Complexity of Generating Mouse Models to Study the Upper Motor Neurons: Let Us Shift Focus from Mice to Neurons



Abstract
:1. Complexity of the Motor Neuron Circuitry
2. Developing Mouse Models to Study Upper Motor Neurons
3. Mouse Models Developed with Genetic Linkage to Upper Motor Neuron Diseases
4. Visualization of CSMN
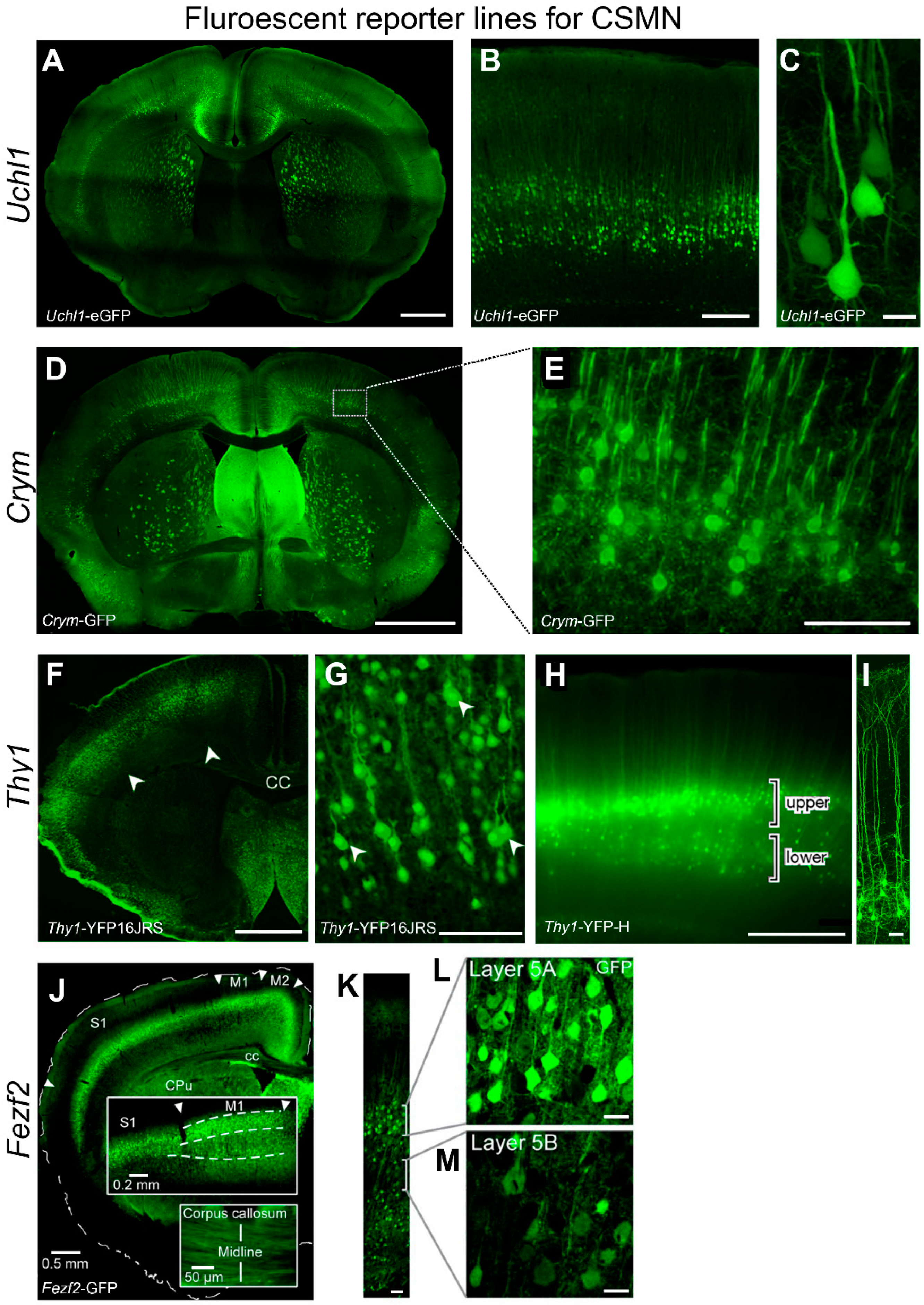
UCHL1 Offers a Unique Opportunity to Study Upper Motor Neuron Biology
5. Shifting Focus from Mice to Neurons Generates Translational Outcomes
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ALS | Amyotrophic Lateral Sclerosis |
| PLS | Primary Lateral Sclerosis |
| HSP | Hereditary Spastic Paraplegia |
| CSMN | corticospinal motor neuron(s) |
| CST | corticospinal tract |
| EM | electron microscopy |
| ER | endoplasmic reticulum |
| UCHL1 | ubiquitin C-terminal hydrolase ligase 1 |
| UMN | upper motor neuron |
| GFP | green fluorescent protein |
| KO | green fluorescent protein |
References
- Shepherd, G.M. Corticostriatal connectivity and its role in disease. Nat. Rev. Neurosci. 2013, 14, 278–291. [Google Scholar] [CrossRef] [PubMed]
- Jara, J.H.; Genc, B.; Klessner, J.L.; Ozdinler, P.H. Retrograde labeling, transduction, and genetic targeting allow cellular analysis of corticospinal motor neurons: Implications in health and disease. Front. Neuroanat. 2014, 8, 16. [Google Scholar] [CrossRef] [PubMed]
- Lemon, R.N. Descending pathways in motor control. Annu. Rev. Neurosci. 2008, 31, 195–218. [Google Scholar] [CrossRef] [PubMed]
- Lemon, R. Recent advances in our understanding of the primate corticospinal system. F1000Research 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Oudega, M.; Perez, M.A. Corticospinal reorganization after spinal cord injury. J. Physiol. 2012, 590, 3647–3663. [Google Scholar] [CrossRef] [PubMed]
- Kanning, K.C.; Kaplan, A.; Henderson, C.E. Motor neuron diversity in development and disease. Annu. Rev. Neurosci. 2010, 33, 409–440. [Google Scholar] [CrossRef]
- Fink, J.K. Progressive spastic paraparesis: Hereditary spastic paraplegia and its relation to primary and amyotrophic lateral sclerosis. Semin. Neurol. 2001, 21, 199–207. [Google Scholar] [CrossRef]
- Udaka, F.; Kameyama, M.; Tomonaga, M. Degeneration of Betz cells in motor neuron disease. A Golgi study. Acta Neuropathol. 1986, 70, 289–295. [Google Scholar] [CrossRef]
- Brown, R.H., Jr.; Robberecht, W. Amyotrophic lateral sclerosis: Pathogenesis. Semin. Neurol. 2001, 21, 131–139. [Google Scholar] [CrossRef]
- Baker, M.R. ALS—dying forward, backward or outward? Nat. Rev. Neurosci. 2014, 10, 660. [Google Scholar] [CrossRef]
- Ravits, J.; Paul, P.; Jorg, C. Focality of upper and lower motor neuron degeneration at the clinical onset of ALS. Neurology 2007, 68, 1571–1575. [Google Scholar] [CrossRef] [PubMed]
- Eisen, A.; Weber, M. The motor cortex and amyotrophic lateral sclerosis. Muscle Nerve 2001, 24, 564–573. [Google Scholar] [CrossRef] [PubMed]
- Fink, J.K. Hereditary spastic paraplegia: Clinico-pathologic features and emerging molecular mechanisms. Acta Neuropathol. 2013, 126, 307–328. [Google Scholar] [CrossRef] [PubMed]
- Blackstone, C. Converging cellular themes for the hereditary spastic paraplegias. Curr. Opin. Neuropathol. 2018, 51, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Blackstone, C. Cellular pathways of hereditary spastic paraplegia. Annu. Rev. Neurosci. 2012, 35, 25–47. [Google Scholar] [CrossRef] [PubMed]
- Boutry, M.; Morais, S.; Stevanin, G. Update on the Genetics of Spastic Paraplegias. Curr. Neurol. Neurosci. Rep. 2019, 19, 18. [Google Scholar] [CrossRef] [PubMed]
- Bis-Brewer, D.M.; Zuchner, S. Perspectives on the Genomics of HSP Beyond Mendelian Inheritance. Front. Neurol. 2018, 9, 958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dervishi, I.; Gozutok, O.; Murnan, K.; Gautam, M.; Heller, D.; Bigio, E.; Ozdinler, P.H. Protein-protein interactions reveal key canonical pathways, upstream regulators, interactome domains, and novel targets in ALS. Sci. Rep. 2018, 8, 14732. [Google Scholar] [CrossRef] [PubMed]
- Chia, R.; Chio, A.; Traynor, B.J. Novel genes associated with amyotrophic lateral sclerosis: Diagnostic and clinical implications. Lancet Neurol. 2018, 17, 94–102. [Google Scholar] [CrossRef]
- Ghasemi, M.; Brown, R.H., Jr. Genetics of Amyotrophic Lateral Sclerosis. Cold Spring Harb. Perspect. Med. 2018, 8. [Google Scholar] [CrossRef]
- Taylor, J.P.; Brown, R.H., Jr.; Cleveland, D.W. Decoding ALS: From genes to mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Gurney, M.E.; Pu, H.; Chiu, A.Y.; Dal Canto, M.C.; Polchow, C.Y.; Alexander, D.D.; Caliendo, J.; Hentati, A.; Kwon, Y.W.; Deng, H.X.; et al. Motor neuron degeneration in mice that express a human CuZn superoxide dismutase mutation. Science 1994, 264, 1772–1775. [Google Scholar] [CrossRef] [PubMed]
- Dobrovic, B.; Curic, G.; Petanjek, Z.; Heffer, M. Dendritic morphology and spine density is not altered in motor cortex and dentate granular cells in mice lacking the ganglioside biosynthetic gene B4galnt1—A quantitative Golgi cox study. Coll. Antropol. 2011, 35, 25–30. [Google Scholar] [PubMed]
- Karle, K.N.; Mockel, D.; Reid, E.; Schols, L. Axonal transport deficit in a KIF5A(-/-) mouse model. Neurogenetics 2012, 13, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Soderblom, C.; Stadler, J.; Jupille, H.; Blackstone, C.; Shupliakov, O.; Hanna, M.C. Targeted disruption of the Mast syndrome gene SPG21 in mice impairs hind limb function and alters axon branching in cultured cortical neurons. Neurogenetics 2010, 11, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Winrow, C.J.; Hemming, M.L.; Allen, D.M.; Quistad, G.B.; Casida, J.E.; Barlow, C. Loss of neuropathy target esterase in mice links organophosphate exposure to hyperactivity. Nat. Genet. 2003, 33, 477–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yagi, T.; Ito, D.; Nihei, Y.; Ishihara, T.; Suzuki, N. N88S seipin mutant transgenic mice develop features of seipinopathy/BSCL2-related motor neuron disease via endoplasmic reticulum stress. Hum. Mol. Genet. 2011, 20, 3831–3840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gautam, M.; Jara, J.H.; Sekerkova, G.; Yasvoina, M.V.; Martina, M.; Ozdinler, P.H. Absence of alsin function leads to corticospinal motor neuron vulnerability via novel disease mechanisms. Hum. Mol. Genet. 2016, 25, 1074–1087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Of men, not mice. Nat. Med. 2013, 19, 379. [CrossRef] [PubMed]
- Janus, C.; Welzl, H. Mouse models of neurodegenerative diseases: Criteria and general methodology. Methods Mol. Boil. 2010, 602, 323–345. [Google Scholar] [CrossRef]
- Ransohoff, R.M. All (animal) models (of neurodegeneration) are wrong. Are they also useful? J. Exp. Med. 2018, 215, 2955–2958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fassier, C.; Hazan, J.; Melki, J. Mouse Models of Autosomal Dominant Spastic Paraplegia. In Movement Disorders: Genetics and Models, 2nd ed.; Academic Press: Cambridge, MA, USA, 2015; pp. 1073–1086. [Google Scholar]
- Blackstone, C. Murine Models of Autosomal Recessive Hereditary Spastic Paraplegia. In Movement Disorders: Genetics and Models, 2nd ed.; Academic Press: Cambridge, MA, USA, 2015; pp. 1087–1093. [Google Scholar]
- Hentati, A.; Pericak-Vance, M.A.; Hung, W.Y.; Belal, S.; Laing, N.; Boustany, R.M.; Hentati, F.; Ben Hamida, M.; Siddique, T. Linkage of ‘pure’ autosomal recessive familial spastic paraplegia to chromosome 8 markers and evidence of genetic locus heterogeneity. Hum. Mol. Genet. 1994, 3, 1263–1267. [Google Scholar] [CrossRef] [PubMed]
- Tsaousidou, M.K.; Ouahchi, K.; Warner, T.T.; Yang, Y.; Simpson, M.A.; Laing, N.G.; Wilkinson, P.A.; Madrid, R.E.; Patel, H.; Hentati, F.; et al. Sequence alterations within CYP7B1 implicate defective cholesterol homeostasis in motor-neuron degeneration. Am. J. Hum. Genet. 2008, 82, 510–515. [Google Scholar] [CrossRef] [PubMed]
- Biddinger, S.B.; Haas, J.T.; Yu, B.B.; Bezy, O.; Jing, E.; Zhang, W.; Unterman, T.G.; Carey, M.C.; Kahn, C.R. Hepatic insulin resistance directly promotes formation of cholesterol gallstones. Nat. Med. 2008, 14, 778–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li-Hawkins, J.; Lund, E.G.; Turley, S.D.; Russell, D.W. Disruption of the oxysterol 7alpha-hydroxylase gene in mice. J. Biol. Chem. 2000, 275, 16536–16542. [Google Scholar] [CrossRef] [PubMed]
- Garner, C.C.; Garner, A.; Huber, G.; Kozak, C.; Matus, A. Molecular cloning of microtubule-associated protein 1 (MAP1A) and microtubule-associated protein 5 (MAP1B): Identification of distinct genes and their differential expression in developing brain. J. Neurochem. 1990, 55, 146–154. [Google Scholar] [CrossRef]
- De Michele, G.; De Fusco, M.; Cavalcanti, F.; Filla, A.; Marconi, R.; Volpe, G.; Monticelli, A.; Ballabio, A.; Casari, G.; Cocozza, S. A new locus for autosomal recessive hereditary spastic paraplegia maps to chromosome 16q24.3. Am. J. Hum. Genet. 1998, 63, 135–139. [Google Scholar] [CrossRef] [PubMed]
- Casari, G.; De Fusco, M.; Ciarmatori, S.; Zeviani, M.; Mora, M.; Fernandez, P.; De Michele, G.; Filla, A.; Cocozza, S.; Marconi, R.; et al. Spastic paraplegia and OXPHOS impairment caused by mutations in paraplegin, a nuclear-encoded mitochondrial metalloprotease. Cell 1998, 93, 973–983. [Google Scholar] [CrossRef]
- Koyama, K.; Emi, M.; Nakamura, Y. The cell adhesion regulator (CAR) gene, TaqI and insertion/deletion polymorphisms, and regional assignment to the peritelomeric region of 16q by linkage analysis. Genomics 1993, 16, 264–265. [Google Scholar] [CrossRef]
- Settasatian, C.; Whitmore, S.A.; Crawford, J.; Bilton, R.L.; Cleton-Jansen, A.M.; Sutherland, G.R.; Callen, D.F. Genomic structure and expression analysis of the spastic paraplegia gene, SPG7. Hum. Genet. 1999, 105, 139–144. [Google Scholar] [CrossRef]
- Ferreirinha, F.; Quattrini, A.; Pirozzi, M.; Valsecchi, V.; Dina, G.; Broccoli, V.; Auricchio, A.; Piemonte, F.; Tozzi, G.; Gaeta, L.; et al. Axonal degeneration in paraplegin-deficient mice is associated with abnormal mitochondria and impairment of axonal transport. J. Clin. Investig. 2004, 113, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Pirozzi, M.; Quattrini, A.; Andolfi, G.; Dina, G.; Malaguti, M.C.; Auricchio, A.; Rugarli, E.I. Intramuscular viral delivery of paraplegin rescues peripheral axonopathy in a model of hereditary spastic paraplegia. J. Clin. Investig. 2006, 116, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, P.; La Mattina, V.; Bernacchia, A.; Magnoni, R.; Cerri, F.; Cox, G.; Quattrini, A.; Casari, G.; Rugarli, E.I. Genetic interaction between the m-AAA protease isoenzymes reveals novel roles in cerebellar degeneration. Hum. Mol. Genet. 2009, 18, 2001–2013. [Google Scholar] [CrossRef] [PubMed]
- Martinez Murillo, F.; Kobayashi, H.; Pegoraro, E.; Galluzzi, G.; Creel, G.; Mariani, C.; Farina, E.; Ricci, E.; Alfonso, G.; Pauli, R.M.; et al. Genetic localization of a new locus for recessive familial spastic paraparesis to 15q13-15. Neurology 1999, 53, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Branchu, J.; Boutry, M.; Sourd, L.; Depp, M.; Leone, C.; Corriger, A.; Vallucci, M.; Esteves, T.; Matusiak, R.; Dumont, M.; et al. Loss of spatacsin function alters lysosomal lipid clearance leading to upper and lower motor neuron degeneration. Neurobiol. Dis. 2017, 102, 21–37. [Google Scholar] [CrossRef] [PubMed]
- Varga, R.E.; Khundadze, M.; Damme, M.; Nietzsche, S.; Hoffmann, B.; Stauber, T.; Koch, N.; Hennings, J.C.; Franzka, P.; Huebner, A.K.; et al. In Vivo Evidence for Lysosome Depletion and Impaired Autophagic Clearance in Hereditary Spastic Paraplegia Type SPG11. PLoS Genet. 2015, 11, e1005454. [Google Scholar] [CrossRef] [PubMed]
- Hughes, C.A.; Byrne, P.C.; Webb, S.; McMonagle, P.; Patterson, V.; Hutchinson, M.; Parfrey, N.A. SPG15, a new locus for autosomal recessive complicated HSP on chromosome 14q. Neurology 2001, 56, 1230–1233. [Google Scholar] [CrossRef] [PubMed]
- Khundadze, M.; Kollmann, K.; Koch, N.; Biskup, C.; Nietzsche, S.; Zimmer, G.; Hennings, J.C.; Huebner, A.K.; Symmank, J.; Jahic, A.; et al. A hereditary spastic paraplegia mouse model supports a role of ZFYVE26/SPASTIZIN for the endolysosomal system. PLoS Genet. 2013, 9, e1003988. [Google Scholar] [CrossRef] [PubMed]
- Patel, H.; Cross, H.; Proukakis, C.; Hershberger, R.; Bork, P.; Ciccarelli, F.D.; Patton, M.A.; McKusick, V.A.; Crosby, A.H. SPG20 is mutated in Troyer syndrome, an hereditary spastic paraplegia. Nat. Genet. 2002, 31, 347–348. [Google Scholar] [CrossRef] [PubMed]
- Cross, H.E.; McKusick, V.A. The Troyer syndrome. A recessive form of spastic paraplegia with distal muscle wasting. Arch. Neurol. 1967, 16, 473–485. [Google Scholar] [CrossRef]
- Renvoise, B.; Stadler, J.; Singh, R.; Bakowska, J.C.; Blackstone, C. Spg20-/-mice reveal multimodal functions for Troyer syndrome protein spartin in lipid droplet maintenance, cytokinesis and BMP signaling. Hum. Mol. Genet. 2012, 21, 3604–3618. [Google Scholar] [CrossRef] [PubMed]
- Simpson, M.A.; Cross, H.; Proukakis, C.; Pryde, A.; Hershberger, R.; Chatonnet, A.; Patton, M.A.; Crosby, A.H. Maspardin is mutated in mast syndrome, a complicated form of hereditary spastic paraplegia associated with dementia. Am. J. Hum. Genet. 2003, 73, 1147–1156. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, P.A.; Simpson, M.A.; Bastaki, L.; Patel, H.; Reed, J.A.; Kalidas, K.; Samilchuk, E.; Khan, R.; Warner, T.T.; Crosby, A.H. A new locus for autosomal recessive complicated hereditary spastic paraplegia (SPG26) maps to chromosome 12p11.1-12q14. J. Med. Gene. 2005, 42, 80–82. [Google Scholar] [CrossRef] [PubMed]
- Bouslam, N.; Benomar, A.; Azzedine, H.; Bouhouche, A.; Namekawa, M.; Klebe, S.; Charon, C.; Durr, A.; Ruberg, M.; Brice, A.; et al. Mapping of a new form of pure autosomal recessive spastic paraplegia (SPG28). Ann. Neurol. 2005, 57, 567–571. [Google Scholar] [CrossRef] [PubMed]
- Inoue, A.; Arima, N.; Ishiguro, J.; Prestwich, G.D.; Arai, H.; Aoki, J. LPA-producing enzyme PA-PLA(1)alpha regulates hair follicle development by modulating EGFR signalling. EMBO J. 2011, 30, 4248–4260. [Google Scholar] [CrossRef] [PubMed]
- Baba, T.; Kashiwagi, Y.; Arimitsu, N.; Kogure, T.; Edo, A.; Maruyama, T.; Nakao, K.; Nakanishi, H.; Kinoshita, M.; Frohman, M.A.; et al. Phosphatidic acid (PA)-preferring phospholipase A1 regulates mitochondrial dynamics. J. Biol. Chem. 2014, 289, 11497–11511. [Google Scholar] [CrossRef] [PubMed]
- Klebe, S.; Azzedine, H.; Durr, A.; Bastien, P.; Bouslam, N.; Elleuch, N.; Forlani, S.; Charon, C.; Koenig, M.; Melki, J.; et al. Autosomal recessive spastic paraplegia (SPG30) with mild ataxia and sensory neuropathy maps to chromosome 2q37.3. Brain 2006, 129, 1456–1462. [Google Scholar] [CrossRef] [Green Version]
- Kondo, M.; Takei, Y.; Hirokawa, N. Motor protein KIF1A is essential for hippocampal synaptogenesis and learning enhancement in an enriched environment. Neuron 2012, 73, 743–757. [Google Scholar] [CrossRef]
- Yonekawa, Y.; Harada, A.; Okada, Y.; Funakoshi, T.; Kanai, Y.; Takei, Y.; Terada, S.; Noda, T.; Hirokawa, N. Defect in synaptic vesicle precursor transport and neuronal cell death in KIF1A motor protein-deficient mice. J. Cell Biol. 1998, 141, 431–441. [Google Scholar] [CrossRef]
- Meixner, M.; Jungnickel, J.; Grothe, C.; Gieselmann, V.; Eckhardt, M. Myelination in the absence of UDP-galactose:ceramide galactosyl-transferase and fatty acid 2-hydroxylase. BMC Neurosci. 2011, 12, 22. [Google Scholar] [CrossRef]
- Dick, K.J.; Al-Mjeni, R.; Baskir, W.; Koul, R.; Simpson, M.A.; Patton, M.A.; Raeburn, S.; Crosby, A.H. A novel locus for an autosomal recessive hereditary spastic paraplegia (SPG35) maps to 16q21-q23. Neurology 2008, 71, 248–252. [Google Scholar] [CrossRef]
- Potter, K.A.; Kern, M.J.; Fullbright, G.; Bielawski, J.; Scherer, S.S.; Yum, S.W.; Li, J.J.; Cheng, H.; Han, X.; Venkata, J.K.; et al. Central nervous system dysfunction in a mouse model of FA2H deficiency. Glia 2011, 59, 1009–1021. [Google Scholar] [CrossRef]
- Zoller, I.; Meixner, M.; Hartmann, D.; Bussow, H.; Meyer, R.; Gieselmann, V.; Eckhardt, M. Absence of 2-hydroxylated sphingolipids is compatible with normal neural development but causes late-onset axon and myelin sheath degeneration. J. Neurosci. 2008, 28, 9741–9754. [Google Scholar] [CrossRef]
- Rainier, S.; Bui, M.; Mark, E.; Thomas, D.; Tokarz, D.; Ming, L.; Delaney, C.; Richardson, R.J.; Albers, J.W.; Matsunami, N.; et al. Neuropathy target esterase gene mutations cause motor neuron disease. Am. J. Hum. Genet. 2008, 82, 780–785. [Google Scholar] [CrossRef]
- Moser, M.; Li, Y.; Vaupel, K.; Kretzschmar, D.; Kluge, R.; Glynn, P.; Buettner, R. Placental failure and impaired vasculogenesis result in embryonic lethality for neuropathy target esterase-deficient mice. Mol. Cell Biol. 2004, 24, 1667–1679. [Google Scholar] [CrossRef]
- Orthmann-Murphy, J.L.; Salsano, E.; Abrams, C.K.; Bizzi, A.; Uziel, G.; Freidin, M.M.; Lamantea, E.; Zeviani, M.; Scherer, S.S.; Pareyson, D. Hereditary spastic paraplegia is a novel phenotype for GJA12/GJC2 mutations. Brain 2009, 132, 426–438. [Google Scholar] [CrossRef]
- Georgiou, E.; Sidiropoulou, K.; Richter, J.; Papaneophytou, C.; Sargiannidou, I.; Kagiava, A.; von Jonquieres, G.; Christodoulou, C.; Klugmann, M.; Kleopa, K.A. Gene therapy targeting oligodendrocytes provides therapeutic benefit in a leukodystrophy model. Brain 2017, 140, 599–616. [Google Scholar] [CrossRef]
- Tress, O.; Maglione, M.; Zlomuzica, A.; May, D.; Dicke, N.; Degen, J.; Dere, E.; Kettenmann, H.; Hartmann, D.; Willecke, K. Pathologic and phenotypic alterations in a mouse expressing a connexin47 missense mutation that causes Pelizaeus-Merzbacher-like disease in humans. PLoS Genet. 2011, 7, e1002146. [Google Scholar] [CrossRef]
- Nelles, E.; Butzler, C.; Jung, D.; Temme, A.; Gabriel, H.D.; Dahl, U.; Traub, O.; Stumpel, F.; Jungermann, K.; Zielasek, J.; et al. Defective propagation of signals generated by sympathetic nerve stimulation in the liver of connexin32-deficient mice. Proc. Natl. Acad. Sci. USA 1996, 93, 9565–9570. [Google Scholar] [CrossRef]
- Odermatt, B.; Wellershaus, K.; Wallraff, A.; Seifert, G.; Degen, J.; Euwens, C.; Fuss, B.; Bussow, H.; Schilling, K.; Steinhauser, C.; et al. Connexin 47 (Cx47)-deficient mice with enhanced green fluorescent protein reporter gene reveal predominant oligodendrocytic expression of Cx47 and display vacuolized myelin in the CNS. J. Neurosci. 2003, 23, 4549–4559. [Google Scholar] [CrossRef]
- Dursun, U.; Koroglu, C.; Kocasoy Orhan, E.; Ugur, S.A.; Tolun, A. Autosomal recessive spastic paraplegia (SPG45) with mental retardation maps to 10q24.3–q25.1. Neurogenetics 2009, 10, 325–331. [Google Scholar] [CrossRef]
- Kviklyte, S.; Vertommen, D.; Yerna, X.; Andersen, H.; Xu, X.; Gailly, P.; Bohlooly, Y.M.; Oscarsson, J.; Rider, M.H. Effects of genetic deletion of soluble 5’-nucleotidases NT5C1A and NT5C2 on AMPK activation and nucleotide levels in contracting mouse skeletal muscles. Am. J. Physiol. Endocrinol. Metab. 2017, 313, E48–E62. [Google Scholar] [CrossRef]
- Boukhris, A.; Feki, I.; Elleuch, N.; Miladi, M.I.; Boland-Auge, A.; Truchetto, J.; Mundwiller, E.; Jezequel, N.; Zelenika, D.; Mhiri, C.; et al. A new locus (SPG46) maps to 9p21.2–q21.12 in a Tunisian family with a complicated autosomal recessive hereditary spastic paraplegia with mental impairment and thin corpus callosum. Neurogenetics 2010, 11, 441–448. [Google Scholar] [CrossRef]
- Matern, H.; Boermans, H.; Lottspeich, F.; Matern, S. Molecular cloning and expression of human bile acid beta-glucosidase. J. Biol. Chem. 2001, 276, 37929–37933. [Google Scholar] [CrossRef]
- Massimo, A.; Maura, S.; Nicoletta, L.; Giulia, M.; Valentina, M.; Elena, C.; Alessandro, P.; Rosaria, B.; Sandro, S. Current and Novel Aspects on the Non-lysosomal beta-Glucosylceramidase GBA2. Neurochem. Res. 2016, 41, 210–220. [Google Scholar] [CrossRef]
- Sultana, S.; Reichbauer, J.; Schule, R.; Mochel, F.; Synofzik, M.; van der Spoel, A.C. Lack of enzyme activity in GBA2 mutants associated with hereditary spastic paraplegia/cerebellar ataxia (SPG46). Biochem. Biophys. Res. Commun. 2015, 465, 35–40. [Google Scholar] [CrossRef]
- Yildiz, Y.; Matern, H.; Thompson, B.; Allegood, J.C.; Warren, R.L.; Ramirez, D.M.; Hammer, R.E.; Hamra, F.K.; Matern, S.; Russell, D.W. Mutation of beta-glucosidase 2 causes glycolipid storage disease and impaired male fertility. J. Clin. Investig. 2006, 116, 2985–2994. [Google Scholar] [CrossRef]
- Martin, E.; Schule, R.; Smets, K.; Rastetter, A.; Boukhris, A.; Loureiro, J.L.; Gonzalez, M.A.; Mundwiller, E.; Deconinck, T.; Wessner, M.; et al. Loss of function of glucocerebrosidase GBA2 is responsible for motor neuron defects in hereditary spastic paraplegia. Am. J. Hum. Genet. 2013, 92, 238–244. [Google Scholar] [CrossRef]
- Blumkin, L.; Lerman-Sagie, T.; Lev, D.; Yosovich, K.; Leshinsky-Silver, E. A new locus (SPG47) maps to 1p13.2-1p12 in an Arabic family with complicated autosomal recessive hereditary spastic paraplegia and thin corpus callosum. J. Neurol. Sci. 2011, 305, 67–70. [Google Scholar] [CrossRef]
- Matsuda, S.; Miura, E.; Matsuda, K.; Kakegawa, W.; Kohda, K.; Watanabe, M.; Yuzaki, M. Accumulation of AMPA receptors in autophagosomes in neuronal axons lacking adaptor protein AP-4. Neuron 2008, 57, 730–745. [Google Scholar] [CrossRef]
- Slabicki, M.; Theis, M.; Krastev, D.B.; Samsonov, S.; Mundwiller, E.; Junqueira, M.; Paszkowski-Rogacz, M.; Teyra, J.; Heninger, A.K.; Poser, I.; et al. A genome-scale DNA repair RNAi screen identifies SPG48 as a novel gene associated with hereditary spastic paraplegia. PLoS Biol. 2010, 8, e1000408. [Google Scholar] [CrossRef]
- Khundadze, M.; Ribaudo, F.; Hussain, A.; Rosentreter, J.; Nietzsche, S.; Thelen, M.; Winter, D.; Hoffmann, B.; Afzal, M.A.; Hermann, T.; et al. A mouse model for SPG48 reveals a block of autophagic flux upon disruption of adaptor protein complex five. Neurobiol. Dis. 2019, 127, 419–431. [Google Scholar] [CrossRef]
- Al-Yahyaee, S.; Al-Gazali, L.I.; De Jonghe, P.; Al-Barwany, H.; Al-Kindi, M.; De Vriendt, E.; Chand, P.; Koul, R.; Jacob, P.C.; Gururaj, A.; et al. A novel locus for hereditary spastic paraplegia with thin corpus callosum and epilepsy. Neurology 2006, 66, 1230–1234. [Google Scholar] [CrossRef]
- Inloes, J.M.; Kiosses, W.B.; Wang, H.; Walther, T.C.; Farese, R.V., Jr.; Cravatt, B.F. Functional Contribution of the Spastic Paraplegia-Related Triglyceride Hydrolase DDHD2 to the Formation and Content of Lipid Droplets. Biochemistry 2018, 57, 827–838. [Google Scholar] [CrossRef]
- Inloes, J.M.; Hsu, K.L.; Dix, M.M.; Viader, A.; Masuda, K.; Takei, T.; Wood, M.R.; Cravatt, B.F. The hereditary spastic paraplegia-related enzyme DDHD2 is a principal brain triglyceride lipase. Proc. Natl. Acad. Sci. USA 2014, 111, 14924–14929. [Google Scholar] [CrossRef] [Green Version]
- Novarino, G.; Fenstermaker, A.G.; Zaki, M.S.; Hofree, M.; Silhavy, J.L.; Heiberg, A.D.; Abdellateef, M.; Rosti, B.; Scott, E.; Mansour, L.; et al. Exome sequencing links corticospinal motor neuron disease to common neurodegenerative disorders. Science 2014, 343, 506–511. [Google Scholar] [CrossRef]
- Helmering, J.; Juan, T.; Li, C.M.; Chhoa, M.; Baron, W.; Gyuris, T.; Richards, W.G.; Turk, J.R.; Lawrence, J.; Cosgrove, P.A.; et al. A mutation in Ampd2 is associated with nephrotic syndrome and hypercholesterolemia in mice. Lipids Health Dis. 2014, 13, 167. [Google Scholar] [CrossRef]
- Akizu, N.; Cantagrel, V.; Schroth, J.; Cai, N.; Vaux, K.; McCloskey, D.; Naviaux, R.K.; Van Vleet, J.; Fenstermaker, A.G.; Silhavy, J.L.; et al. AMPD2 regulates GTP synthesis and is mutated in a potentially treatable neurodegenerative brainstem disorder. Cell 2013, 154, 505–517. [Google Scholar] [CrossRef]
- Li, P.; Ogino, K.; Hoshikawa, Y.; Morisaki, H.; Toyama, K.; Morisaki, T.; Morikawa, K.; Ninomiya, H.; Yoshida, A.; Hashimoto, K.; et al. AMP deaminase 3 plays a critical role in remote reperfusion lung injury. Biochem. Biophys. Res. Commun. 2013, 434, 131–136. [Google Scholar] [CrossRef]
- Friedman, D.J.; Kunzli, B.M.; Yi, A.R.; Sevigny, J.; Berberat, P.O.; Enjyoji, K.; Csizmadia, E.; Friess, H.; Robson, S.C. From the Cover: CD39 deletion exacerbates experimental murine colitis and human polymorphisms increase susceptibility to inflammatory bowel disease. Proc. Natl. Acad. Sci. USA 2009, 106, 16788–16793. [Google Scholar] [CrossRef]
- Friedman, D.J.; Rennke, H.G.; Csizmadia, E.; Enjyoji, K.; Robson, S.C. The vascular ectonucleotidase ENTPD1 is a novel renoprotective factor in diabetic nephropathy. Diabetes 2007, 56, 2371–2379. [Google Scholar] [CrossRef]
- Enjyoji, K.; Sevigny, J.; Lin, Y.; Frenette, P.S.; Christie, P.D.; Esch, J.S., 2nd; Imai, M.; Edelberg, J.M.; Rayburn, H.; Lech, M.; et al. Targeted disruption of cd39/ATP diphosphohydrolase results in disordered hemostasis and thromboregulation. Nat. Med. 1999, 5, 1010–1017. [Google Scholar] [CrossRef]
- Lossos, A.; Elazar, N.; Lerer, I.; Schueler-Furman, O.; Fellig, Y.; Glick, B.; Zimmerman, B.E.; Azulay, H.; Dotan, S.; Goldberg, S.; et al. Myelin-associated glycoprotein gene mutation causes Pelizaeus-Merzbacher disease-like disorder. Brain 2015, 138, 2521–2536. [Google Scholar] [CrossRef]
- Bartsch, S.; Montag, D.; Schachner, M.; Bartsch, U. Increased number of unmyelinated axons in optic nerves of adult mice deficient in the myelin-associated glycoprotein (MAG). Brain Res. 1997, 762, 231–234. [Google Scholar] [CrossRef]
- Cafferty, W.B.; Duffy, P.; Huebner, E.; Strittmatter, S.M. MAG and OMgp synergize with Nogo-A to restrict axonal growth and neurological recovery after spinal cord trauma. J. Neurosci. 2010, 30, 6825–6837. [Google Scholar] [CrossRef]
- Jones, M.V.; Nguyen, T.T.; Ewaleifoh, O.; Lebson, L.; Whartenby, K.A.; Griffin, J.W.; Calabresi, P.A. Accelerated axon loss in MOG35-55 experimental autoimmune encephalomyelitis (EAE) in myelin-associated glycoprotein-deficient (MAGKO) mice. J. Neuroimmunol. 2013, 262, 53–61. [Google Scholar] [CrossRef]
- Li, M.; Shibata, A.; Li, C.; Braun, P.E.; McKerracher, L.; Roder, J.; Kater, S.B.; David, S. Myelin-associated glycoprotein inhibits neurite/axon growth and causes growth cone collapse. J. Neurosci. Res. 1996, 46, 404–414. [Google Scholar] [CrossRef]
- Lopez, P.H.; Ahmad, A.S.; Mehta, N.R.; Toner, M.; Rowland, E.A.; Zhang, J.; Dore, S.; Schnaar, R.L. Myelin-associated glycoprotein protects neurons from excitotoxicity. J. Neurochem. 2011, 116, 900–908. [Google Scholar] [CrossRef] [Green Version]
- Marcus, J.; Dupree, J.L.; Popko, B. Myelin-associated glycoprotein and myelin galactolipids stabilize developing axo-glial interactions. J. Cell Biol. 2002, 156, 567–577. [Google Scholar] [CrossRef]
- Montag, D.; Giese, K.P.; Bartsch, U.; Martini, R.; Lang, Y.; Bluthmann, H.; Karthigasan, J.; Kirschner, D.A.; Wintergerst, E.S.; Nave, K.A.; et al. Mice deficient for the myelin-associated glycoprotein show subtle abnormalities in myelin. Neuron 1994, 13, 229–246. [Google Scholar] [CrossRef]
- Pan, B.; Fromholt, S.E.; Hess, E.J.; Crawford, T.O.; Griffin, J.W.; Sheikh, K.A.; Schnaar, R.L. Myelin-associated glycoprotein and complementary axonal ligands, gangliosides, mediate axon stability in the CNS and PNS: Neuropathology and behavioral deficits in single- and double-null mice. Exp. Neurol. 2005, 195, 208–217. [Google Scholar] [CrossRef] [Green Version]
- Gan-Or, Z.; Bouslam, N.; Birouk, N.; Lissouba, A.; Chambers, D.B.; Veriepe, J.; Androschuk, A.; Laurent, S.B.; Rochefort, D.; Spiegelman, D.; et al. Mutations in CAPN1 Cause Autosomal-Recessive Hereditary Spastic Paraplegia. Am. J. Hum. Genet. 2016, 98, 1038–1046. [Google Scholar] [CrossRef] [Green Version]
- Stifanese, R.; Averna, M.; De Tullio, R.; Pedrazzi, M.; Milanese, M.; Bonifacino, T.; Bonanno, G.; Salamino, F.; Pontremoli, S.; Melloni, E. Role of calpain-1 in the early phase of experimental ALS. Arch. Biochem. Biophys. 2014, 562, 1–8. [Google Scholar] [CrossRef]
- Wang, Y.; Hersheson, J.; Lopez, D.; Hammer, M.; Liu, Y.; Lee, K.H.; Pinto, V.; Seinfeld, J.; Wiethoff, S.; Sun, J.; et al. Defects in the CAPN1 Gene Result in Alterations in Cerebellar Development and Cerebellar Ataxia in Mice and Humans. Cell Rep. 2016, 16, 79–91. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.G.; Li, Y.; Raza, K.; Yu, X.X.; Ghoshal, S.; Geddes, J.W. Calpain 1 knockdown improves tissue sparing and functional outcomes after spinal cord injury in rats. J. Neurotrauma 2013, 30, 427–433. [Google Scholar] [CrossRef]
- Arthur, J.S.; Elce, J.S.; Hegadorn, C.; Williams, K.; Greer, P.A. Disruption of the murine calpain small subunit gene, Capn4: Calpain is essential for embryonic development but not for cell growth and division. Mol. Cell. Biol. 2000, 20, 4474–4481. [Google Scholar] [CrossRef]
- Kara, E.; Tucci, A.; Manzoni, C.; Lynch, D.S.; Elpidorou, M.; Bettencourt, C.; Chelban, V.; Manole, A.; Hamed, S.A.; Haridy, N.A.; et al. Genetic and phenotypic characterization of complex hereditary spastic paraplegia. Brain 2016, 139, 1904–1918. [Google Scholar] [CrossRef]
- Estrada-Cuzcano, A.; Martin, S.; Chamova, T.; Synofzik, M.; Timmann, D.; Holemans, T.; Andreeva, A.; Reichbauer, J.; De Rycke, R.; Chang, D.I.; et al. Loss-of-function mutations in the ATP13A2/PARK9 gene cause complicated hereditary spastic paraplegia (SPG78). Brain 2017, 140, 287–305. [Google Scholar] [CrossRef]
- Fleming, S.M.; Santiago, N.A.; Mullin, E.J.; Pamphile, S.; Karkare, S.; Lemkuhl, A.; Ekhator, O.R.; Linn, S.C.; Holden, J.G.; Aga, D.S.; et al. The effect of manganese exposure in Atp13a2-deficient mice. Neurotoxicology 2018, 64, 256–266. [Google Scholar] [CrossRef]
- Gusdon, A.M.; Zhu, J.; Van Houten, B.; Chu, C.T. ATP13A2 regulates mitochondrial bioenergetics through macroautophagy. Neurobiol. Dis. 2012, 45, 962–972. [Google Scholar] [CrossRef] [Green Version]
- Kett, L.R.; Stiller, B.; Bernath, M.M.; Tasset, I.; Blesa, J.; Jackson-Lewis, V.; Chan, R.B.; Zhou, B.; Di Paolo, G.; Przedborski, S.; et al. alpha-Synuclein-independent histopathological and motor deficits in mice lacking the endolysosomal Parkinsonism protein Atp13a2. J. Neurosci. 2015, 35, 5724–5742. [Google Scholar] [CrossRef]
- Qiao, C.; Yin, N.; Gu, H.Y.; Zhu, J.L.; Ding, J.H.; Lu, M.; Hu, G. Atp13a2 Deficiency Aggravates Astrocyte-Mediated Neuroinflammation via NLRP3 Inflammasome Activation. CNS Neurosci. Ther. 2016, 22, 451–460. [Google Scholar] [CrossRef]
- Sato, S.; Koike, M.; Funayama, M.; Ezaki, J.; Fukuda, T.; Ueno, T.; Uchiyama, Y.; Hattori, N. Lysosomal Storage of Subunit c of Mitochondrial ATP Synthase in Brain-Specific Atp13a2-Deficient Mice. Am. J. Pathol. 2016, 186, 3074–3082. [Google Scholar] [CrossRef] [Green Version]
- Schultheis, P.J.; Fleming, S.M.; Clippinger, A.K.; Lewis, J.; Tsunemi, T.; Giasson, B.; Dickson, D.W.; Mazzulli, J.R.; Bardgett, M.E.; Haik, K.L.; et al. Atp13a2-deficient mice exhibit neuronal ceroid lipofuscinosis, limited alpha-synuclein accumulation and age-dependent sensorimotor deficits. Hum. Mol. Genet. 2013, 22, 2067–2082. [Google Scholar] [CrossRef]
- Tsunemi, T.; Hamada, K.; Krainc, D. ATP13A2/PARK9 regulates secretion of exosomes and alpha-synuclein. J. Neurosci. 2014, 34, 15281–15287. [Google Scholar] [CrossRef]
- Usenovic, M.; Tresse, E.; Mazzulli, J.R.; Taylor, J.P.; Krainc, D. Deficiency of ATP13A2 leads to lysosomal dysfunction, alpha-synuclein accumulation, and neurotoxicity. J. Neurosci. 2012, 32, 4240–4246. [Google Scholar] [CrossRef]
- Bilguvar, K.; Tyagi, N.K.; Ozkara, C.; Tuysuz, B.; Bakircioglu, M.; Choi, M.; Delil, S.; Caglayan, A.O.; Baranoski, J.F.; Erturk, O.; et al. Recessive loss of function of the neuronal ubiquitin hydrolase UCHL1 leads to early-onset progressive neurodegeneration. Proc. Natl. Acad. Sci. USA 2013, 110, 3489–3494. [Google Scholar] [CrossRef] [Green Version]
- Jara, J.H.; Genc, B.; Cox, G.A.; Bohn, M.C.; Roos, R.P.; Macklis, J.D.; Ulupinar, E.; Ozdinler, P.H. Corticospinal Motor Neurons Are Susceptible to Increased ER Stress and Display Profound Degeneration in the Absence of UCHL1 Function. Cereb. Cortex 2015, 25, 4259–4272. [Google Scholar] [CrossRef] [Green Version]
- Saigoh, K.; Wang, Y.L.; Suh, J.G.; Yamanishi, T.; Sakai, Y.; Kiyosawa, H.; Harada, T.; Ichihara, N.; Wakana, S.; Kikuchi, T.; et al. Intragenic deletion in the gene encoding ubiquitin carboxy-terminal hydrolase in gad mice. Nat. Genet. 1999, 23, 47–51. [Google Scholar] [CrossRef]
- Walters, B.J.; Campbell, S.L.; Chen, P.C.; Taylor, A.P.; Schroeder, D.G.; Dobrunz, L.E.; Artavanis-Tsakonas, K.; Ploegh, H.L.; Wilson, J.A.; Cox, G.A.; et al. Differential effects of Usp14 and Uch-L1 on the ubiquitin proteasome system and synaptic activity. Mol. Cell. Neurosci. 2008, 39, 539–548. [Google Scholar] [CrossRef] [Green Version]
- Coulombe, J.; Gamage, P.; Gray, M.T.; Zhang, M.; Tang, M.Y.; Woulfe, J.; Saffrey, M.J.; Gray, D.A. Loss of UCHL1 promotes age-related degenerative changes in the enteric nervous system. Front. Aging Neurosci. 2014, 6, 129. [Google Scholar] [CrossRef]
- Reinicke, A.T.; Laban, K.; Sachs, M.; Kraus, V.; Walden, M.; Damme, M.; Sachs, W.; Reichelt, J.; Schweizer, M.; Janiesch, P.C.; et al. Ubiquitin C-terminal hydrolase L1 (UCH-L1) loss causes neurodegeneration by altering protein turnover in the first postnatal weeks. Proc. Natl. Acad. Sci. USA 2019, 116, 7963–7972. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Sugiura, Y.; Myers, K.G.; Liu, Y.; Lin, W. Ubiquitin carboxyl-terminal hydrolase L1 is required for maintaining the structure and function of the neuromuscular junction. Proc. Natl. Acad. Sci. USA 2010, 107, 1636–1641. [Google Scholar] [CrossRef] [Green Version]
- Yamazaki, K.; Wakasugi, N.; Tomita, T.; Kikuchi, T.; Mukoyama, M.; Ando, K. Gracile axonal dystrophy (GAD), a new neurological mutant in the mouse. Proc. Soc. Exp. Biol. Med. 1988, 187, 209–215. [Google Scholar] [CrossRef]
- Suh, J.G.; Yamanishi, T.; Matsui, K.; Tanaka, K.; Wada, K. Mapping of the gracile axonal dystrophy (gad) gene to a region between D5Mit197 and D5Mit113 on proximal mouse chromosome 5. Genomics 1995, 27, 549–551. [Google Scholar] [CrossRef]
- Genc, B.; Jara, J.H.; Schultz, M.C.; Manuel, M.; Stanford, M.J.; Gautam, M.; Klessner, J.L.; Sekerkova, G.; Heller, D.B.; Cox, G.A.; et al. Absence of UCHL 1 function leads to selective motor neuropathy. Ann. Clin. Transl. Neurol. 2016, 3, 331–345. [Google Scholar] [CrossRef]
- Miura, H.; Oda, K.; Endo, C.; Yamazaki, K.; Shibasaki, H.; Kikuchi, T. Progressive degeneration of motor nerve terminals in GAD mutant mouse with hereditary sensory axonopathy. Neuropathol. Appl. Neurobiol. 1993, 19, 41–51. [Google Scholar] [CrossRef]
- Hazan, J.; Lamy, C.; Melki, J.; Munnich, A.; de Recondo, J.; Weissenbach, J. Autosomal dominant familial spastic paraplegia is genetically heterogeneous and one locus maps to chromosome 14q. Nat. Genet. 1993, 5, 163–167. [Google Scholar] [CrossRef]
- Boustany, R.M.; Fleischnick, E.; Alper, C.A.; Marazita, M.L.; Spence, M.A.; Martin, J.B.; Kolodny, E.H. The autosomal dominant form of “pure” familial spastic paraplegia: Clinical findings and linkage analysis of a large pedigree. Neurology 1987, 37, 910–915. [Google Scholar] [CrossRef]
- Zhao, X.; Alvarado, D.; Rainier, S.; Lemons, R.; Hedera, P.; Weber, C.H.; Tukel, T.; Apak, M.; Heiman-Patterson, T.; Ming, L.; et al. Mutations in a newly identified GTPase gene cause autosomal dominant hereditary spastic paraplegia. Nat. Genet. 2001, 29, 326–331. [Google Scholar] [CrossRef]
- Gao, Y.; Jiang, T.; Qu, C.; Tao, H.; Cao, H.; Zhao, Y.; Wang, Y.; Qu, J.; Chen, J.G. Atlastin-1 regulates dendritic morphogenesis in mouse cerebral cortex. Neurosci. Res. 2013, 77, 137–142. [Google Scholar] [CrossRef]
- Shih, Y.T.; Hsueh, Y.P. VCP and ATL1 regulate endoplasmic reticulum and protein synthesis for dendritic spine formation. Nat. Commun. 2016, 7, 11020. [Google Scholar] [CrossRef] [Green Version]
- Hazan, J.; Fontaine, B.; Bruyn, R.P.; Lamy, C.; van Deutekom, J.C.; Rime, C.S.; Durr, A.; Melki, J.; Lyon-Caen, O.; Agid, Y.; et al. Linkage of a new locus for autosomal dominant familial spastic paraplegia to chromosome 2p. Hum. Mol. Genet. 1994, 3, 1569–1573. [Google Scholar] [CrossRef]
- Hentati, A.; Pericak-Vance, M.A.; Lennon, F.; Wasserman, B.; Hentati, F.; Juneja, T.; Angrist, M.H.; Hung, W.Y.; Boustany, R.M.; Bohlega, S.; et al. Linkage of a locus for autosomal dominant familial spastic paraplegia to chromosome 2p markers. Hum. Mol. Genet. 1994, 3, 1867–1871. [Google Scholar] [CrossRef]
- Fassier, C.; Tarrade, A.; Peris, L.; Courageot, S.; Mailly, P.; Dalard, C.; Delga, S.; Roblot, N.; Lefevre, J.; Job, D.; et al. Microtubule-targeting drugs rescue axonal swellings in cortical neurons from spastin knockout mice. Dis. Models Mech. 2013, 6, 72–83. [Google Scholar] [CrossRef]
- Kasher, P.R.; De Vos, K.J.; Wharton, S.B.; Manser, C.; Bennett, E.J.; Bingley, M.; Wood, J.D.; Milner, R.; McDermott, C.J.; Miller, C.C.; et al. Direct evidence for axonal transport defects in a novel mouse model of mutant spastin-induced hereditary spastic paraplegia (HSP) and human HSP patients. J. Neurochem. 2009, 110, 34–44. [Google Scholar] [CrossRef]
- Qiang, L.; Piermarini, E.; Muralidharan, H.; Yu, W.; Leo, L.; Hennessy, L.E.; Fernandes, S.; Connors, T.; Yates, P.L.; Swift, M.; et al. Hereditary spastic paraplegia: Gain-of-function mechanisms revealed by new transgenic mouse. Hum. Mol. Genet. 2019, 28, 1136–1152. [Google Scholar] [CrossRef]
- Tarrade, A.; Fassier, C.; Courageot, S.; Charvin, D.; Vitte, J.; Peris, L.; Thorel, A.; Mouisel, E.; Fonknechten, N.; Roblot, N.; et al. A mutation of spastin is responsible for swellings and impairment of transport in a region of axon characterized by changes in microtubule composition. Hum. Mol. Genet. 2006, 15, 3544–3558. [Google Scholar] [CrossRef] [Green Version]
- Fink, J.K.; Sharp, G.B.; Lange, B.M.; Wu, C.B.; Haley, T.; Otterud, B.; Peacock, M.; Leppert, M. Autosomal dominant, familial spastic paraplegia, type I: Clinical and genetic analysis of a large North American family. Neurology 1995, 45, 325–331. [Google Scholar] [CrossRef]
- Watanabe, F.; Arnold, W.D.; Hammer, R.E.; Ghodsizadeh, O.; Moti, H.; Schumer, M.; Hashmi, A.; Hernandez, A.; Sneh, A.; Sahenk, Z.; et al. Pathogenesis of autosomal dominant hereditary spastic paraplegia (SPG6) revealed by a rat model. J. Neuropathol. Exp. Neurol. 2013, 72, 1016–1028. [Google Scholar] [CrossRef]
- Hedera, P.; DiMauro, S.; Bonilla, E.; Wald, J.; Eldevik, O.P.; Fink, J.K. Phenotypic analysis of autosomal dominant hereditary spastic paraplegia linked to chromosome 8q. Neurology 1999, 53, 44–50. [Google Scholar] [CrossRef]
- Hedera, P.; Rainier, S.; Alvarado, D.; Zhao, X.; Williamson, J.; Otterud, B.; Leppert, M.; Fink, J.K. Novel locus for autosomal dominant hereditary spastic paraplegia, on chromosome 8q. Am. J. Hum. Genet. 1999, 64, 563–569. [Google Scholar] [CrossRef]
- Rocco, P.; Vainzof, M.; Froehner, S.C.; Peters, M.F.; Marie, S.K.; Passos-Bueno, M.R.; Zatz, M. Brazilian family with pure autosomal dominant spastic paraplegia maps to 8q: Analysis of muscle beta 1 syntrophin. Am. J. Med. Genet. 2000, 92, 122–127. [Google Scholar] [CrossRef]
- Jahic, A.; Khundadze, M.; Jaenisch, N.; Schule, R.; Klimpe, S.; Klebe, S.; Frahm, C.; Kassubek, J.; Stevanin, G.; Schols, L.; et al. The spectrum of KIAA0196 variants, and characterization of a murine knockout: Implications for the mutational mechanism in hereditary spastic paraplegia type SPG8. Orphanet J. Rare Dis. 2015, 10, 147. [Google Scholar] [CrossRef]
- Reid, E.; Dearlove, A.M.; Rhodes, M.; Rubinsztein, D.C. A new locus for autosomal dominant “pure” hereditary spastic paraplegia mapping to chromosome 12q13, and evidence for further genetic heterogeneity. Am. J. Hum. Genet. 1999, 65, 757–763. [Google Scholar] [CrossRef]
- Xia, C.H.; Roberts, E.A.; Her, L.S.; Liu, X.; Williams, D.S.; Cleveland, D.W.; Goldstein, L.S. Abnormal neurofilament transport caused by targeted disruption of neuronal kinesin heavy chain KIF5A. J. Cell Biol. 2003, 161, 55–66. [Google Scholar] [CrossRef] [Green Version]
- Reid, E.; Dearlove, A.M.; Osborn, O.; Rogers, M.T.; Rubinsztein, D.C. A locus for autosomal dominant “pure” hereditary spastic paraplegia maps to chromosome 19q13. Am. J. Hum. Genet. 2000, 66, 728–732. [Google Scholar] [CrossRef]
- Ikemoto, T.; Hosoya, T.; Takata, K.; Aoyama, H.; Hiramatsu, T.; Onoe, H.; Suzuki, M.; Endo, M. Functional role of neuroendocrine-specific protein-like 1 in membrane translocation of GLUT4. Diabetes 2009, 58, 2802–2812. [Google Scholar] [CrossRef]
- Fontaine, B.; Davoine, C.S.; Durr, A.; Paternotte, C.; Feki, I.; Weissenbach, J.; Hazan, J.; Brice, A. A new locus for autosomal dominant pure spastic paraplegia, on chromosome 2q24-q34. Am. J. Hum. Genet. 2000, 66, 702–707. [Google Scholar] [CrossRef]
- Blackstone, C.; O’Kane, C.J.; Reid, E. Hereditary spastic paraplegias: Membrane traffic and the motor pathway. Nat. Rev. Neurosci. 2011, 12, 31–42. [Google Scholar] [CrossRef]
- Bross, P.; Magnoni, R.; Bie, A.S. Molecular chaperone disorders: Defective Hsp60 in neurodegeneration. Curr. Top. Med. Chem. 2012, 12, 2491–2503. [Google Scholar] [CrossRef]
- Christensen, J.H.; Nielsen, M.N.; Hansen, J.; Fuchtbauer, A.; Fuchtbauer, E.M.; West, M.; Corydon, T.J.; Gregersen, N.; Bross, P. Inactivation of the hereditary spastic paraplegia-associated Hspd1 gene encoding the Hsp60 chaperone results in early embryonic lethality in mice. Cell Stress Chaperones 2010, 15, 851–863. [Google Scholar] [CrossRef] [Green Version]
- Magnoni, R.; Palmfeldt, J.; Christensen, J.H.; Sand, M.; Maltecca, F.; Corydon, T.J.; West, M.; Casari, G.; Bross, P. Late onset motoneuron disorder caused by mitochondrial Hsp60 chaperone deficiency in mice. Neurobiol. Dis. 2013, 54, 12–23. [Google Scholar] [CrossRef]
- Patel, H.; Hart, P.E.; Warner, T.T.; Houlston, R.S.; Patton, M.A.; Jeffery, S.; Crosby, A.H. The Silver syndrome variant of hereditary spastic paraplegia maps to chromosome 11q12-q14, with evidence for genetic heterogeneity within this subtype. Am. J. Hum. Genet. 2001, 69, 209–215. [Google Scholar] [CrossRef]
- Silver, J.R. Familial spastic paraplegia with amyotrophy of the hands. Ann. Hum. Genet. 1966, 30, 69–75. [Google Scholar] [CrossRef]
- Guo, J.; Qiu, W.; Soh, S.L.; Wei, S.; Radda, G.K.; Ong, W.Y.; Pang, Z.P.; Han, W. Motor neuron degeneration in a mouse model of seipinopathy. Cell Death Dis. 2013, 4, e535. [Google Scholar] [CrossRef]
- Cui, X.; Wang, Y.; Tang, Y.; Liu, Y.; Zhao, L.; Deng, J.; Xu, G.; Peng, X.; Ju, S.; Liu, G.; et al. Seipin ablation in mice results in severe generalized lipodystrophy. Hum. Mol. Genet. 2011, 20, 3022–3030. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Yin, J.; Wang, C.; Liao, J.; Liu, G.; Chen, L. Lack of seipin in neurons results in anxiety- and depression-like behaviors via down regulation of PPARgamma. Hum. Mol. Genet. 2014, 23, 4094–4102. [Google Scholar] [CrossRef]
- Zuchner, S.; Kail, M.E.; Nance, M.A.; Gaskell, P.C.; Svenson, I.K.; Marchuk, D.A.; Pericak-Vance, M.A.; Ashley-Koch, A.E. A new locus for dominant hereditary spastic paraplegia maps to chromosome 2p12. Neurogenetics 2006, 7, 127–129. [Google Scholar] [CrossRef]
- Zuchner, S.; Wang, G.; Tran-Viet, K.N.; Nance, M.A.; Gaskell, P.C.; Vance, J.M.; Ashley-Koch, A.E.; Pericak-Vance, M.A. Mutations in the novel mitochondrial protein REEP1 cause hereditary spastic paraplegia type 31. Am. J. Hum. Genet. 2006, 79, 365–369. [Google Scholar] [CrossRef]
- Deutch, A.Y.; Hedera, P.; Colbran, R.J. REEPing the benefits of an animal model of hereditary spastic paraplegia. J. Clin. Investig. 2013, 123, 4134–4136. [Google Scholar] [CrossRef] [Green Version]
- Lim, Y.; Cho, I.T.; Schoel, L.J.; Cho, G.; Golden, J.A. Hereditary spastic paraplegia-linked REEP1 modulates endoplasmic reticulum/mitochondria contacts. Ann. Neurol. 2015, 78, 679–696. [Google Scholar] [CrossRef]
- Renvoise, B.; Malone, B.; Falgairolle, M.; Munasinghe, J.; Stadler, J.; Sibilla, C.; Park, S.H.; Blackstone, C. Reep1 null mice reveal a converging role for hereditary spastic paraplegia proteins in lipid droplet regulation. Hum. Mol. Genet. 2016, 25, 5111–5125. [Google Scholar] [CrossRef]
- Lin, P.; Li, J.; Liu, Q.; Mao, F.; Li, J.; Qiu, R.; Hu, H.; Song, Y.; Yang, Y.; Gao, G.; et al. A missense mutation in SLC33A1, which encodes the acetyl-CoA transporter, causes autosomal-dominant spastic paraplegia (SPG42). Am. J. Hum. Genet. 2008, 83, 752–759. [Google Scholar] [CrossRef]
- Lin, P.; Mao, F.; Liu, Q.; Shao, C.; Yan, C.; Gong, Y. Prenatal diagnosis of autosomal dominant hereditary spastic paraplegia (SPG42) caused by SLC33A1 mutation in a Chinese kindred. Prenat. Diagn. 2010, 30, 485–486. [Google Scholar] [CrossRef]
- Liu, P.; Jiang, B.; Ma, J.; Lin, P.; Zhang, Y.; Shao, C.; Sun, W.; Gong, Y. S113R mutation in SLC33A1 leads to neurodegeneration and augmented BMP signaling in a mouse model. Dis. Model Mech. 2017, 10, 53–62. [Google Scholar] [CrossRef]
- Rinaldi, C.; Schmidt, T.; Situ, A.J.; Johnson, J.O.; Lee, P.R.; Chen, K.L.; Bott, L.C.; Fado, R.; Harmison, G.H.; Parodi, S.; et al. Mutation in CPT1C Associated With Pure Autosomal Dominant Spastic Paraplegia. JAMA Neurol. 2015, 72, 561–570. [Google Scholar] [CrossRef]
- Carrasco, P.; Jacas, J.; Sahun, I.; Muley, H.; Ramirez, S.; Puisac, B.; Mezquita, P.; Pie, J.; Dierssen, M.; Casals, N. Carnitine palmitoyltransferase 1C deficiency causes motor impairment and hypoactivity. Behav. Brain Res. 2013, 256, 291–297. [Google Scholar] [CrossRef]
- Casals, N.; Zammit, V.; Herrero, L.; Fado, R.; Rodriguez-Rodriguez, R.; Serra, D. Carnitine palmitoyltransferase 1C: From cognition to cancer. Prog. Lipid Res. 2016, 61, 134–148. [Google Scholar] [CrossRef]
- Jouet, M.; Rosenthal, A.; Armstrong, G.; MacFarlane, J.; Stevenson, R.; Paterson, J.; Metzenberg, A.; Ionasescu, V.; Temple, K.; Kenwrick, S. X-linked spastic paraplegia (SPG1), MASA syndrome and X-linked hydrocephalus result from mutations in the L1 gene. Nat. Genet. 1994, 7, 402–407. [Google Scholar] [CrossRef]
- Demyanenko, G.P.; Tsai, A.Y.; Maness, P.F. Abnormalities in neuronal process extension, hippocampal development, and the ventricular system of L1 knockout mice. J. Neurosci. 1999, 19, 4907–4920. [Google Scholar] [CrossRef]
- Guseva, D.; Angelov, D.N.; Irintchev, A.; Schachner, M. Ablation of adhesion molecule L1 in mice favours Schwann cell proliferation and functional recovery after peripheral nerve injury. Brain 2009, 132, 2180–2195. [Google Scholar] [CrossRef] [Green Version]
- Tapanes-Castillo, A.; Weaver, E.J.; Smith, R.P.; Kamei, Y.; Caspary, T.; Hamilton-Nelson, K.L.; Slifer, S.H.; Martin, E.R.; Bixby, J.L.; Lemmon, V.P. A modifier locus on chromosome 5 contributes to L1 cell adhesion molecule X-linked hydrocephalus in mice. Neurogenetics 2010, 11, 53–71. [Google Scholar] [CrossRef]
- Kobayashi, H.; Hoffman, E.P.; Marks, H.G. The rumpshaker mutation in spastic paraplegia. Nat. Genet. 1994, 7, 351–352. [Google Scholar] [CrossRef]
- Saugier-Veber, P.; Munnich, A.; Bonneau, D.; Rozet, J.M.; Le Merrer, M.; Gil, R.; Boespflug-Tanguy, O. X-linked spastic paraplegia and Pelizaeus-Merzbacher disease are allelic disorders at the proteolipid protein locus. Nat. Genet. 1994, 6, 257–262. [Google Scholar] [CrossRef]
- Cambi, F.; Tang, X.M.; Cordray, P.; Fain, P.R.; Keppen, L.D.; Barker, D.F. Refined genetic mapping and proteolipid protein mutation analysis in X-linked pure hereditary spastic paraplegia. Neurology 1996, 46, 1112–1117. [Google Scholar] [CrossRef]
- Bonneau, D.; Rozet, J.M.; Bulteau, C.; Berthier, M.; Mettey, R.; Gil, R.; Munnich, A.; Le Merrer, M. X linked spastic paraplegia (SPG2): Clinical heterogeneity at a single gene locus. J. Med. Genet. 1993, 30, 381–384. [Google Scholar] [CrossRef]
- Al-Saktawi, K.; McLaughlin, M.; Klugmann, M.; Schneider, A.; Barrie, J.A.; McCulloch, M.C.; Montague, P.; Kirkham, D.; Nave, K.A.; Griffiths, I.R. Genetic background determines phenotypic severity of the Plp rumpshaker mutation. J. Neurosci. Res. 2003, 72, 12–24. [Google Scholar] [CrossRef]
- Edgar, J.M.; McLaughlin, M.; Barrie, J.A.; McCulloch, M.C.; Garbern, J.; Griffiths, I.R. Age-related axonal and myelin changes in the rumpshaker mutation of the Plp gene. Acta Neuropathol. 2004, 107, 331–335. [Google Scholar] [CrossRef]
- Griffiths, I.; Klugmann, M.; Anderson, T.; Yool, D.; Thomson, C.; Schwab, M.H.; Schneider, A.; Zimmermann, F.; McCulloch, M.; Nadon, N.; et al. Axonal swellings and degeneration in mice lacking the major proteolipid of myelin. Science 1998, 280, 1610–1613. [Google Scholar] [CrossRef]
- Klugmann, M.; Schwab, M.H.; Puhlhofer, A.; Schneider, A.; Zimmermann, F.; Griffiths, I.R.; Nave, K.A. Assembly of CNS myelin in the absence of proteolipid protein. Neuron 1997, 18, 59–70. [Google Scholar] [CrossRef]
- Luders, K.A.; Patzig, J.; Simons, M.; Nave, K.A.; Werner, H.B. Genetic dissection of oligodendroglial and neuronal Plp1 function in a novel mouse model of spastic paraplegia type 2. Glia 2017, 65, 1762–1776. [Google Scholar] [CrossRef]
- Yool, D.A.; Klugmann, M.; McLaughlin, M.; Vouyiouklis, D.A.; Dimou, L.; Barrie, J.A.; McCulloch, M.C.; Nave, K.A.; Griffiths, I.R. Myelin proteolipid proteins promote the interaction of oligodendrocytes and axons. J. Neurosci. Res. 2001, 63, 151–164. [Google Scholar] [CrossRef]
- Nixon, C.W.; Connelly, M.E. Hind-leg paralysis: A new sex-linked mutation in the Syrian hamster. J. Hered. 1968, 59, 276–278. [Google Scholar] [CrossRef]
- Schneider, A.; Montague, P.; Griffiths, I.; Fanarraga, M.; Kennedy, P.; Brophy, P.; Nave, K.A. Uncoupling of hypomyelination and glial cell death by a mutation in the proteolipid protein gene. Nature 1992, 358, 758–761. [Google Scholar] [CrossRef]
- Eymard-Pierre, E.; Yamanaka, K.; Haeussler, M.; Kress, W.; Gauthier-Barichard, F.; Combes, P.; Cleveland, D.W.; Boespflug-Tanguy, O. Novel missense mutation in ALS2 gene results in infantile ascending hereditary spastic paralysis. Ann. Neurol. 2006, 59, 976–980. [Google Scholar] [CrossRef]
- Gros-Louis, F.; Meijer, I.A.; Hand, C.K.; Dube, M.P.; MacGregor, D.L.; Seni, M.H.; Devon, R.S.; Hayden, M.R.; Andermann, F.; Andermann, E.; et al. An ALS2 gene mutation causes hereditary spastic paraplegia in a Pakistani kindred. Ann. Neurol. 2003, 53, 144–145. [Google Scholar] [CrossRef]
- Herzfeld, T.; Wolf, N.; Winter, P.; Hackstein, H.; Vater, D.; Muller, U. Maternal uniparental heterodisomy with partial isodisomy of a chromosome 2 carrying a splice acceptor site mutation (IVS9-2A>T) in ALS2 causes infantile-onset ascending spastic paralysis (IAHSP). Neurogenetics 2009, 10, 59–64. [Google Scholar] [CrossRef]
- Ben Hamida, M.; Hentati, F.; Ben Hamida, C. Hereditary motor system diseases (chronic juvenile amyotrophic lateral sclerosis). Conditions combining a bilateral pyramidal syndrome with limb and bulbar amyotrophy. Brain 1990, 113, 347–363. [Google Scholar] [CrossRef]
- Hentati, A.; Bejaoui, K.; Pericak-Vance, M.A.; Hentati, F.; Speer, M.C.; Hung, W.Y.; Figlewicz, D.A.; Haines, J.; Rimmler, J.; Ben Hamida, C.; et al. Linkage of recessive familial amyotrophic lateral sclerosis to chromosome 2q33-q35. Nat. Genet. 1994, 7, 425–428. [Google Scholar] [CrossRef]
- Deng, H.X.; Zhai, H.; Fu, R.; Shi, Y.; Gorrie, G.H.; Yang, Y.; Liu, E.; Dal Canto, M.C.; Mugnaini, E.; Siddique, T. Distal axonopathy in an alsin-deficient mouse model. Hum. Mol. Genet. 2007, 16, 2911–2920. [Google Scholar] [CrossRef] [Green Version]
- Devon, R.S.; Orban, P.C.; Gerrow, K.; Barbieri, M.A.; Schwab, C.; Cao, L.P.; Helm, J.R.; Bissada, N.; Cruz-Aguado, R.; Davidson, T.L.; et al. Als2-deficient mice exhibit disturbances in endosome trafficking associated with motor behavioral abnormalities. Proc. Natl. Acad. Sci. USA 2006, 103, 9595–9600. [Google Scholar] [CrossRef]
- Yamanaka, K.; Miller, T.M.; McAlonis-Downes, M.; Chun, S.J.; Cleveland, D.W. Progressive spinal axonal degeneration and slowness in ALS2-deficient mice. Ann. Neurol. 2006, 60, 95–104. [Google Scholar] [CrossRef]
- Hadano, S.; Benn, S.C.; Kakuta, S.; Otomo, A.; Sudo, K.; Kunita, R.; Suzuki-Utsunomiya, K.; Mizumura, H.; Shefner, J.M.; Cox, G.A.; et al. Mice deficient in the Rab5 guanine nucleotide exchange factor ALS2/alsin exhibit age-dependent neurological deficits and altered endosome trafficking. Hum. Mol. Genet. 2006, 15, 233–250. [Google Scholar] [CrossRef]
- Jacquier, A.; Bellouze, S.; Blanchard, S.; Bohl, D.; Haase, G. Astrocytic protection of spinal motor neurons but not cortical neurons against loss of Als2/alsin function. Hum. Mol. Genet. 2009, 18, 2127–2139. [Google Scholar] [CrossRef]
- Coutelier, M.; Goizet, C.; Durr, A.; Habarou, F.; Morais, S.; Dionne-Laporte, A.; Tao, F.; Konop, J.; Stoll, M.; Charles, P.; et al. Alteration of ornithine metabolism leads to dominant and recessive hereditary spastic paraplegia. Brain 2015, 138, 2191–2205. [Google Scholar] [CrossRef] [Green Version]
- Slavotinek, A.M.; Pike, M.; Mills, K.; Hurst, J.A. Cataracts, motor system disorder, short stature, learning difficulties, and skeletal abnormalities: A new syndrome? Am. J. Med. Genet. 1996, 62, 42–47. [Google Scholar] [CrossRef]
- Yildirim, Y.; Orhan, E.K.; Iseri, S.A.; Serdaroglu-Oflazer, P.; Kara, B.; Solakoglu, S.; Tolun, A. A frameshift mutation of ERLIN2 in recessive intellectual disability, motor dysfunction and multiple joint contractures. Hum. Mol. Genet. 2011, 20, 1886–1892. [Google Scholar] [CrossRef] [Green Version]
- Oz-Levi, D.; Ben-Zeev, B.; Ruzzo, E.K.; Hitomi, Y.; Gelman, A.; Pelak, K.; Anikster, Y.; Reznik-Wolf, H.; Bar-Joseph, I.; Olender, T.; et al. Mutation in TECPR2 reveals a role for autophagy in hereditary spastic paraparesis. Am. J. Hum. Genet. 2012, 91, 1065–1072. [Google Scholar] [CrossRef]
- Shimazaki, H.; Takiyama, Y.; Ishiura, H.; Sakai, C.; Matsushima, Y.; Hatakeyama, H.; Honda, J.; Sakoe, K.; Naoi, T.; Namekawa, M.; et al. A homozygous mutation of C12orf65 causes spastic paraplegia with optic atrophy and neuropathy (SPG55). J. Med. Genet. 2012, 49, 777–784. [Google Scholar] [CrossRef]
- Beetz, C.; Johnson, A.; Schuh, A.L.; Thakur, S.; Varga, R.E.; Fothergill, T.; Hertel, N.; Bomba-Warczak, E.; Thiele, H.; Nurnberg, G.; et al. Inhibition of TFG function causes hereditary axon degeneration by impairing endoplasmic reticulum structure. Proc. Natl. Acad. Sci. USA 2013, 110, 5091–5096. [Google Scholar] [CrossRef] [Green Version]
- Esteves, T.; Durr, A.; Mundwiller, E.; Loureiro, J.L.; Boutry, M.; Gonzalez, M.A.; Gauthier, J.; El-Hachimi, K.H.; Depienne, C.; Muriel, M.P.; et al. Loss of association of REEP2 with membranes leads to hereditary spastic paraplegia. Am. J. Hum. Genet. 2014, 94, 268–277. [Google Scholar] [CrossRef]
- Lossos, A.; Stumpfig, C.; Stevanin, G.; Gaussen, M.; Zimmerman, B.E.; Mundwiller, E.; Asulin, M.; Chamma, L.; Sheffer, R.; Misk, A.; et al. Fe/S protein assembly gene IBA57 mutation causes hereditary spastic paraplegia. Neurology 2015, 84, 659–667. [Google Scholar] [CrossRef]
- Vernon, H.J.; McClellan, R.; Batista, D.A.; Naidu, S. Mutations in FARS2 and non-fatal mitochondrial dysfunction in two siblings. Am. J. Med. Genet. A 2015, 167A, 1147–1151. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, W.; Fang, Z.; Shi, J.; Che, F.; He, C.; Yao, L.; Wang, E.; Wu, Y. A Newly Identified Missense Mutation in FARS2 Causes Autosomal-Recessive Spastic Paraplegia. Hum. Mutat. 2016, 37, 165–169. [Google Scholar] [CrossRef]
- Mannan, A.U.; Krawen, P.; Sauter, S.M.; Boehm, J.; Chronowska, A.; Paulus, W.; Neesen, J.; Engel, W. ZFYVE27 (SPG33), a novel spastin-binding protein, is mutated in hereditary spastic paraplegia. Am. J. Hum. Genet. 2006, 79, 351–357. [Google Scholar] [CrossRef]
- Dupre, N.; Valdmanis, P.N.; Bouchard, J.P.; Rouleau, G.A. Autosomal dominant primary lateral sclerosis. Neurology 2007, 68, 1156–1157. [Google Scholar] [CrossRef]
- Valdmanis, P.N.; Dupre, N.; Rouleau, G.A. A locus for primary lateral sclerosis on chromosome 4ptel-4p16.1. Arch. Neurol. 2008, 65, 383–386. [Google Scholar] [CrossRef]
- Enjyoji, K.; Kotani, K.; Thukral, C.; Blumel, B.; Sun, X.; Wu, Y.; Imai, M.; Friedman, D.; Csizmadia, E.; Bleibel, W.; et al. Deletion of cd39/entpd1 results in hepatic insulin resistance. Diabetes 2008, 57, 2311–2320. [Google Scholar] [CrossRef]
- Connell, J.W.; Allison, R.; Reid, E. Quantitative Gait Analysis Using a Motorized Treadmill System Sensitively Detects Motor Abnormalities in Mice Expressing ATPase Defective Spastin. PLoS ONE 2016, 11, e0152413. [Google Scholar] [CrossRef]
- Beetz, C.; Koch, N.; Khundadze, M.; Zimmer, G.; Nietzsche, S.; Hertel, N.; Huebner, A.K.; Mumtaz, R.; Schweizer, M.; Dirren, E.; et al. A spastic paraplegia mouse model reveals REEP1-dependent ER shaping. J. Clin. Investig. 2013, 123, 4273–4282. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Hentati, A.; Deng, H.X.; Dabbagh, O.; Sasaki, T.; Hirano, M.; Hung, W.Y.; Ouahchi, K.; Yan, J.; Azim, A.C.; et al. The gene encoding alsin, a protein with three guanine-nucleotide exchange factor domains, is mutated in a form of recessive amyotrophic lateral sclerosis. Nat. Genet. 2001, 29, 160–165. [Google Scholar] [CrossRef]
- Arlotta, P.; Molyneaux, B.J.; Chen, J.; Inoue, J.; Kominami, R.; Macklis, J.D. Neuronal subtype-specific genes that control corticospinal motor neuron development in vivo. Neuron 2005, 45, 207–221. [Google Scholar] [CrossRef]
- Jara, J.H.; Stanford, M.J.; Zhu, Y.; Tu, M.; Hauswirth, W.W.; Bohn, M.C.; DeVries, S.H.; Ozdinler, P.H. Healthy and diseased corticospinal motor neurons are selectively transduced upon direct AAV2-2 injection into the motor cortex. Gene. Ther. 2016, 23, 272–282. [Google Scholar] [CrossRef] [Green Version]
- Jara, J.H.; Villa, S.R.; Khan, N.A.; Bohn, M.C.; Ozdinler, P.H. AAV2 mediated retrograde transduction of corticospinal motor neurons reveals initial and selective apical dendrite degeneration in ALS. Neurobiol. Dis. 2012, 47, 174–183. [Google Scholar] [CrossRef] [Green Version]
- Fink, K.L.; Strittmatter, S.M.; Cafferty, W.B. Comprehensive Corticospinal Labeling with mu-crystallin Transgene Reveals Axon Regeneration after Spinal Cord Trauma in ngr1-/- Mice. J. Neurosci. 2015, 35, 15403–15418. [Google Scholar] [CrossRef]
- Schaefer, A.M.; Sanes, J.R.; Lichtman, J.W. A compensatory subpopulation of motor neurons in a mouse model of amyotrophic lateral sclerosis. J. Comp. Neurol. 2005, 490, 209–219. [Google Scholar] [CrossRef]
- Wong, F.; Fan, L.; Wells, S.; Hartley, R.; Mackenzie, F.E.; Oyebode, O.; Brown, R.; Thomson, D.; Coleman, M.P.; Blanco, G.; et al. Axonal and neuromuscular synaptic phenotypes in Wld(S), SOD1(G93A) and ostes mutant mice identified by fiber-optic confocal microendoscopy. Mol. Cell. Neurosci. 2009, 42, 296–307. [Google Scholar] [CrossRef]
- Richter, M.W.; Roskams, A.J. Corticospinal neurons respond differentially to neurotrophins and myelin-associated glycoprotein in vitro. J. Neurosci. Res. 2009, 87, 2222–2236. [Google Scholar] [CrossRef]
- Feng, G.; Mellor, R.H.; Bernstein, M.; Keller-Peck, C.; Nguyen, Q.T.; Wallace, M.; Nerbonne, J.M.; Lichtman, J.W.; Sanes, J.R. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron 2000, 28, 41–51. [Google Scholar] [CrossRef]
- Yu, J.; Anderson, C.T.; Kiritani, T.; Sheets, P.L.; Wokosin, D.L.; Wood, L.; Shepherd, G.M. Local-Circuit Phenotypes of Layer 5 Neurons in Motor-Frontal Cortex of YFP-H Mice. Front. Neural Circuits 2008, 2, 6. [Google Scholar] [CrossRef]
- Bareyre, F.M.; Kerschensteiner, M.; Misgeld, T.; Sanes, J.R. Transgenic labeling of the corticospinal tract for monitoring axonal responses to spinal cord injury. Nat. Med. 2005, 11, 1355–1360. [Google Scholar] [CrossRef]
- Ozdinler, P.H.; Benn, S.; Yamamoto, T.H.; Guzel, M.; Brown, R.H., Jr.; Macklis, J.D. Corticospinal motor neurons and related subcerebral projection neurons undergo early and specific neurodegeneration in hSOD1G93A transgenic ALS mice. J. Neurosci. 2011, 31, 4166–4177. [Google Scholar] [CrossRef]
- Tantirigama, M.L.; Oswald, M.J.; Clare, A.J.; Wicky, H.E.; Day, R.C.; Hughes, S.M.; Empson, R.M. Fezf2 expression in layer 5 projection neurons of mature mouse motor cortex. J. Comp. Neurol. 2016, 524, 829–845. [Google Scholar] [CrossRef]
- Yasvoina, M.V.; Genc, B.; Jara, J.H.; Sheets, P.L.; Quinlan, K.A.; Milosevic, A.; Shepherd, G.M.; Heckman, C.J.; Ozdinler, P.H. eGFP expression under UCHL1 promoter genetically labels corticospinal motor neurons and a subpopulation of degeneration-resistant spinal motor neurons in an ALS mouse model. J. Neurosci. 2013, 33, 7890–7904. [Google Scholar] [CrossRef]
- Bishop, P.; Rocca, D.; Henley, J.M. Ubiquitin C-terminal hydrolase L1 (UCH-L1): Structure, distribution and roles in brain function and dysfunction. Biochem. J. 2016, 473, 2453–2462. [Google Scholar] [CrossRef]
- Day, I.N.; Thompson, R.J. UCHL1 (PGP 9.5): Neuronal biomarker and ubiquitin system protein. Prog. Neuropathol. 2010, 90, 327–362. [Google Scholar] [CrossRef]
- Liu, Y.; Fallon, L.; Lashuel, H.A.; Liu, Z.; Lansbury, P.T., Jr. The UCH-L1 gene encodes two opposing enzymatic activities that affect alpha-synuclein degradation and Parkinson’s disease susceptibility. Cell 2002, 111, 209–218. [Google Scholar] [CrossRef]
- Cartier, A.E.; Djakovic, S.N.; Salehi, A.; Wilson, S.M.; Masliah, E.; Patrick, G.N. Regulation of synaptic structure by ubiquitin C-terminal hydrolase L1. J. Neurosci. 2009, 29, 7857–7868. [Google Scholar] [CrossRef]
- Leroy, E.; Boyer, R.; Auburger, G.; Leube, B.; Ulm, G.; Mezey, E.; Harta, G.; Brownstein, M.J.; Jonnalagada, S.; Chernova, T.; et al. The ubiquitin pathway in Parkinson’s disease. Nature 1998, 395, 451–452. [Google Scholar] [CrossRef]
- Hussain, S.; Bedekovics, T.; Liu, Q.; Hu, W.; Jeon, H.; Johnson, S.H.; Vasmatzis, G.; May, D.G.; Roux, K.J.; Galardy, P.J. UCH-L1 bypasses mTOR to promote protein biosynthesis and is required for MYC-driven lymphomagenesis in mice. Blood 2018, 132, 2564–2574. [Google Scholar] [CrossRef] [Green Version]
- Hussain, S.; Foreman, O.; Perkins, S.L.; Witzig, T.E.; Miles, R.R.; van Deursen, J.; Galardy, P.J. The de-ubiquitinase UCH-L1 is an oncogene that drives the development of lymphoma in vivo by deregulating PHLPP1 and Akt signaling. Leukemia 2010, 24, 1641–1655. [Google Scholar] [CrossRef]
- Jara, J.H.; Frank, D.D.; Ozdinler, P.H. Could dysregulation of UPS be a common underlying mechanism for cancer and neurodegeneration? Lessons from UCHL1. Cell Biochem. Biophys. 2013, 67, 45–53. [Google Scholar] [CrossRef]
- Das, C.; Hoang, Q.Q.; Kreinbring, C.A.; Luchansky, S.J.; Meray, R.K.; Ray, S.S.; Lansbury, P.T.; Ringe, D.; Petsko, G.A. Structural basis for conformational plasticity of the Parkinson’s disease-associated ubiquitin hydrolase UCH-L1. Proc. Natl. Acad. Sci. USA 2006, 103, 4675–4680. [Google Scholar] [CrossRef]
- Das Bhowmik, A.; Patil, S.J.; Deshpande, D.V.; Bhat, V.; Dalal, A. Novel splice-site variant of UCHL1 in an Indian family with autosomal recessive spastic paraplegia-79. J. Hum. Genet. 2018, 63, 927–933. [Google Scholar] [CrossRef]
- Rydning, S.L.; Backe, P.H.; Sousa, M.M.L.; Iqbal, Z.; Oye, A.M.; Sheng, Y.; Yang, M.; Lin, X.; Slupphaug, G.; Nordenmark, T.H.; et al. Novel UCHL1 mutations reveal new insights into ubiquitin processing. Hum. Mol. Genet. 2017, 26, 1217–1218. [Google Scholar] [CrossRef]
- Mukoyama, M.; Yamazaki, K.; Kikuchi, T.; Tomita, T. Neuropathology of gracile axonal dystrophy (GAD) mouse. An animal model of central distal axonopathy in primary sensory neurons. Acta Neuropathol. 1989, 79, 294–299. [Google Scholar] [CrossRef]
- Ittner, L.M.; Halliday, G.M.; Kril, J.J.; Gotz, J.; Hodges, J.R.; Kiernan, M.C. FTD and ALS--translating mouse studies into clinical trials. Nat. Rev. Neurosci. 2015, 11, 360–366. [Google Scholar] [CrossRef]
- Philips, T.; Rothstein, J.D. Rodent Models of Amyotrophic Lateral Sclerosis. Curr. Protoc. Pharmacol. 2015, 69, 1–21. [Google Scholar] [CrossRef]
- Ahmed, R.M.; Irish, M.; van Eersel, J.; Ittner, A.; Ke, Y.D.; Volkerling, A.; van der Hoven, J.; Tanaka, K.; Karl, T.; Kassiou, M.; et al. Mouse models of frontotemporal dementia: A comparison of phenotypes with clinical symptomatology. Neurosci. Biobehav. Rev. 2017, 74, 126–138. [Google Scholar] [CrossRef] [Green Version]
- Picher-Martel, V.; Valdmanis, P.N.; Gould, P.V.; Julien, J.P.; Dupre, N. From animal models to human disease: A genetic approach for personalized medicine in ALS. Acta Neuropathol. Commun. 2016, 4, 70. [Google Scholar] [CrossRef]
- De Giorgio, F.; Maduro, C.; Fisher, E.M.C.; Acevedo-Arozena, A. Transgenic and physiological mouse models give insights into different aspects of amyotrophic lateral sclerosis. Dis. Models Mech. 2019, 12. [Google Scholar] [CrossRef]
- Morrice, J.R.; Gregory-Evans, C.Y.; Shaw, C.A. Animal models of amyotrophic lateral sclerosis: A comparison of model validity. Neural. Regen. Res. 2018, 13, 2050–2054. [Google Scholar] [CrossRef]
- Lutz, C. Mouse models of ALS: Past, present and future. Brain Res. 2018, 1693, 1–10. [Google Scholar] [CrossRef]
- Batra, R.; Lee, C.W. Mouse Models of C9orf72 Hexanucleotide Repeat Expansion in Amyotrophic Lateral Sclerosis/Frontotemporal Dementia. Front. Cell Neurosci. 2017, 11, 196. [Google Scholar] [CrossRef]
- Fisher, E.M.C.; Bannerman, D.M. Mouse models of neurodegeneration: Know your question, know your mouse. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef]
- Geevasinga, N.; Menon, P.; Ozdinler, P.H.; Kiernan, M.C.; Vucic, S. Pathophysiological and diagnostic implications of cortical dysfunction in ALS. Nat. Rev. Neurosci. 2016, 12, 651–661. [Google Scholar] [CrossRef]
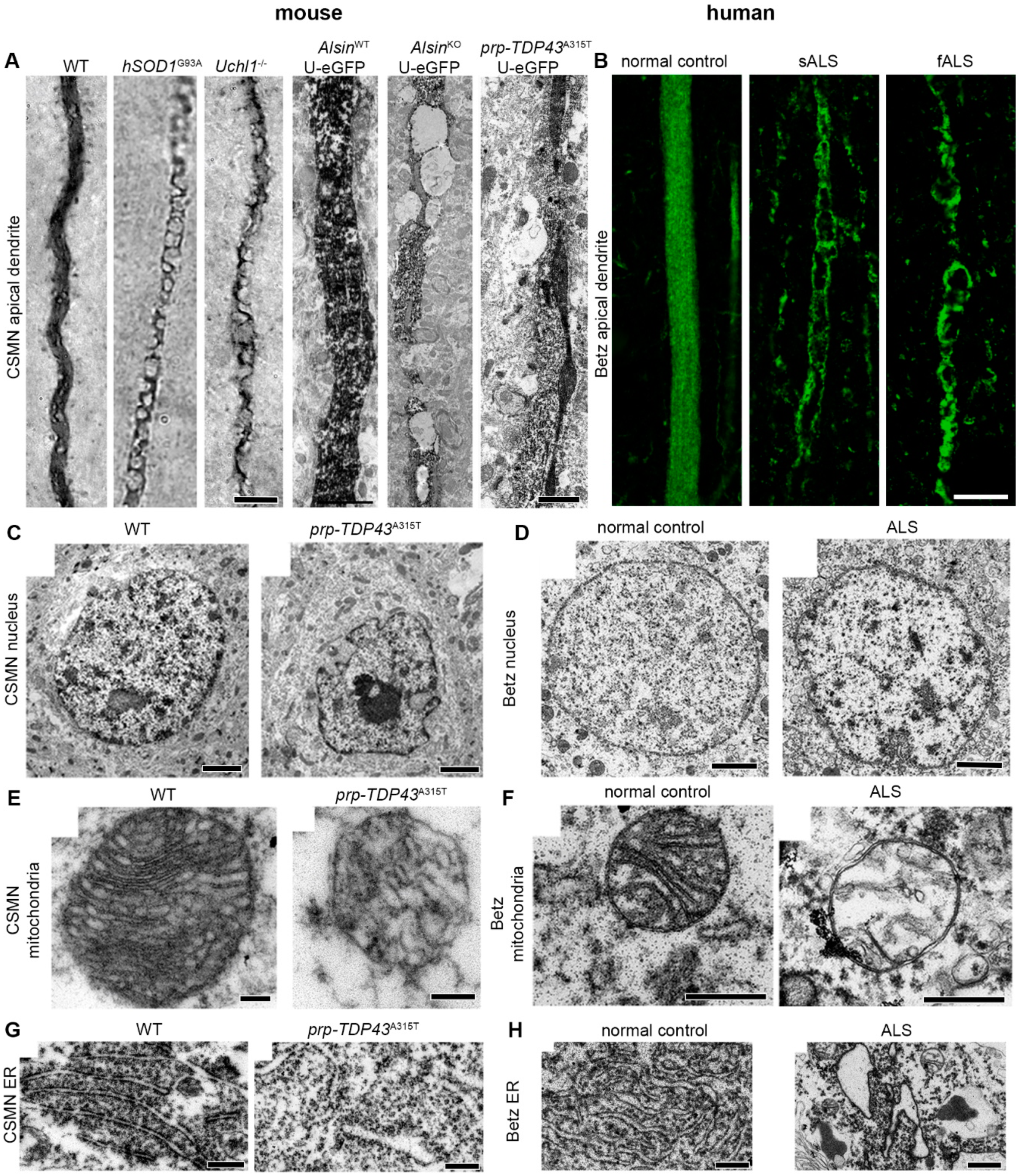
- Gautam, M.; Jara, J.H.; Kocak, N.; Rylaarsdam, L.E.; Kim, K.D.; Bigio, E.H.; Hande Ozdinler, P. Mitochondria, ER, and nuclear membrane defects reveal early mechanisms for upper motor neuron vulnerability with respect to TDP-43 pathology. Acta Neuropathol. 2019, 137, 47–69. [Google Scholar] [CrossRef]
- Genc, B.; Ozdinler, P.H. Moving forward in clinical trials for ALS: Motor neurons lead the way please. Drug Discov. Today 2014, 19, 441–449. [Google Scholar] [CrossRef]
- Dervishi, I.; Ozdinler, P.H. Incorporating upper motor neuron health in ALS drug discovery. Drug Discov. Today 2018, 23, 696–703. [Google Scholar] [CrossRef]
- Gerfen, C.R.; Paletzki, R.; Heintz, N. GENSAT BAC cre-recombinase driver lines to study the functional organization of cerebral cortical and basal ganglia circuits. Neuron 2013, 80, 1368–1383. [Google Scholar] [CrossRef]
- Kim, J.; Hughes, E.G.; Shetty, A.S.; Arlotta, P.; Goff, L.A.; Bergles, D.E.; Brown, S.P. Changes in the Excitability of Neocortical Neurons in a Mouse Model of Amyotrophic Lateral Sclerosis Are Not Specific to Corticospinal Neurons and Are Modulated by Advancing Disease. J. Neurosci. 2017, 37, 9037–9053. [Google Scholar] [CrossRef] [Green Version]
- Leone, D.P.; Heavner, W.E.; Ferenczi, E.A.; Dobreva, G.; Huguenard, J.R.; Grosschedl, R.; McConnell, S.K. Satb2 Regulates the Differentiation of Both Callosal and Subcerebral Projection Neurons in the Developing Cerebral Cortex. Cereb. Cortex 2015, 25, 3406–3419. [Google Scholar] [CrossRef]
- Woodworth, M.B.; Greig, L.C.; Liu, K.X.; Ippolito, G.C.; Tucker, H.O.; Macklis, J.D. Ctip1 Regulates the Balance between Specification of Distinct Projection Neuron Subtypes in Deep Cortical Layers. Cell Rep. 2016, 15, 999–1012. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Disease | Gene | Mouse Model Available | Motor Cortex Involvement |
|---|---|---|---|
| SPG5A | CYP7B1 [34,35] | Y [36,37] | |
| SPG7 | PARAPLEGIN [38,39,40,41,42] | Y [43,44,45] | |
| SPG11 | SPATACSIN [46] | Y [47,48] | Y [47,48] |
| SPG15 | ZFYVE26 (SPASTIZIN) [49] | Y [50] | Y [50] |
| SPG20 | SPARTIN [51,52] | Y [53] | |
| SPG21 | MASPARDIN [54] | Y [25] | N [25] |
| SPG26 | B4GALNT1 (GM1, GALNACT) [55] | Y [23] | N [23] |
| SPG28 | DDHD1 (PAPLA) [56] | Y [57,58] | |
| SPG30 | KIF1A [59] | Y [60,61,62] | |
| SPG35 | FA2H [63] | Y [64,65] | |
| SPG39 | PNPLA6 (NTE) [66] | Y [26,67] | N [26] |
| SPG44 | GJC2 (CX47) [68] | Y [69,70,71,72] | |
| SPG45 | NT5C2 [73] | Y [74] | |
| SPG46 | GBA2 [75,76] | Y [77,78,79,80] | |
| SPG47 | AP-4 [81] | Y [82] | |
| SPG48 | KIAA0415 (AP-5Z1) [83] | Y [84] | Y [84] |
| SPG54 | DDHD2 (KIAA0725P, IPLA1γ) [85] | Y [86,87] | |
| SPG63 | AMPD2 [88] | Y [89,90,91] | |
| SPG64 | ENTPD1 (CD39) [88] | Y [92,93,94] | |
| SPG75 | MAG [88,95] | Y [96,97,98,99,100,101,102,103] | |
| SPG76 | CAPN1 [104] | Y [105,106,107,108] | |
| SPG78 | ATP13A2 [109,110] | Y [111,112,113,114,115,116,117,118] | |
| SPG79 | UCHL1 [119] | Y [120,121,122,123,124,125,126,127] | Y [120,124,125,128,129] |
| SPG3A | ATL1 [130,131,132] | Y [133,134] | |
| SPG4 | SPAST [135,136] | Y [137,138,139,140] | Y [140] |
| SPG6 | NIPA1 (CXFIP1) [141] | Y [142] | |
| SPG8 | KIAA0196 (WASH C5, STRUMPELLIN, RTSC1) [143,144,145] | Y [146] | |
| SPG10 | KIF5A [147] | Y [24,148] | N [24] |
| SPG12 | RTN2 (NSPL1) [149] | Y [150] | |
| SPG13 | SSPD1 (HSP60, HSPD1) [151] | Y [152,153,154,155] | Y [153,155] |
| SPG17 | BSCL2/SEIPIN [156,157] | Y [27,158,159,160] | N [27] |
| SPG31 | REEP1 [161,162] | Y [163,164,165] | Y [163,165] |
| SPG42 | SLC33A1 [166,167] | Y [168] | |
| SPG73 | CPT1C [169] | Y [170,171] | Y [170] |
| SPG1 | L1CAM [172] | Y [173,174,175] | Y [173,174] |
| SPG2 | PLP1 [176,177,178,179] | Y [176,180,181,182,183,184,185,186,187] | |
| PLS | ALS2 [188,189,190,191,192] | Y [28,193,194,195,196,197] | Y [28] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Genc, B.; Gozutok, O.; Ozdinler, P.H. Complexity of Generating Mouse Models to Study the Upper Motor Neurons: Let Us Shift Focus from Mice to Neurons. Int. J. Mol. Sci. 2019, 20, 3848. https://doi.org/10.3390/ijms20163848
Genc B, Gozutok O, Ozdinler PH. Complexity of Generating Mouse Models to Study the Upper Motor Neurons: Let Us Shift Focus from Mice to Neurons. International Journal of Molecular Sciences. 2019; 20(16):3848. https://doi.org/10.3390/ijms20163848
Chicago/Turabian StyleGenc, Baris, Oge Gozutok, and P. Hande Ozdinler. 2019. "Complexity of Generating Mouse Models to Study the Upper Motor Neurons: Let Us Shift Focus from Mice to Neurons" International Journal of Molecular Sciences 20, no. 16: 3848. https://doi.org/10.3390/ijms20163848
APA StyleGenc, B., Gozutok, O., & Ozdinler, P. H. (2019). Complexity of Generating Mouse Models to Study the Upper Motor Neurons: Let Us Shift Focus from Mice to Neurons. International Journal of Molecular Sciences, 20(16), 3848. https://doi.org/10.3390/ijms20163848