P.S. Dysregulation of Lipids in Alzheimer’s Disease and Their Role as Potential Biomarkers. Alzheimer’s Dement. 2017, 13, 810–827. [Google Scholar] [CrossRef]
- Naudí, A.; Cabré, R.; Jové, M.; Ayala, V.; Gonzalo, H.; Portero-Otín, M.; Ferrer, I.; Pamplona, R. Lipidomics of Human Brain Aging and Alzheimer’s Disease Pathology. Int. Rev. Neurobiol. 2015, 122, 133–189. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, J.A.; Brunaldi, K. A Model for Fatty Acid Transport into the Brain. J. Mol. Neurosci. 2007, 33, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, J.A.; Guo, W.; Kamp, F. Mechanism of Cellular Uptake of Long-Chain Fatty Acids: Do We Need Cellular Proteins? Mol. Cell. Biochem. 2002, 239, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, R.W.; Hatch, G.M. Fatty Acid Transport into the Brain: Of Fatty Acid Fables and Lipid Tails. Prostaglandins Leukot. Essent. Fat. Acids 2011, 85, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Trigatti, B.L.; Anderson, R.G.W.; Gerber, G.E. Identification of Caveolin-1 as a Fatty Acid Binding Protein. Biochem. Biophys. Res. Commun. 1999, 255, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, R.W.; On, N.H.; Del Bigio, M.R.; Miller, D.W.; Hatch, G.M. Fatty Acid Transport Protein Expression in Human Brain and Potential Role in Fatty Acid Transport across Human Brain Microvessel Endothelial Cells. J. Neurochem. 2011, 117, 735–746. [Google Scholar] [CrossRef] [PubMed]
- Hooijmans, C.R.; Kiliaan, A.J. Fatty Acids, Lipid Metabolism and Alzheimer Pathology. Eur. J. Pharmacol. 2008, 585, 176–196. [Google Scholar] [CrossRef] [PubMed]
- Fester, L.; Zhou, L.; Bütow, A.; Huber, C.; Von Lossow, R.; Prange-Kiel, J.; Jarry, H.; Rune, G.M. Cholesterol-Promoted Synaptogenesis Requires the Conversion of Cholesterol to Estradiol in the Hippocampus. Hippocampus 2009, 19, 692–705. [Google Scholar] [CrossRef] [PubMed]
- Hadders-Algra, M. Prenatal Long-Chain Polyunsaturated Fatty Acid Status: The Importance of a Balanced Intake of Docosahexaenoic Acid and Arachidonic Acid. J. Perinat. Med. 2008, 36, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Ruipérez, V.; Darios, F.; Davletov, B. Alpha-Synuclein, Lipids and Parkinson’s Disease. Prog. Lipid Res. 2010, 49, 420–428. [Google Scholar] [CrossRef] [PubMed]
- Tillman, T.S.; Cascio, M. Effects of Membrane Lipids on Ion Channel Structure and Function. Cell Biochem. Biophys. 2003, 38, 161–190. [Google Scholar] [CrossRef]
- Berkecz, R.; Kovács, Z.; Penke, B.; VÍgh, L.; Crul, T.; Paragi, G.; Gera, J. The Role of Lipids and Membranes in the Pathogenesis of Alzheimer’s Disease: A Comprehensive View. Curr. Alzheimer Res. 2018, 15, 1191–1212. [Google Scholar] [CrossRef]
- Sonnino, S.; Prinetti, A. Membrane Domains and the “Lipid Raft” Concept. Curr. Med. Chem. 2012, 20, 4–21. [Google Scholar] [CrossRef]
- Dart, C. Lipid Microdomains and the Regulation of Ion Channel Function. J. Physiol. 2010, 588, 3169–3178. [Google Scholar] [CrossRef] [PubMed]
- Michel, V.; Bakovic, M. Lipid Rafts in Health and Disease. Biol. Cell 2007, 99, 129–140. [Google Scholar] [CrossRef]
- Levental, I.; Grzybek, M.; Simons, K. Greasing Their Way: Lipid Modifications Determine Protein Association with Membrane Rafts. Biochemistry 2010, 6305–6316. [Google Scholar] [CrossRef]
- Pike, L.J. The Challenge of Lipid Rafts. J. Lipid Res. 2008, 50, S323–S328. [Google Scholar] [CrossRef]
- Marin, R.; Fabelo, N.; Fernández-Echevarría, C.; Canerina-Amaro, A.; Rodríguez-Barreto, D.; Quinto-Alemany, D.; Mesa-Herrera, F.; Díaz, M. Lipid Raft Alterations in Aged-Associated Neuropathologies. Curr. Alzheimer Res. 2016, 13, 973–984. [Google Scholar] [CrossRef] [Green Version]
- Díaz, M.; Fabelo, N.; Martín, V.; Ferrer, I.; Gómez, T.; Marín, R. Biophysical Alterations in Lipid Rafts from Human Cerebral Cortex Associate with Increased BACE1/AβPP Interaction in Early Stages of Alzheimer’s Disease. J. Alzheimer’s Dis. 2014, 43, 1185–1198. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Michaelis, M.L.; Michaelis, E.K. Functional genomics of brain aging and Alzheimer’s disease: focus on selective neuronal vulnerability. Curr. Genom. 2010, 11, 618–633. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P.; Magnus, T. Ageing and Neuronal Vulnerability. Nat. Rev. Neurosci. 2006, 7, 278–294. [Google Scholar] [CrossRef]
- Burger, M.; Seidel, K. Chemical Biomorphosis of the Human Brain and Sciatic Nerve; a Survey. Z. Alternsforsch. 1958, 12, 52–79. [Google Scholar] [PubMed]
- Rouser, G.; Yamamoto, A. Curvilinear Regression Course of Human Brain Lipid Composition Changes with Age. Lipids 1968, 3, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Söderberg, M.; Edlund, C.; Kristensson, K.; Dallner, G. Lipid Compositions of Different Regions of the Human Brain During Aging. J. Neurochem. 1990, 54, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Svennerholm, L.; Boström, K.; Helander, C.G.; Jungbjer, B. Membrane Lipids in the Aging Human Brain. J. Neurochem. 1991, 56, 2051–2059. [Google Scholar] [CrossRef] [PubMed]
- Thelen, K.M.; Falkai, P.; Bayer, T.A.; Lütjohann, D. Cholesterol Synthesis Rate in Human Hippocampus Declines with Aging. Neurosci. Lett. 2006, 403, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Dietschy, J.M.; Turley, S.D. Thematic Review Series: Brain Lipids. Cholesterol Metabolism in the Central Nervous System during Early Development and in the Mature Animal. J. Lipid Res. 2004, 45, 1375–1397. [Google Scholar] [CrossRef] [PubMed]
- Martín, V.; Fabelo, N.; Santpere, G.; Puig, B.; Marín, R.; Ferrer, I.; Díaz, M. Lipid Alterations in Lipid Rafts from Alzheimer’s Disease Human Brain Cortex. J. Alzheimer’s Dis. 2010, 19, 489–502. [Google Scholar] [CrossRef]
- Chou, Y.; Lin, S.; Hsin, L.; Tsai, H.; Mei, C. Cholesterol Deficiency Increases the Vulnerability of Hippocampal Glia in Primary Culture to Glutamate-Induced Excitotoxicity. Neurochem. Int. 2003, 43, 197–209. [Google Scholar] [CrossRef]
- Brinton, R.D. Neurosteroids as Regenerative Agents in the Brain: Therapeutic Implications. Nat. Publ. Gr. 2013, 9, 241–250. [Google Scholar] [CrossRef]
- Farooqui, A.A.; Liss, L.; Horrocks, L.A. Neurochemical Aspects of Alzheimer’s Disease: Involvement of Membrane Phospholipids. Metab. Brain Dis. 1988, 3, 19–35. [Google Scholar] [CrossRef]
- Svennerholm, L.; Boström, K.; Jungbjer, B.; Olsson, L. Membrane Lipids of Adult Human Brain: Lipid Composition of Frontal and Temporal Lobe in Subjects of Age 20 to 100 Years. J. Neurochem. 2010, 63, 1802–1811. [Google Scholar] [CrossRef]
- Norris, S.E.; Friedrich, M.G.; Mitchell, T.W.; Truscott, R.J.W.; Else, P.L. Human Prefrontal Cortex Phospholipids Containing Docosahexaenoic Acid Increase during Normal Adult Aging, Whereas Those Containing Arachidonic Acid Decrease. Neurobiol. Aging 2015, 36, 1659–1669. [Google Scholar] [CrossRef]
- Hancock, S.E.; Friedrich, M.G.; Mitchell, T.W.; Truscott, R.J.W.; Else, P.L. Decreases in Phospholipids Containing Adrenic and Arachidonic Acids Occur in the Human Hippocampus over the Adult Lifespan. Lipids 2015, 50, 861–872. [Google Scholar] [CrossRef]
- Else, P.L.; Hancock, S.E.; Friedrich, M.G.; Mitchell, T.W.; Truscott, R.J.W. The Phospholipid Composition of the Human Entorhinal Cortex Remains Relatively Stable over 80 Years of Adult Aging. GeroScience 2017, 39, 73–82. [Google Scholar] [CrossRef]
- Rider, T.; Tso, P.; Liu, Y.; Jandacek, R.; McNamara, R.K. The Aging Human Orbitofrontal Cortex: Decreasing Polyunsaturated Fatty Acid Composition and Associated Increases in Lipogenic Gene Expression and Stearoyl-CoA Desaturase Activity. Prostaglandins Leukot. Essent. Fat. Acids 2008, 78, 293–304. [Google Scholar] [CrossRef]
- Venable, M.E.; Webb-Froehlich, L.M.; Sloan, E.F.; Thomley, J.E. Shift in Sphingolipid Metabolism Leads to an Accumulation of Ceramide in Senescence. Mech. Ageing Dev. 2006, 127, 473–480. [Google Scholar] [CrossRef]
- Kracun, I.; Rosner, H.; Drnovsek, V.; Vukelic, Z.; Cosovic, C.; Trbojevic-Cepe, M.; Kubat, M. Gangliosides in the Human Brain Development and Aging. Neurochem. Int. 1992, 20, 421–431. [Google Scholar] [CrossRef]
- Vanmierlo, T.; Lütjohann, D.; Mulder, M. Brain Cholesterol in Normal and Pathological Aging. OCL—Ol. Corps Gras Lipides 2011, 18, 214–217. [Google Scholar] [CrossRef]
- Colin, J.; Gregory-Pauron, L.; Lanhers, M.C.; Claudepierre, T.; Corbier, C.; Yen, F.T.; Malaplate-Armand, C.; Oster, T. Membrane Raft Domains and Remodeling in Aging Brain. Biochimie 2016, 130, 178–187. [Google Scholar] [CrossRef]
- Marin, R.; Fabelo, N.; Martín, V.; Garcia-Esparcia, P.; Ferrer, I.; Quinto-Alemany, D.; Díaz, M. Anomalies Occurring in Lipid Profiles and Protein Distribution in Frontal Cortex Lipid Rafts in Dementia with Lewy Bodies Disclose Neurochemical Traits Partially Shared by Alzheimer’s and Parkinson’s Diseases. Neurobiol. Aging 2017, 49, 52–59. [Google Scholar] [CrossRef]
- Yamamoto, N.; Igbabvoa, U.; Shimada, Y.; Ohno-iwashita, Y.; Kobayashi, M.; Wood, W.G.; Fujita, S.C.; Yanagisawa, K. Accelerated Aβ Aggregation in the Presence of GM1-Ganglioside-Accumulated Synaptosomes of Aged ApoE4-Knock-in Mouse Brain. FEBS Lett. 2004, 569, 135–139. [Google Scholar] [CrossRef]
- Echeverría, F.; Valenzuela, R.; Catalina Hernandez-Rodas, M.; Valenzuela, A. Docosahexaenoic Acid (DHA), a Fundamental Fatty Acid for the Brain: New Dietary Sources. Prostaglandins Leukot. Essent. Fat. Acids 2017, 124, 1–10. [Google Scholar] [CrossRef]
- Díaz, M.; Fabelo, N.; Ferrer, I.; Marín, R. “Lipid raft aging” in the human frontal cortex during nonpathological aging: gender influences and potential implications in Alzheimer’s disease. Neurobiol. Aging 2018. [Google Scholar] [CrossRef]
- Cutuli, D. Functional and Structural Benefits Induced by Omega-3 Polyunsaturated Fatty Acids During Aging. Curr. Neuropharmacol. 2017, 15 , 534–542. [Google Scholar] [CrossRef]
- Cabré, R.; Naudí, A.; Dominguez-Gonzalez, M.; Jové, M.; Ayala, V.; Mota-Martorell, N.; Pradas, I.; Nogueras, L.; Rué, M.; Portero-Otín, M.; et al. Lipid Profile in Human Frontal Cortex Is Sustained Throughout Healthy Adult Life Span to Decay at Advanced Ages. J. Gerontol.—Ser. A Biol. Sci. Med. Sci. 2018, 73, 703–710. [Google Scholar] [CrossRef]
- Castellani, R.J.; Rolston, R.K.; Smith, M.A. Alzheimer disease. Disease-a-month DM 2010, 56. [Google Scholar] [CrossRef]
- Rauk, A. The Chemistry of Alzheimer’s Disease. Chem. Soc. Rev. 2009, 38, 2698–2715. [Google Scholar] [CrossRef]
- John, H.; Gerald, H. Alzheimer’ s Disease: The Amyloid Cascade Hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef]
- Haass, C.; Selkoe, D.J. Soluble Protein Oligomers in Neurodegeneration: Lessons from the Alzheimer’s Amyloid β-Peptide. Nat. Rev. Mol. Cell Biol. 2007, 8, 101–112. [Google Scholar] [CrossRef]
- Walsh, D.M.; Selkoe, D.J. Aβ Oligomers—A Decade of Discovery. J. Neurochem. 2007, 101, 1172–1184. [Google Scholar] [CrossRef]
- Sakono, M.; Zako, T. Amyloid Oligomers: Formation and Toxicity of Aβ Oligomers. FEBS J. 2010, 277, 1348–1358. [Google Scholar] [CrossRef]
- Osborn, L.M.; Kamphuis, W.; Wadman, W.J.; Hol, E.M. Progress in Neurobiology Astrogliosis: An Integral Player in the Pathogenesis of Alzheimer’s Disease. Prog. Neurobiol. 2016. [Google Scholar] [CrossRef]
- Heneka, M.T.; Carson, M.J.; Khoury, J.E.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s Disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef]
- Huang, W.; Zhang, X.; Chen, W. Role of Oxidative Stress in Alzheimer’s Disease (Review). Biomed. Rep. 2016, 4, 519–522. [Google Scholar] [CrossRef]
- Sisodia, S.S.; Gallagher, M. A Role for the β-Amyloid Precursor Protein in Memory? PNAS 1998, 95, 12074–12076. [Google Scholar] [CrossRef]
- Supnet, C.; Bezprozvanny, I. The Dysregulation of Intracellular Calcium in Alzheimer Disease. Cell Calcium 2010, 47, 183–189. [Google Scholar] [CrossRef]
- Area-Gomez, E.; Schon, E.A. Alzheimer Disease. In Organelle Contact Sites: From Molecular Mechanism to Disease; Tagaya, M., Simmen, T., Eds.; Springer: Singapore, 2017; pp. 149–156. [Google Scholar] [CrossRef]
- Ullah, G.; Demuro, A.; Parker, I.; Pearson, J.E. Analyzing and Modeling the Kinetics of Amyloid Beta Pores Associated with Alzheimer’s Disease Pathology. PLoS ONE 2015, 10, e0137357. [Google Scholar] [CrossRef]
- Allinson, T.M.J.; Parkin, E.T.; Turner, A.J.; Hooper, N.M. ADAMs Family Members as Amyloid Precursor Protein α-Secretases. 2003, 74, 342–352. 2003, 74, 342–352. [Google Scholar]
- Zhou, Z.D.; Chan, C.H.S.; Ma, Q.H.; Xu, X.H.; Xiao, Z.C.; Tan, E.K. The Roles of Amyloid Precursor Protein (APP) in Neurogenesis, Implications to Pathogenesis and Therapy of Alzheimer Disease (AD). Cell Adhes. Migr. 2011, 5, 280–292. [Google Scholar] [CrossRef]
- Parkin, E.T.; Watt, N.T.; Hussain, I.; Eckman, E.A.; Eckman, C.B.; Manson, J.C.; Baybutt, H.N.; Turner, A.J.; Hooper, N.M. Cellular Prion Protein Regulates Beta-Secretase Cleavage of the Alzheimer’s Amyloid Precursor Protein. Proc. Natl. Acad. Sci. USA 2007, 104, 11062–11067. [Google Scholar] [CrossRef]
- Cheng, H.; Vetrivel, K.S.; Gong, P.; Meckler, X.; Parent, A.; Thinakaran, G. Mechanisms of Disease: New Therapeutic Strategies for Alzheimer’s Disease—Targeting APP Processing in Lipid Rafts. Nat. Clin. Pract. Neurol. 2007, 3, 374–382. [Google Scholar] [CrossRef]
- Lemkul, J.A.; Bevan, D.R. Lipid Composition Influences the Release of Alzheimer’s Amyloid β-Peptide from Membranes. Protein Sci. 2011, 20, 1530–1545. [Google Scholar] [CrossRef]
- Ikeda, K.; Yamaguchi, T.; Fukunaga, S.; Hoshino, M.; Matsuzaki, K. Mechanism of Amyloid β-Protein Aggregation Mediated by GM1 Ganglioside Clusters. Biochemistry 2011, 50, 6433–6440. [Google Scholar] [CrossRef]
- Tong, B.C.K.; Wu, A.J.; Li, M.; Cheung, K.H. Calcium Signaling in Alzheimer’s Disease & Therapies. Biochim. Biophys. Acta—Mol. Cell Res. 2018, 1865, 1745–1760. [Google Scholar] [CrossRef]
- Simons, M.; Keller, P.; De Strooper, B.; Beyreuther, K.; Dotti, C.G.; Simons, K. Cholesterol Depletion Inhibits the Generation of Beta-Amyloid in Hippocampal Neurons. Proc. Natl. Acad. Sci. USA 1998, 95, 6460–6464. [Google Scholar] [CrossRef]
- Sparks, D.L. Cholesterol Metabolism and Brain Amyloidosis: Evidence for a Role of Copper in the Clearance of Abeta through the Liver. Curr. Alzheimer Res. 2007, 4, 165–169. [Google Scholar] [CrossRef]
- Fahrenholz, F.; Kojro, E.; Gimpl, G.; Lammich, S.; Ma, W. Low Cholesterol Stimulates the Nonamyloidogenic Pathway by Its Effect on the α-Secretase ADAM 10. Proc. Natl. Acad. Sci. USA 2001, 98, 5815–5820. [Google Scholar]
- Wahrle, S.; Das, P.; Nyborg, A.C.; McLendon, C.; Shoji, M.; Kawarabayashi, T.; Younkin, L.H.; Younkin, S.G.; Golde, T.E. Cholesterol-Dependent γ-Secretase Activity in Buoyant Cholesterol-Rich Membrane Microdomains. Neurobiol. Dis. 2002, 9, 11–23. [Google Scholar] [CrossRef]
- Marquer, C.; Devauges, V.; Cossec, J.; Duyckaerts, C.; Le, S. Local Cholesterol Increase Triggers Amyloid Precursor Protein-Bace1 Clustering in Lipid Rafts and Rapid Endocytosis. FASEB J. 2011, 25, 1295–1305. [Google Scholar] [CrossRef]
- Beel, A.J.; Sakakura, M.; Barrett, P.J.; Sanders, C.R. Direct Binding of Cholesterol to the Amyloid Precursor Protein: An Important Interaction in Lipid-Alzheimer’s Disease Relationships? Biochim. Biophys. Acta—Mol. Cell Biol. Lipids 2010, 1801, 975–982. [Google Scholar] [CrossRef]
- Mayeux, R. Epidemiology of Neurodegeneration. Annu. Rev. Neurosci. 2003, 26, 81–104. [Google Scholar] [CrossRef]
- Cutler, R.G.; Kelly, J.; Storie, K.; Pedersen, W.A.; Tammara, A.; Hatanpaa, K.; Troncoso, J.C.; Mattson, M.P. Involvement of Oxidative Stress-Induced Abnormalities in Ceramide and Cholesterol Metabolism in Brain Aging and Alzheimer’s Disease. Proc. Natl. Acad. Sci. USA 2004, 101, 2070–2075. [Google Scholar] [CrossRef]
- Sparks, D.L. Coronary Artery Disease, Hypertension, ApoE, and Cholesterol: A Link to Alzheimer’s Disease? Ann. N. Y. Acad. Sci. 1997, 826, 128–146. [Google Scholar] [CrossRef]
- Vance, J.E. MAM (Mitochondria-Associated Membranes) in Mammalian Cells: Lipids and Beyond. Biochim. Biophys. Acta—Mol. Cell Biol. Lipids 2014, 1841, 595–609. [Google Scholar] [CrossRef]
- Tambini, M.D.; Pera, M.; Kanter, E.; Yang, H.; Guardia-Laguarta, C.; Holtzman, D.; Sulzer, D.; Area-Gomez, E.; Schon, E.A. ApoE4 Upregulates the Activity of Mitochondria-Associated ER Membranes. EMBO Rep. 2016, 17, 27–36. [Google Scholar] [CrossRef]
- Vaya, J.; Schipper, H.M. Oxysterols, Cholesterol Homeostasis, and Alzheimer Disease. J. Neurochem. 2007, 102, 1727–1737. [Google Scholar] [CrossRef]
- Marwarha, G.; Raza, S.; Prasanthi, J.R.P.; Ghribi, O. Gadd153 and NF-ΚB Crosstalk Regulates 27-Hydroxycholesterol-Induced Increase in BACE1 and β-Amyloid Production in Human Neuroblastoma SH-SY5Y Cells. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Brown, J.; Theisler, C.; Silberman, S.; Magnuson, D.; Gottardi-Littell, N.; Lee, J.M.; Yager, D.; Crowley, J.; Sambamurti, K.; Rahman, M.M.; et al. Differential Expression of Cholesterol Hydroxylases in Alzheimer’s Disease. J. Biol. Chem. 2004, 279, 34674–34681. [Google Scholar] [CrossRef]
- Bieschke, J.A.N.; Zhang, Q.; Bosco, D.A.; Lerner, R.A.; Powers, E.T.; Wentworth, P.; Kelly, J.W. Small Molecule Oxidation Products Trigger Disease-Associated Protein Misfolding. Acc. Chem. Res. 2006, 39, 611–619. [Google Scholar] [CrossRef]
- Famer, D.; Meaney, S.; Mousavi, M.; Nordberg, A.; Björkhem, I.; Crisby, M. Regulation of α- and β-Secretase Activity by Oxysterols: Cerebrosterol Stimulates Processing of APP via the α-Secretase Pathway. Biochem. Biophys. Res. Commun. 2007, 359, 46–50. [Google Scholar] [CrossRef]
- Hao, M.; Mukherjee, S.; Maxfield, F.R. Cholesterol Depletion Induces Large Scale Domain Segregation in Living Cell Membranes. Proc. Natl. Acad. Sci. USA 2001, 98, 13072–13077. [Google Scholar] [CrossRef]
- Mondal, M.; Mesmin, B.; Mukherjee, S.; Maxfield, F.R. Sterols Are Mainly in the Cytoplasmic Leaflet of the Plasma Membrane and the Endocytic Recycling Compartment in CHO Cells. Mol. Biol. Cell 2009, 20, 581–588. [Google Scholar] [CrossRef] [Green Version]
- Cecchi, C.; Nichino, D.; Zampagni, M.; Bernacchioni, C.; Evangelisti, E.; Pensal, A.; Liguri, G.; Gliozzi, A.; Stefani, M.; Relini, A. A Protective Role for Lipid Raft Cholesterol against Amyloid-Induced Membrane Damage in Human Neuroblastoma Cells. Biochim. Biophys. Acta—Mol. Cell Biol. Lipids 2009, 1788, 2204–2216. [Google Scholar] [CrossRef]
- Han, X.; Rozen, S.; Boyle, S.H.; Hellegers, C.; Cheng, H.; Burke, J.R.; Welsh-Bohmer, K.A.; Doraiswamy, P.M.; Kaddurah-Daouk, R. Metabolomics in Early Alzheimer’s Disease: Identification of Altered Plasma Sphingolipidome Using Shotgun Lipidomics. PLoS ONE 2011, 6, e21643. [Google Scholar] [CrossRef]
- Soreghan, B.; Thomas, S.N.; Yang, A.J. Aberrant Sphingomyelin/Ceramide Metabolic-Induced Neuronal Endosomal/Lysosomal Dysfunction: Potential Pathological Consequences in Age-Related Neurodegeneration. Adv. Drug Deliv. Rev. 2003, 55, 1515–1524. [Google Scholar] [CrossRef]
- Farooqui, A.A. Lipid Mediators and Their Metabolism in the Nucleus: Implications for Alzheimer’s Disease. J. Alzheimer’s Dis. 2012, 30, 163–178. [Google Scholar] [CrossRef]
- Ditaranto-Desimone, K.; Saito, M.; Tekirian, T.L.; Saito, M.; Berg, M.; Dubowchik, G.; Soreghan, B.; Thomas, S.; Marks, N.; Yang, A.J. Neuronal Endosomal/Lysosomal Membrane Destabilization Activates Caspases and Induces Abnormal Accumulation of the Lipid Secondary Messenger Ceramide. Brain Res. Bull. 2003, 59, 523–531. [Google Scholar] [CrossRef]
- Frisardi, V.; Panza, F.; Seripa, D.; Farooqui, T.; Farooqui, A.A. Glycerophospholipids and Glycerophospholipid-Derived Lipid Mediators: A Complex Meshwork in Alzheimer’s Disease Pathology. Prog. Lipid Res. 2011, 50, 313–330. [Google Scholar] [CrossRef]
- Haughey, N.J.; Bandaru, V.V.R.; Bae, M.; Mattson, M.P. Roles for Dysfunctional Sphingolipid Metabolism in Alzheimer’s Disease Neuropathogenesis. Biochim. Biophys. Acta—Mol. Cell Biol. Lipids 2010, 1801, 878–886. [Google Scholar] [CrossRef]
- Mielke, M.; Haughey, N. Could Plasma Sphingolipids Be Diagnostic or Prognostic Biomarkers for Alzheimer’s Disease? Clin. Lipidol. 2013, 7, 525–536. [Google Scholar] [CrossRef]
- Furukawa, K.; Ohmi, Y.; Noriyo, T.; Yuji, K.; Orie, T.; Keiko, F. Regulatory Mechanisms of Nervous Systems with Glycosphingolipids. Neurochem. Res. 2011, 36, 1578–1586. [Google Scholar] [CrossRef]
- Wu, G.; Neil, Z.L.; Amin, R.; Ledeen, R.W. Mice Lacking Major Brain Gangliosides Develop Parkinsonism. Neurochem. Res. 2011, 36, 1706–1714. [Google Scholar] [CrossRef] [Green Version]
- Zha, Q.; Ruan, Y.; Hartmann, T.; Beyreuther, K.; Zhang, D. GM1 Ganglioside Regulates the Proteolysis of Amyloid Precursor Protein. Mol. Psychiatry 2004, 9, 946–952. [Google Scholar] [CrossRef]
- Ariga, T.; Mcdonald, M.P.; Yu, R.K. Role of Ganglioside Metabolism in the Pathogenesis of Alzheimer’s Disease—A Review. J. Lipid Res. 2008, 49, 1157–1175. [Google Scholar] [CrossRef]
- Parton, R.G. Ultrastructural Localization of Gangliosides; GM1 Is Concentrated in Caveolae. J. Histochem. Cytochem. 1994, 42, 155–166. [Google Scholar] [CrossRef]
- Okada, T.; Ikeda, K.; Wakabayashi, M.; Ogawa, M.; Matsuzaki, K. Formation of Toxic Aβ (1–40) Fibrils on GM1 Ganglioside-Containing Membranes Mimicking Lipid Rafts: Polymorphisms in Aβ (1–40) Fibrils. J. Mol. Biol. 2008, 382, 1066–1074. [Google Scholar] [CrossRef]
- Zhang, Q.; Ohmi, Y.; Yamaguchi, T.; Furukawa, K.; Furukawa, K.; Yamauchi, Y.; Okajima, T.; Ohkawa, Y. Expression of B4GALNT1, an Essential Glycosyltransferase for the Synthesis of Complex Gangliosides, Suppresses BACE1 Degradation and Modulates APP Processing. Sci. Rep. 2016, 6, 34505. [Google Scholar] [CrossRef]
- Posse de Chaves, E.; Sipione, S. Sphingolipids and Gangliosides of the Nervous System in Membrane Function and Dysfunction. FEBS Lett. 2010, 584, 1748–1759. [Google Scholar] [CrossRef]
- Ledeen, R.W.; Wu, G. Nuclear Sphingolipids: Metabolism and Signaling. J. Lipid Res. 2008, 49, 1176–1186. [Google Scholar] [CrossRef]
- Mocchetti, I. Exogenous Gangliosides, Neuronal Plasticity and Repair, and the Neurotrophins. Cell. Mol. Life Sci. 2005, 62, 2283–2294. [Google Scholar] [CrossRef]
- Saavedra, L.; Mohamed, A.; Kar, S.; De Chaves, E.P. Internalization of Beta-Amyloid Peptide by Primary Neurons in the Absence of Apolipoprotein E. J. Biol. Chem. 2007, 282, 35722–35732. [Google Scholar] [CrossRef]
- Fabelo, N.; Martín, V.; Marín, R.; Moreno, D.; Ferrer, I.; Díaz, M. Altered Lipid Composition in Cortical Lipid Rafts Occurs at Early Stages of Sporadic Alzheimer’s Disease and Facilitates APP/BACE1 Interactions. Neurobiol. Aging 2014, 35, 1801–1812. [Google Scholar] [CrossRef]
- Navarro-Pardo, E.; Holland, C.A.; Cano, A. Sex Hormones and Healthy Psychological Aging in Women. Front. Aging Neurosci. 2018, 9, 439. [Google Scholar] [CrossRef] [Green Version]
- Mario, D. Hippocampal Lipid Homeostasis in APP/PS1 Mice Is Modulated by a Complex Interplay Between Dietary DHA and Estrogens: Relevance for Alzheimer’s Disease. J. Alzheimer’s Dis. 2016, 49, 459–481. [Google Scholar] [CrossRef]
- Canerina-Amaro, A.; Hernandez-Abad, L.G.; Ferrer, I.; Quinto-Alemany, D.; Mesa-Herrera, F.; Ferri, C.; Puertas-Avendano, R.A.; Diaz, M.; Marin, R. Lipid Raft ER Signalosome Malfunctions in Menopause and Alzheimer’s Disease. Front. Biosci. (Schol. Ed.) 2017, 9, 111–126. [Google Scholar] [CrossRef]
- Lees, A.J.; Hardy, J.; Revesz, T. Parkinson’s Disease. Lancet 2009, 373, 2055–2066. [Google Scholar] [CrossRef]
- Broen, M.P.G.; Narayen, N.E.; Kuijf, M.L.; Dissanayaka, N.N.W.; Leentjens, A.F.G. Prevalence of Anxiety in Parkinson’s Disease: A Systematic Review and Meta-Analysis. Mov. Disord. 2016, 31, 1125–1133. [Google Scholar] [CrossRef]
- Ascherio, A.; Schwarzschild, M.A. The Epidemiology of Parkinson’s Disease: Risk Factors and Prevention. Lancet Neurol. 2016, 15, 1257–1272. [Google Scholar] [CrossRef]
- Reich, S.G.; Savitt, J.M. Parkinson’s Disease. Med. Clin. N. Am. 2019, 103, 337–350. [Google Scholar] [CrossRef]
- Hely, M.A.; Reid, W.G.J.; Adena, M.A.; Halliday, G.M.; Morris, J.G.L. The Sydney Multicenter Study of Parkinson’s Disease: The Inevitability of Dementia at 20 Years. Mov. Disord. 2008, 23, 837–844. [Google Scholar] [CrossRef]
- Pacheco, C.; Aguayo, L.G.; Opazo, C. An Extracellular Mechanism That Can Explain the Neur Effects of α-Synuclein Aggregates in the Brain. Front. Physiol. 2012, 3, 297. [Google Scholar] [CrossRef]
- Burré, J. The Synaptic Function of α-Synuclein. J. Parkinson’s. Dis. 2015, 5, 699–713. [Google Scholar] [CrossRef]
- Sharon, R.; Goldberg, M.S.; Bar-josef, I.; Betensky, R.A.; Shen, J.; Selkoe, D.J. α-Synuclein Occurs in Lipid-Rich High Molecular Weight Complexes, Binds Fatty Acids, and Shows Homology to the Fatty Acid-Binding Proteins. Proc. Natl. Acad. Sci. USA 2001, 98, 9110–9115. [Google Scholar] [CrossRef]
- Colebc, N.B.; Murphy, D.D.; Grider, T.; Rueter, S.; Brasaemle, D.; Nussbaum, R.L. Lipid Droplet Binding and Oligomerization Properties of the Parkinson’s Disease Protein α-Synuclein. J. Biol. Chem. 2002, 277, 6344–6352. [Google Scholar] [CrossRef]
- Schoonenboom, N.S.M.; Mulder, C.; Vanderstichele, H.; Van Elk, E.J.; Kok, A.; Van Kamp, G.J.; Scheltens, P.; Blankenstein, M.A. Effects of Processing and Storage Conditions on Amyloid β (1-42) and Tau Concentrations in Cerebrospinal Fluid: Implications for Use in Clinical Practice. Clin. Chem. 2005, 51, 189–195. [Google Scholar] [CrossRef]
- Varkey, J.; Isas, J.M.; Mizuno, N.; Jensen, M.B.; Bhatia, V.K.; Jao, C.C.; Petrlova, J.; Voss, J.C.; Stamou, D.G.; Steven, A.C.; et al. Membrane Curvature Induction and Tubulation Are Common Features of Synucleins and Apolipoproteins. J. Biol. Chem. 2010, 285, 32486–32493. [Google Scholar] [CrossRef] [Green Version]
- Mizuno, N.; Varkey, J.; Kegulian, N.C.; Hegde, B.G.; Cheng, N.; Langen, R.; Steven, A.C. Remodeling of Lipid Vesicles into Cylindrical Micelles by α-Synuclein in an Extended α-Helical Conformation. J. Biol. Chem. 2012, 287, 29301–29311. [Google Scholar] [CrossRef]
- Westphal, C.H.; Chandra, S.S. Monomeric Synucleins Generate Membrane Curvature. J. Biol. Chem. 2013, 288, 1829–1840. [Google Scholar] [CrossRef] [Green Version]
- Shi, Z.; Sachs, J.; Rhoades, E.; Baumgart, T.; Haven, N. Biophysics of α-Synuclein Induced Membrane Remodelling. Phys. Chem. Chem. Phys. 2015, 17, 15561–15568. [Google Scholar] [CrossRef]
- Braun, A.R.; Lacy, M.M.; Ducas, V.C.; Rhoades, E.; Sachs, J.N. A-Synuclein-Induced Membrane Remodeling Is Driven by Binding. J. Am. Chem. Soc. 2014, 136, 9962–9972. [Google Scholar] [CrossRef]
- Madine, J.; Doig, A.J.; Middleton, D.A. A Study of the Regional Effects of α-Synuclein on the Organization and Stability of Phospholipid Bilayers. Biochemistry 2006, 45, 5783–5792. [Google Scholar] [CrossRef]
- Payton, J.E.; Perrin, R.J.; Woods, W.S.; George, J.M. Structural Determinants of PLD2 Inhibition by α-Synuclein. J. Mol. Biol. 2004, 337, 1001–1009. [Google Scholar] [CrossRef]
- Gorbatyuk, O.S.; Li, S.; Nguyen, F.N.; Manfredsson, F.P.; Kondrikova, G.; Sullivan, L.F.; Meyers, C.; Chen, W.; Mandel, R.J.; Muzyczka, N. α -Synuclein Expression in Rat Substantia Nigra Suppresses Phospholipase D2 Toxicity and Nigral Neurodegeneration. Mol. Ther. 2009, 18, 1758–1768. [Google Scholar] [CrossRef]
- Vendruscolo, M.; Dobson, C.M.; Galvagnion, C.; Meisl, G.; Michaels, T.C.T.; Buell, A.K.; Knowles, T.P.J. Lipid Vesicles Trigger α-Synuclein Aggregation by Stimulating Primary Nucleation. Nat. Chem. Biol. 2015, 11, 229–234. [Google Scholar] [CrossRef]
- Nath, S.; Giraldo, A.M.V.; Eriksson, I.; Öllinger, K.; Bornefall, P. Impact of High Cholesterol in a Parkinson’s Disease Model: Prevention of Lysosomal Leakage versus Stimulation of α-Synuclein Aggregation. Eur. J. Cell Biol. 2017, 96, 99–109. [Google Scholar] [CrossRef]
- Cheng, D.; Kim, W.S.; Garner, B. Regulation of α-Synuclein Expression by Liver X Receptor Ligands in Vitro. Neuroreport 2008, 19, 1685–1689. [Google Scholar] [CrossRef]
- Ghribi, O.; Schommer, E.; Feist, G.; Thomasson, S.; Thompson, A.; Rantham Prabhakara, J.P. Differential Effects of 24-Hydroxycholesterol and 27-Hydroxycholesterol on Tyrosine Hydroxylase and α-Synuclein in Human Neuroblastoma SH-SY5Y Cells. J. Neurochem. 2008, 107, 1722–1729. [Google Scholar] [CrossRef]
- Fan, X.; Kim, H.-J.; Parini, P.; Gabbi, C.; Warner, M.; Yakimchuk, K.; Gustafsson, J.-A. Liver X Receptor (LXR): A Link between -Sitosterol and Amyotrophic Lateral Sclerosis-Parkinson’s Dementia. Proc. Natl. Acad. Sci. USA 2008, 105, 2094–2099. [Google Scholar] [CrossRef]
- Tansey, M.G.; Frank-Cannon, T.C.; Li, H.; Valasek, M.A.; Dietschy, J.M.; Turley, S.D.; Repa, J.J. Liver X Receptor Activation Enhances Cholesterol Loss from the Brain, Decreases Neuroinflammation, and Increases Survival of the NPC1 Mouse. J. Neurosci. 2007, 27, 14470–14480. [Google Scholar] [CrossRef] [Green Version]
- Hultenby, K.; Gustafsson, J.-A.; Wang, L.; Andersson, S.; Schuster, G.U.; Zhang, Q. Liver X Receptors in the Central Nervous System: From Lipid Homeostasis to Neuronal Degeneration. Proc. Natl. Acad. Sci. USA 2002, 99, 13878–13883. [Google Scholar] [CrossRef]
- Barceló-Coblijn, G.; Golovko, M.Y.; Weinhofer, I.; Berger, J.; Murphy, E.J. Brain Neutral Lipids Mass Is Increased in α-Synuclein Gene-Ablated Mice. J. Neurochem. 2007, 101, 132–141. [Google Scholar] [CrossRef]
- Perrin, R.J.; Woods, W.S.; Clayton, D.F.; George, J.M. Interaction of Human α-Synuclein and Parkinson’s Disease Variants with Phospholipids. J. Biol. Chem. 2002, 275, 34393–34398. [Google Scholar] [CrossRef]
- Broersen, K.; Brink, D.V.D.; Fraser, G.; Goedert, M.; Davletov, B. Alpha-Synuclein Adopts an Alpha-Helical Conformation in the Presence of Polyunsaturated Fatty Acids to Hinder Micelle Formation. Biochemistry 2006, 45, 15610–15616. [Google Scholar] [CrossRef]
- Sharon, R.; Bar-joseph, I.; Frosch, M.P.; Walsh, D.M.; Hamilton, J.A.; Selkoe, D.J. The Formation of Highly Soluble Oligomers of Alpha-Synuclein Is Regulated by Fatty Acids and Enhanced in Parkinson’s Disease. Neuron 2003, 37, 583–595. [Google Scholar] [CrossRef]
- Lin, P.Y.; Huang, S.Y.; Su, K.P. A Meta-Analytic Review of Polyunsaturated Fatty Acid Compositions in Patients with Depression. Biol. Psychiatry 2010, 68, 140–147. [Google Scholar] [CrossRef]
- El-Agnaf, O.M.A.; Jakes, R.; Curran, M.D.; Wallace, A. Effects of the Mutations Ala30 to Pro and Ala53 to Thr on the Physical and Morphological Properties of α-Synuclein Protein Implicated in Parkinson’s Disease. FEBS Lett. 1998, 440, 67–70. [Google Scholar] [CrossRef]
- Giasson, B.I.; Uryu, K.; Trojanowski, J.Q.; Lee, V.M.Y. Mutant and Wild Type Human α-Synucleins Assemble into Elongated Filaments with Distinct Morphologies in Vitro. J. Biol. Chem. 1999, 274, 7619–7622. [Google Scholar] [CrossRef]
- Pyszko, J.A.; Strosznajder, J.B. Original Article the Key Role of Sphingosine Kinases in the Molecular Mechanism of Neuronal Cell Survival and Death in an Experimental Model of Parkinson’s Disease. Folia Neuropathol. 2014, 3, 260–269. [Google Scholar] [CrossRef]
- Canerina-amaro, A.; Pereda, D.; Diaz, M.; Rodriguez-barreto, D.; Casañas-sánchez, V.; Heffer, M.; Garcia-esparcia, P.; Ferrer, I.; George, S. Differential Aggregation and Phosphorylation of Alpha Synuclein in Membrane Compartments Associated with Parkinson Disease. Front. Neurosci. 2019, 13, 382. [Google Scholar] [CrossRef]
- Martinez, Z.; Zhu, M.; Han, S.; Fink, A.L. GM1 Specifically Interacts with Alpha-Synuclein and Inhibits Fibrillation. Biochem. Pharmacol. 2007, 46, 1868–1877. [Google Scholar]
- Ariga, T. Pathogenic Role of Ganglioside Metabolism in Neurodegenerative Diseases. J. Neurosci. Res. 2014, 1242, 1227–1242. [Google Scholar] [CrossRef]
- Badawy, M.M.S.; Okada, T.; Kajimoto, T.; Hirase, M.; Matovelo, S.; Nakamura, S.; Yoshida, D.; Ijuin, T.; Nakamura, S. Extracellular α-Synuclein Drives Sphingosine 1-Phosphate Receptor Subtype 1 out of Lipid Rafts, Leading to Impaired Inhibitory G-Protein Signaling. J. Biol. Chem. 2018, 293, 8208–8216. [Google Scholar] [CrossRef]
- Fabelo, N.; Martin, V.; Santpere, G.; Marín, R.; Torrent, L.; Ferrer, I.; Díaz, M. Severe Alterations in Lipid Composition of Frontal Cortex Lipid Rafts from Parkinson’s Disease and Incidental Parkinson’s Disease. Mol. Med. 2011, 17. [Google Scholar] [CrossRef]
- Davidson, W.S.; Jonas, A.; Clayton, D.F.; George, J.M. Stabilization of Alpha-Synuclein Secondary Structure upon Binding to Synthetic Membranes. J. Biol. Chem. 1998, 273, 9443–9449. [Google Scholar] [CrossRef]
- Kubo, S.; Nemani, V.M.; Chalkley, R.J.; Anthony, M.D.; Hattori, N.; Mizuno, Y.; Edwards, R.H.; Fortin, D.L. A Combinatorial Code for the Interaction of Alpha-Synuclein with Membranes. J. Biol. Chem. 2005, 280, 31664–31672. [Google Scholar] [CrossRef]
- Gedalya, T.B.; Loeb, V.; Israeli, E.; Altschuler, Y.; Selkoe, D.J.; Sharon, R. α-Synuclein and Polyunsaturated Fatty Acids Promote Clathrin-Mediated Endocytosis and Synaptic Vesicle Recycling. Traffic 2009, 10, 218–234. [Google Scholar] [CrossRef]
- Madeira, A.; Yang, J.; Zhang, X.; Vikeved, E.; Nilsson, A.; Andrén, P.E.; Svenningsson, P. Caveolin-1 Interacts with Alpha-Synuclein and Mediates Toxic Actions of Cellular Alpha-Synuclein Overexpression. Neurochem. Int. 2011, 59, 280–289. [Google Scholar] [CrossRef]
- Fortin, D.L. Lipid Rafts Mediate the Synaptic Localization of α-Synuclein. J. Neurosci. 2004, 24, 6715–6723. [Google Scholar] [CrossRef]
- Ferrucci, L.; Giallauria, F.; Guralnik, J.M. Epidemiology of Aging. Radiol. Clin. N. Am. 2008, 46. [Google Scholar] [CrossRef]
- Garn, H.; Coronel, C.; Waser, M.; Caravias, G.; Ransmayr, G. Differential Diagnosis between Patients with Probable Alzheimer’s Disease, Parkinson’s Disease Dementia, or Dementia with Lewy Bodies and Frontotemporal Dementia, Behavioral Variant, Using Quantitative Electroencephalographic Features. J. Neural Transm. 2017, 124, 569–581. [Google Scholar] [CrossRef]
- Jack, C.R.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a Biological Definition of Alzheimer’s Disease. Alzheimer’s Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef]
- Gibb, W.; Lees, A.J. The Relevance of the Lewy Body to the Pathogenesis of Idiopathic Parkinson’s Disease. J. Neurol. Neurosurg. Psychiatry 1988, 51, 745–752. [Google Scholar] [CrossRef]
- Rao, G.; Eaton, C. Does This Patient Have Parkinson Disease? Mov. Disord. 2015, 289, 347–353. [Google Scholar] [CrossRef]
- Tolosa, E.; Wenning, G.W.P.W. The Diagnosis of Parkinson’s Disease. Lancet Neurol. 2006, 5, 75–86. [Google Scholar] [CrossRef]
- DeMaagd, G. Parkinson’s Disease and Its Management. Munic. Solid Waste Manag. Dev. Ctries. 2016, 40, 504–532. [Google Scholar] [CrossRef]
- Han, X.; Fagan, A.M.; Cheng, H.; Morris, J.C.; Xiong, C.; Holtzman, D.M. Cerebrospinal Fluid Sulfatide Is Decreased in Subjects with Incipient Dementia. Ann. Neurol. 2003, 54, 115–119. [Google Scholar] [CrossRef]
- Mulder, C.; Wahlund, L.O.; Teerlink, T.; Blomberg, M.; Veerhuis, R.; Van Kamp, G.J.; Scheltens, P.; Scheffer, P.G. Decreased Lysophosphatidylcholine/Phosphatidylcholine Ratio in Cerebrospinal Fluid in Alzheimer’s Disease. J. Neural Transm. 2003, 110, 949–955. [Google Scholar] [CrossRef]
- Walter, A.; Korth, U.; Hilgert, M.; Hartmann, J.; Weichel, O.; Hilgert, M.; Fassbender, K.; Schmitt, A.; Klein, J. Glycerophosphocholine Is Elevated in Cerebrospinal Fluid of Alzheimer Patients. Neurobiol. Aging 2004, 25, 1299–1303. [Google Scholar] [CrossRef]
- Mielke, M.M.; Haughey, N.J.; Bandaru, V.V.R.; Zetterberg, H.; Blennow, K.; Andreasson, U.; Johnson, S.C.; Gleason, C.E.; Blazel, H.M.; Puglielli, L.; et al. Cerebrospinal Fluid Sphingolipids, β-Amyloid, and Tau in Adults at Risk for Alzheimer’s Disease. Neurobiol. Aging 2014, 35, 2486–2494. [Google Scholar] [CrossRef]
- Testa, G.; Staurenghi, E.; Zerbinati, C.; Gargiulo, S.; Iuliano, L.; Giaccone, G.; Fantò, F.; Poli, G.; Leonarduzzi, G.; Gamba, P. Changes in Brain Oxysterols at Different Stages of Alzheimer’s Disease: Their Involvement in Neuroinflammation. Redox Biol. 2016, 10, 24–33. [Google Scholar] [CrossRef]
- Heverin, M.; Bogdanovic, N.; Lütjohann, D.; Bayer, T.; Pikuleva, I.; Bretillon, L.; Diczfalusy, U.; Winblad, B.; Björkhem, I. Changes in the Levels of Cerebral and Extracerebral Sterols in the Brain of Patients with Alzheimer’s Disease. J. Lipid Res. 2004, 45, 186–193. [Google Scholar] [CrossRef]
- Catalá, A.; Díaz, M. Impact of Lipid Peroxidation on the Physiology and Pathophysiology of Cell Membranes; Frontiers Media: Lausanne, Switzerland, 2017. [Google Scholar] [CrossRef]
- Shinto, L.; Quinn, J.; Montine, T.; Dodge, H.H.; Woodward, W.; Baldauf-Wagner, S.; Waichunas, D.; Bumgarner, L.; Bourdette, D.; Silbert, L.; et al. A Randomized Placebo-Controlled Pilot Trial of Omega-3 Fatty Acids and Alpha Lipoic Acid in Alzheimer’s Disease. J. Alzheimer’s Dis. 2014, 38, 111–120. [Google Scholar] [CrossRef]
- Yuan, L.; Liu, J.; Ma, W.; Dong, L.; Wang, W.; Che, R.; Xiao, R. Dietary Pattern and Antioxidants in Plasma and Erythrocyte in Patients with Mild Cognitive Impairment from China. Nutrition 2016, 32, 193–198. [Google Scholar] [CrossRef]
- Monacelli, F.; Borghi, R.; Cammarata, S.; Nencioni, A.; Piccini, A.; Tabaton, M.; Odetti, P. Amnestic Mild Cognitive Impairment and Conversion to Alzheimer’s Disease: Insulin Resistance and Glycoxidation as Early Biomarker Clusters. J. Alzheimer’s Dis. 2015, 45, 89–95. [Google Scholar] [CrossRef]
- Scheff, S.W.; Ansari, M.A.; Mufson, E.J. Oxidative Stress and Hippocampal Synaptic Protein Levels in Elderly Cognitively Intact Individuals with Alzheimer’s Disease Pathology. Neurobiol. Aging 2016, 42, 1–12. [Google Scholar] [CrossRef]
- Rosén, C.; Mattsson, N.; Johansson, P.M.; Andreasson, U.; Wallin, A.; Hansson, O.; Johansson, J.O.; Lamont, J.; Svensson, J.; Blennow, K.; et al. Discriminatory Analysis of Biochip-Derived Protein Patterns in CSF and Plasma in Neurodegenerative Diseases. Front. Aging Neurosci. 2011, 3, 1–10. [Google Scholar] [CrossRef]
- Sepe, F.N.; Chiasserini, D.; Parnetti, L. Role of FABP3 as Biomarker in Alzheimer’s Disease and Synucleinopathies. Future Neurol. 2018, 13, 199–207. [Google Scholar] [CrossRef]
- Chiasserini, D.; Parnetti, L.; Andreasson, U.; Zetterberg, H.; Giannandrea, D.; Calabresi, P.; Blennow, K. CSF Levels of Heart Fatty Acid Binding Protein Are Altered during Early Phases of Alzheimer’s Disease. J. Alzheimer’s Dis. 2010, 22, 1281–1288. [Google Scholar] [CrossRef]
- Harari, O.; Chuchanga, C.; Pickering, E.H.; Bertelsen, S.; Fagan, A.M.; Holtzman, D.M. Phosphorylated Tau-Aβ42 Ratio as a Continuous Trait for Biomarker Discovery for Early-Stage Alzheimer’s Disease in Multiplex Immunoassay Panels of Cerebrospinal Fluid. Biol. Psychiatry 2015, 75, 723–731. [Google Scholar] [CrossRef]
- Chaudhuri, K.R.; Martinez-Martin, P. Quantitation of Non-Motor Symptoms in Parkinson’s Disease. Eur. J. Neurol. 2008, 15 (Suppl. 2), 2–8. [Google Scholar] [CrossRef]
- Braak, H.; Del Tredici, K. Reply to “Controversies over the Staging of α-Synuclein Pathology in Parkinson’s Disease”. Acta Neuropathol. 2008, 116, 129–131. [Google Scholar] [CrossRef]
- Mollenhauer, B.; Zhang, J. Biochemical Premotor Biomarkers for Parkinson’s Disease. Mov. Disord. 2012, 27, 644–650. [Google Scholar] [CrossRef]
- Mollenhauer, B.; Locascio, J.J.; Schulz-Schaeffer, W.; Sixel-Döring, F.; Trenkwalder, C.; Schlossmacher, M.G. α-Synuclein and Tau Concentrations in Cerebrospinal Fluid of Patients Presenting with Parkinsonism: A Cohort Study. Lancet Neurol. 2011, 10, 230–240. [Google Scholar] [CrossRef]
- Woulfe, J.M.; Duke, R.; Middeldorp, J.M.; Stevens, S.; Vervoort, M.; Hashimoto, M.; Masliah, E.; Chan, P.; Monte, D.A.D.; Langston, J.W.; et al. Absence of Elevated Anti-Alpha-Synuclein and Anti-EBV Latent Membrane Protein Antibodies in PD. Neurology 2002, 58, 1436. [Google Scholar] [CrossRef]
- Öhrfelt, A.; Grognet, P.; Andreasen, N.; Wallin, A.; Vanmechelen, E.; Blennow, K.; Zetterberg, H. Cerebrospinal Fluid α-Synuclein in Neurodegenerative Disorders-A Marker of Synapse Loss? Neurosci. Lett. 2009, 450, 332–335. [Google Scholar] [CrossRef]
- Tateno, F.; Sakakibara, R.; Kawai, T. Alpha-Synuclein in the Cerebrospinal Fluid Differentiates Synucleinopathies (Parkinson Disease, Dementia with Lewy Bodies, Multiple System Atrophy) from Alzheimer Disease. Alzheimer Dis. Assoc. Disord. 2012, 26, 213–216. [Google Scholar] [CrossRef]
- Park, M.J.; Cheon, S.M.; Bae, H.R.; Kim, S.H.; Kim, J.W. Elevated Levels of α-Synuclein Oligomer in the Cerebrospinal Fluid of Drug-Naïve Patients with Parkinson’s Disease. J. Clin. Neurol. 2011, 7, 215–222. [Google Scholar] [CrossRef]
- Parnetti, L.; Chiasserini, D.; Bellomo, G.; Giannandrea, D.; de Carlo, C.; Qureshi, M.M.; Ardah, M.T.; Varghese, S.; Bonanni, L.; Borroni, B.; et al. Cerebrospinal Fluid Tau/α-Synuclein Ratio in Parkinson’s Disease and Degenerative Dementias. Mov. Disord. 2011, 26, 1428–1435. [Google Scholar] [CrossRef]
- Wang, Y.; Shi, M.; Chung, K.A.; Zabetian, C.P.; Leverenz, J.B.; Berg, D.; Srulijes, K.; Trojanowski, J.Q.; Lee, V.M.Y.; Siderowf, A.D.; et al. Phosphorylated α-Synuclein in Parkinson’s Disease. Sci. Transl. Med. 2012, 4. [Google Scholar] [CrossRef]
- Kang, J.H.; Irwin, D.J.; Chen-Plotkin, A.S.; Siderowf, A.; Caspell, C.; Coffey, C.S.; Waligórska, T.; Taylor, P.; Pan, S.; Frasier, M.; et al. Association of Cerebrospinal Fluid β-Amyloid 1-42, t-Tau, p-Tau181, and α-Synuclein Levels with Clinical Features of Drug-Naive Patients with Early Parkinson Disease. JAMA Neurol. 2013, 70, 1277–1287. [Google Scholar] [CrossRef]
- Wennström, M.; Surova, Y.; Hall, S.; Nilsson, C.; Minthon, L.; Boström, F.; Hansson, O.; Nielsen, H.M. Low CSF Levels of Both α-Synuclein and the α-Synuclein Cleaving Enzyme Neurosin in Patients with Synucleinopathy. PLoS ONE 2013, 8, e53250. [Google Scholar] [CrossRef]
- Parnetti, L.; Castrioto, A.; Chiasserini, D.; Persichetti, E.; Tambasco, N.; El-agnaf, O.; Calabresi, P. Cerebrospinal Fluid Biomarkers in Parkinson Disease. Nat. Rev. Neurol. 2013, 9, 131–140. [Google Scholar] [CrossRef]
- Parnetti, L.; Farotti, L.; Eusebi, P.; Chiasserini, D.; De Carlo, C.; Giannandrea, D.; Salvadori, N.; Lisetti, V.; Tambasco, N.; Rossi, A.; et al. Differential Role of CSF Alpha-Synuclein Species, Tau, and Aβ42 in Parkinson’s Disease. Front. Aging Neurosci. 2014, 6, 53. [Google Scholar] [CrossRef]
- Van Dijk, K.D.; Persichetti, E.; Chiasserini, D.; Eusebi, P.; Beccari, T.; Calabresi, P.; Berendse, H.W.; Parnetti, L.; van de Berg, W.D.J. Changes in Endolysosomal Enzyme Activities in Cerebrospinal Fluid of Patients with Parkinson’s Disease. Mov. Disord. 2013, 28, 747–754. [Google Scholar] [CrossRef]
- Aerts, M.B.; Esselink, R.A.J.; Abdo, W.F.; Bloem, B.R.; Verbeek, M.M. CSF α-Synuclein Does Not Differentiate between Parkinsonian Disorders. Neurobiol. Aging 2012, 33, 430.e1–430.e3. [Google Scholar] [CrossRef]
- He, R.; Yan, X.; Guo, J.; Xu, Q.; Tang, B.; Sun, Q. Recent Advances in Biomarkers for Parkinson’s Disease. Front. Aging Neurosci. 2018, 10, 305. [Google Scholar] [CrossRef]
- Concannon, R.; Finn, D.P.; Dowd, E. Cannabinoids in Parkinson’s Disease. Cannabinoids Neurol. Ment. Dis. 2015, 2, 35–59. [Google Scholar] [CrossRef]
- Pisani, A.; Fezza, F.; Galati, S.; Battista, N.; Napolitano, S.; Finazzi-Agrò, A.; Bernardi, G.; Brusa, L.; Pierantozzi, M.; Stanzione, P.; et al. High Endogenous Cannabinoid Levels in the Cerebrospinal Fluid of Untreated Parkinson’s Disease Patients. Ann. Neurol. 2005, 57, 777–779. [Google Scholar] [CrossRef]
- Pisani, V.; Moschella, V.; Bari, M.; Fezza, F.; Galati, S.; Bernardi, G.; Stanzione, P.; Pisani, A.; Maccarrone, M. Dynamic Changes of Anandamide in the Cerebrospinal Fluid of Parkinson’s Disease Patients. Mov. Disord. 2010, 25, 920–924. [Google Scholar] [CrossRef]
- Emamzadeh, F.N. Role of Apolipoproteins and α-Synuclein in Parkinson’s Disease. J. Mol. Neurosci. 2017, 62, 344–355. [Google Scholar] [CrossRef]
- Elliott, D.A.; Weickert, C.S.; Garner, B. Apolipoproteins in the Brain: Implications for Neurological and Psychiatric Disorders. Clin. Lipidol. 2010, 5, 555–573. [Google Scholar] [CrossRef]
- Qiang, J.K.; Wong, Y.C.; Siderowf, A.; Hurtig, H.I.; Xie, S.X.; Lee, V.M.; Trojanowski, J.Q.; Yearout, D.; Leverenz, J.; Thomas, J.; et al. Plasma Apolipoprotein A1 as a Biomarker for Parkinson’s Disease. Ann. Neurol. 2013, 74, 119–127. [Google Scholar] [CrossRef]
- Swanson, C.R.; Berlyand, Y.; Xie, S.X.; Alcalay, R.N.; Chahine, L.M.; Chen-Plotkin, A.S. Plasma Apolipoprotein A1 Associates with Age at Onset and Motor Severity in Early Parkinson’s Disease Patients. Mov. Disord. 2015, 30, 1648–1656. [Google Scholar] [CrossRef]
- Chalimoniuk, M.; Snoek, G.T.; Adamczyk, A.; Małecki, A.; Strosznajder, J.B. Phosphatidylinositol Transfer Protein Expression Altered by Aging and Parkinson Disease. Cell. Mol. Neurobiol. 2006, 26, 1153–1166. [Google Scholar] [CrossRef]
- Dexter, D.T.; Carter, C.J.; Wells, F.R.; Javoy-Agid, F.; Agid, Y.; Lees, A.; Jenner, P.; Marsden, C.D. Basal Lipid Peroxidation in Substantia Nigra Is Increased in Parkinson’s Disease. J. Neurochem. 1989, 52, 381–389. [Google Scholar] [CrossRef]
- Yoritaka, A.; Uchida, K.; Stadtman, E.R.; Hattori, N.; Tanaka, M.; Mizuno, Y. Immunohistochemical Detection of 4-Hydroxynonenal Protein Adducts in Parkinson Disease. Proc. Natl. Acad. Sci. USA 1996, 93, 2696–2701. [Google Scholar] [CrossRef]
- Gmitterova, K.; Heinemann, U.; Gawinecka, J.; Varges, D.; Ciesielczyk, B.; Valkovic, P.; Benetin, J.; Zerr, I. 8-OHdG in Cerebrospinal Fluid as a Marker of Oxidative Stress in Various Neurodegenerative Diseases. Neurodegener. Dis. 2009, 6, 263–269. [Google Scholar] [CrossRef]
- Isobe, C.; Abe, T.; Terayama, Y. Levels of Reduced and Oxidized Coenzyme Q-10 and 8-Hydroxy-2′- Deoxyguanosine in the CSF of Patients with Alzheimer’s Disease Demonstrate That Mitochondrial Oxidative Damage and/or Oxidative DNA Damage Contributes to the Neurodegenerative Process. J. Neurol. 2010, 257, 399–404. [Google Scholar] [CrossRef]
- Kikuchi, A.; Takeda, A.; Onodera, H.; Kimpara, T.; Hisanaga, K.; Sato, N.; Nunomura, A.; Castellani, R.J.; Perry, G.; Smith, M.A.; et al. Systemic Increase of Oxidative Nucleic Acid Damage in Parkinson’s Disease and Multiple System Atrophy. Neurobiol. Dis. 2002, 9, 244–248. [Google Scholar] [CrossRef]
- He, X.; Huang, Y.; Li, B.; Gong, C.X.; Schuchman, E.H. Deregulation of Sphingolipid Metabolism in Alzheimer’s Disease. Neurobiol. Aging 2010, 31, 398–408. [Google Scholar] [CrossRef]
- González-Domínguez, R.; García-Barrera, T.; Gómez-Ariza, J.L. Combination of Metabolomic and Phospholipid-Profiling Approaches for the Study of Alzheimer’s Disease. J. Proteom. 2014, 104, 37–47. [Google Scholar] [CrossRef]
- Yanai, H. Effects of N-6 and n-3 Polyunsaturated Fatty Acids on Colorectal Carcinogenesis. J. Clin. Med. Res. 2017, 9, 1–9. [Google Scholar] [CrossRef]
- Thomas, H.; Pelleieux, S.; Vitale, N.; Oliver, J. Arachidonic Acid in Alzheimer’s Disease. J. Neurol. Neuromed. 2016, 1, 1–6. [Google Scholar] [CrossRef]
- Fraser, T.; Tayler, H.; Love, S. Fatty Acid Composition of Frontal, Temporal and Parietal Neocortex in the Normal Human Brain and in Alzheimer’s Disease. Neurochem. Res. 2010, 35, 503–513. [Google Scholar] [CrossRef]
- Patel, D.; Witt, S.N. Ethanolamine and Phosphatidylethanolamine: Partners in Health and Disease. Oxid. Med. Cell. Longev. 2017, 2017, 4829180. [Google Scholar] [CrossRef]
- Farmer, K.; Smith, C.A.; Hayley, S.; Smith, J. Major Alterations of Phosphatidylcholine and Lysophosphotidylcholine Lipids in the Substantia Nigra Using an Early Stage Model of Parkinson’s Disease. Int. J. Mol. Sci. 2015, 16, 18865–18877. [Google Scholar] [CrossRef]




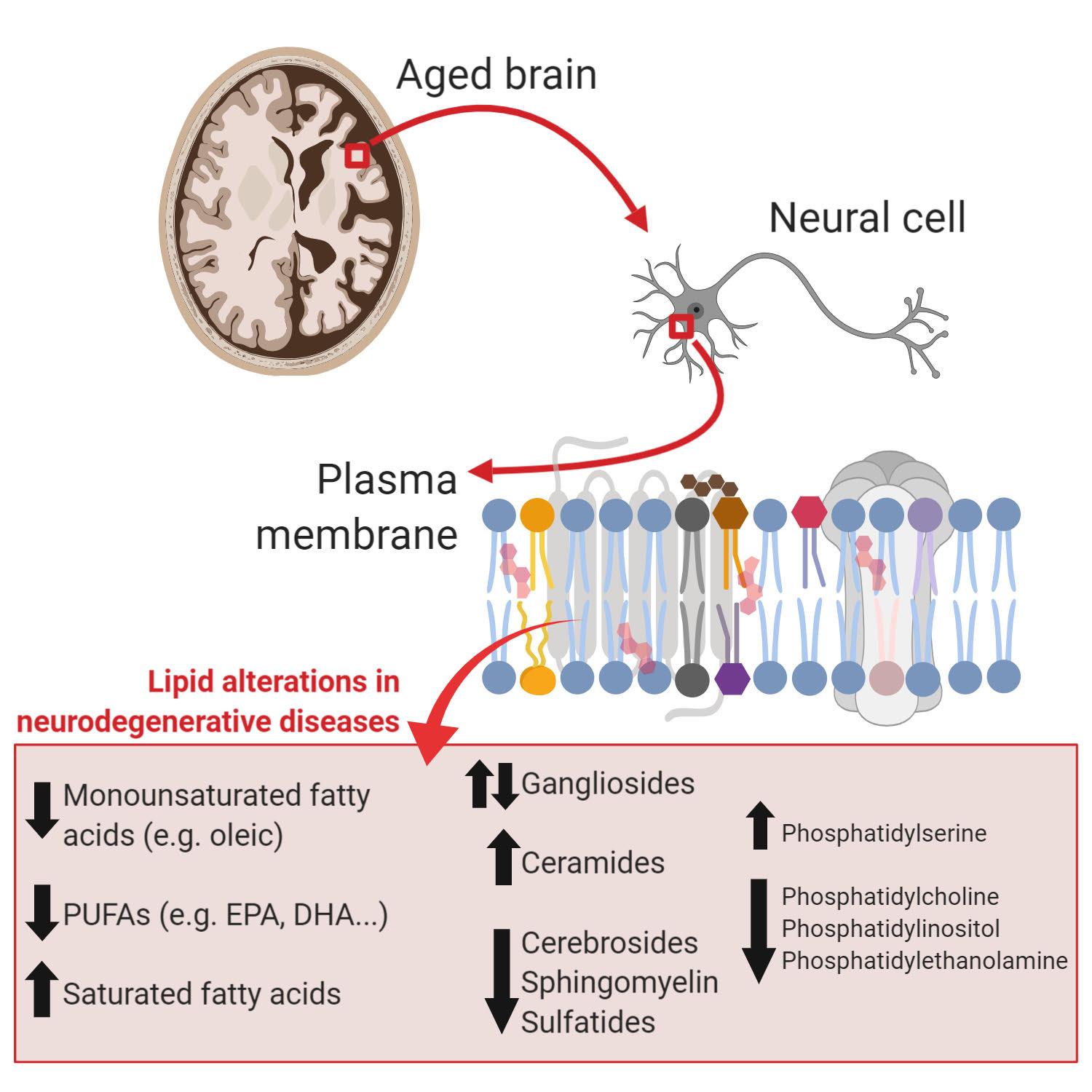{kind=link}
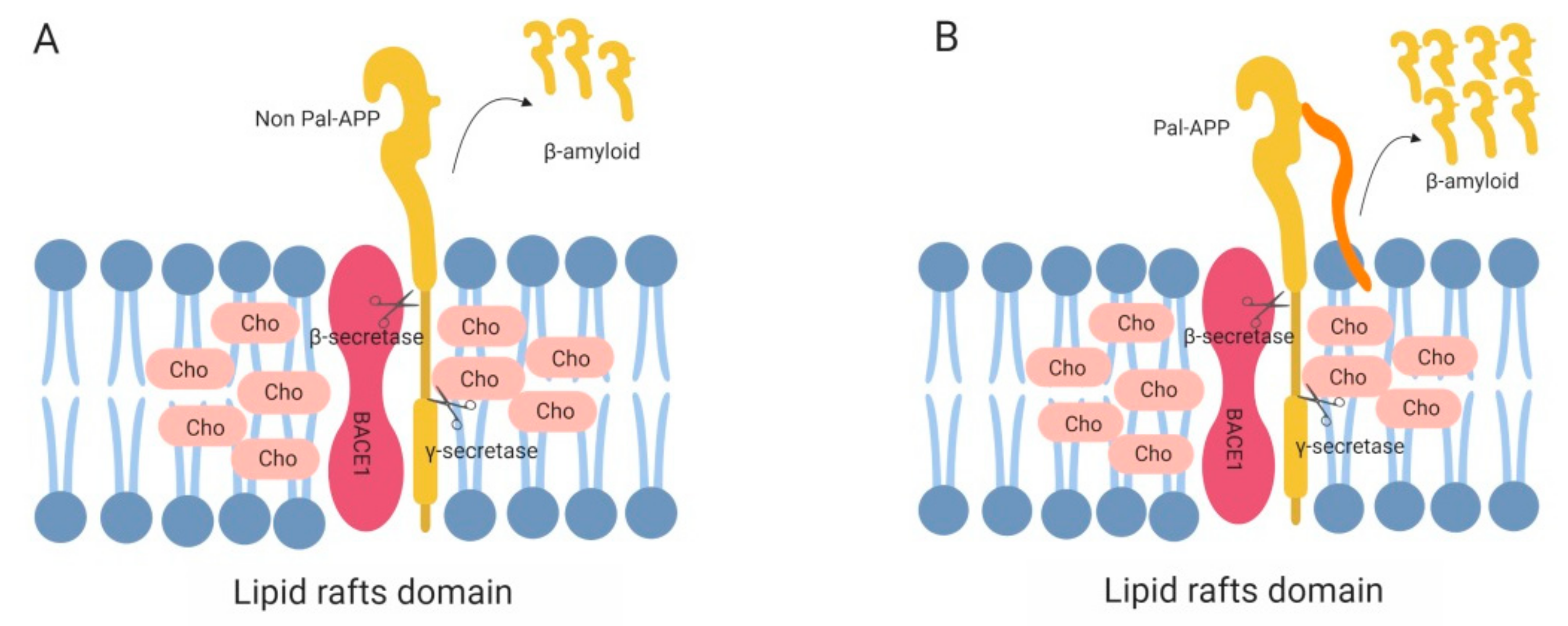{kind=link}
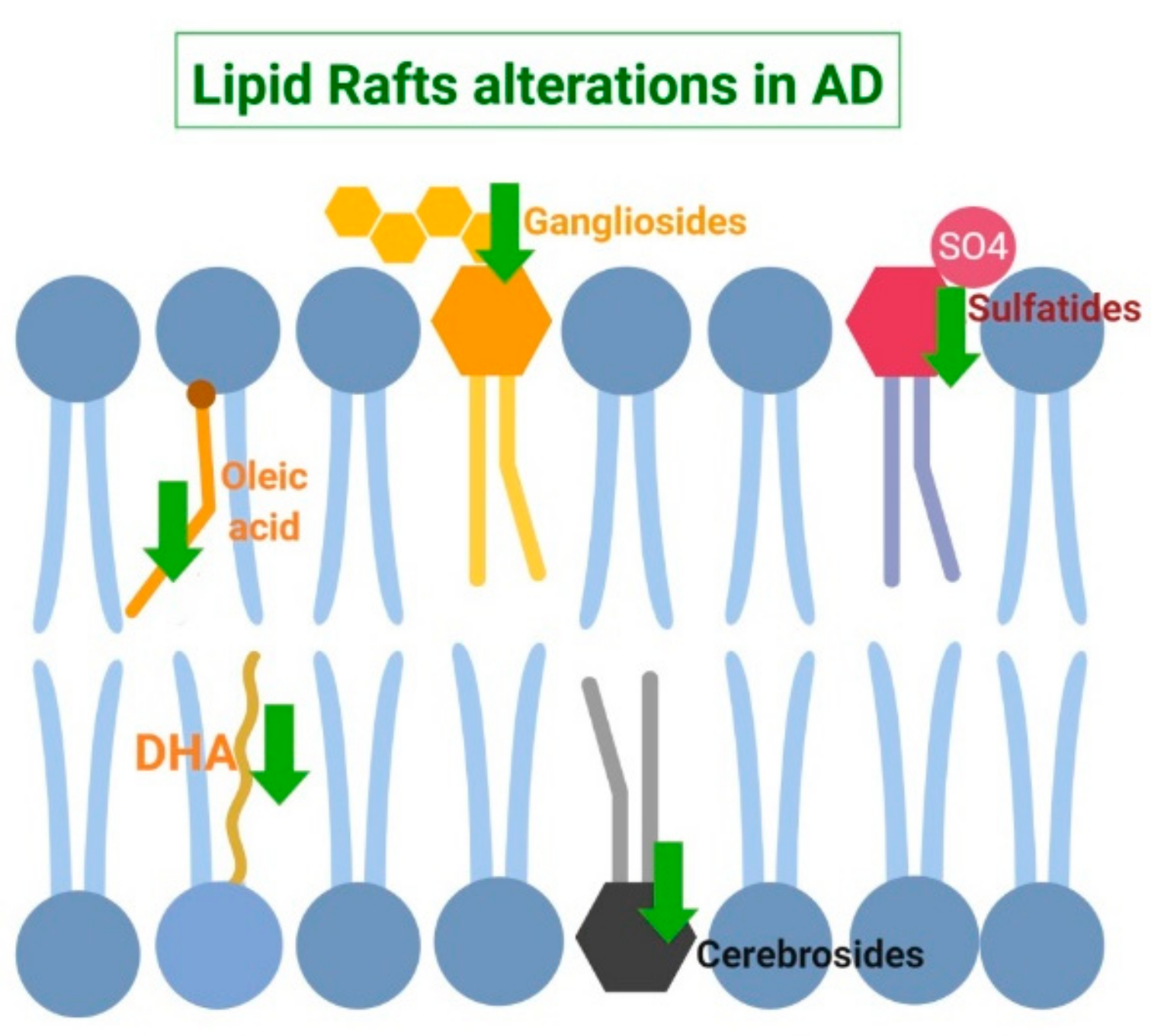{kind=link}
{kind=link}
| Distinct Changes of Lipid Species in Human Brain and Peripheral Fluids | |||
|---|---|---|---|
| Alzheimer’s Disease | |||
| Lipid | Variation | Sample | Reference |
| Ceramide | Increase | Brain tissue | [209] |
| Sphingomyelin | Decrease | Brain tissue | [209] |
| Phosphatidylcholine | Decrease | Serum | [210] |
| Phosphatidylinositol | Decrease | Serum | [210] |
| Phosphatidylethanolamine | Decrease | Serum | [210] |
| DHA | Decrease | Lipid rafts | [36] |
| Linolenic acid | Decrease | Plasma | [211] |
| ARA | Increase | Brain tissue | [212] |
| Palmitic acid | Increase | Brain tissue | [213] |
| Stearic acid | Increase | Brain tissue | [213] |
| Oleic acid | Decrease | Lipid rafts | [36] |
| Gangliosides | Decrease | Brain tissue/Lipid rafts | [36,46] |
| Cerebrosides | Decrease | Lipid rafts | [36] |
| Sulfatides | Decrease | Lipid rafts | [36] |
| Lipid Metabolites | |||
| 24-OH | Increase | Brain tissue | [172] |
| 27-OH | Increase | Brain tissue | [171] |
| Oxidative Stress Markers | |||
| MDA | Increase | Serum/Plasma | [174,175,214] |
| 4-HNE | Increase | Brain tissue | [176] |
| Proteins Related with Lipid Metabolism | |||
| HDL | Decrease | Serum | [77] |
| LDL | Increase | Serum | [77] |
| FABP3 | Increase | CSF | [177] |
| Parkinson’s Disease | |||
| Lipids | Variation | Sample | Reference |
| Ceramide | Increase | Brain tissue | [109] |
| Sphingomyelin | Decrease | Brain tissue | [215] |
| Phosphatidylcholine | Decrease | Brain tissue | [216] |
| Phosphatidylserine | Increase | Lipid rafts | [154] |
| Phosphatidylinositol | Decrease | Brain tissue | [204] |
| Phosphatidylethanolamine | Decreased | Brain tissue | [215] |
| EPA | Decrease | Lipid rafts | [49,154] |
| DHA | Decrease | Lipid rafts | [49,154] |
| Palmitic acid | Increase | Lipid rafts | [154] |
| Stearic acid | Increase | Lipid rafts | [154] |
| Oleic acid | Decrease | CSF | [217] |
| Palmitoleic acid | Decrease | CSF | [217] |
| Linoleic acid | Decrease | CSF | [217] |
| Gangliosides | Increase | Lipid rafts | [150] |
| Lipid Metabolites | |||
| Endocannabinoids | Increase | CSF | [199,200] |
| Apo1 | Decrease | CSF | [204,205] |
| Oxidative Stress Markers | |||
| MDA | Increase | Brain tissue | [205] |
| 4-HNE | Increase | SNPc/CSF | [206] |
| 8-OHdG | Increase | Serum/CSF | [209,210,211] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mesa-Herrera, F.; Taoro-González, L.; Valdés-Baizabal, C.; Diaz, M.; Marín, R. Lipid and Lipid Raft Alteration in Aging and Neurodegenerative Diseases: A Window for the Development of New Biomarkers. Int. J. Mol. Sci. 2019, 20, 3810. https://doi.org/10.3390/ijms20153810
Mesa-Herrera F, Taoro-González L, Valdés-Baizabal C, Diaz M, Marín R. Lipid and Lipid Raft Alteration in Aging and Neurodegenerative Diseases: A Window for the Development of New Biomarkers. International Journal of Molecular Sciences. 2019; 20(15):3810. https://doi.org/10.3390/ijms20153810
Chicago/Turabian StyleMesa-Herrera, Fátima, Lucas Taoro-González, Catalina Valdés-Baizabal, Mario Diaz, and Raquel Marín. 2019. "Lipid and Lipid Raft Alteration in Aging and Neurodegenerative Diseases: A Window for the Development of New Biomarkers" International Journal of Molecular Sciences 20, no. 15: 3810. https://doi.org/10.3390/ijms20153810
APA StyleMesa-Herrera, F., Taoro-González, L., Valdés-Baizabal, C., Diaz, M., & Marín, R. (2019). Lipid and Lipid Raft Alteration in Aging and Neurodegenerative Diseases: A Window for the Development of New Biomarkers. International Journal of Molecular Sciences, 20(15), 3810. https://doi.org/10.3390/ijms20153810