Matrix Metalloproteases as Influencers of the Cells’ Social Media


Abstract
:1. Introduction: MMPs Act as Emojis in Cell–Cell Communication
2. MMPs Process the Gatekeepers of Cell Signaling
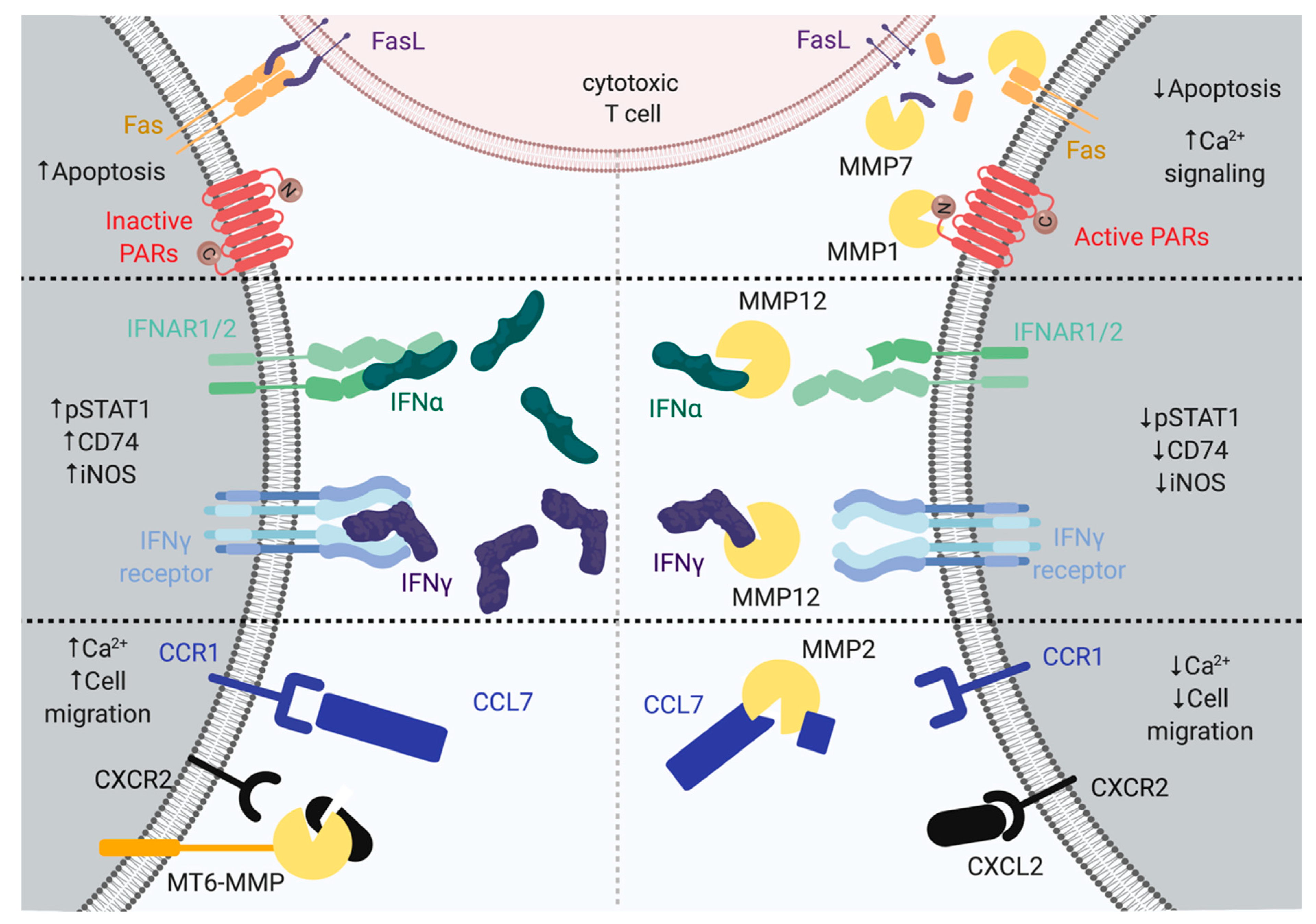
3. MMP7 Modulates CD95/Fas Signaling in Cell Death
4. Co-Expression of MMP14/MT1–MMP and DR6 in Cell Death
5. Processing of Ephrin B2 Receptor Mediates Cell Motility
6. Processing of CD44 Decreases Cell Adhesion
7. MMP Processing of Chemokines
8. MMPs–Orchestrator of the Fine Tuning of Cytokine Signaling
9. How Can We Stop MMPs Influencing the Cells’ Social Media Communications?
Conflicts of Interest
References
- Dufour, A. Degradomics of matrix metalloproteinases in inflammatory diseases. Front. Biosci. 2015, 7, 150–167. [Google Scholar] [CrossRef]
- Butler, G.S.; Overall, C.M. Proteomic identification of multitasking proteins in unexpected locations complicates drug targeting. Nat. Rev. Drug Discov. 2009, 8, 935–948. [Google Scholar] [CrossRef]
- Hu, J.; Van den Steen, P.E.; Sang, Q.-X.A.; Opdenakker, G. Matrix metalloproteinase inhibitors as therapy for inflammatory and vascular diseases. Nat. Rev. Drug Discov. 2007, 6, 480–498. [Google Scholar] [CrossRef] [PubMed]
- Dufour, A.; Overall, C.M. Subtracting Matrix Out of the Equation: New Key Roles of Matrix Metalloproteinases in Innate Immunity and Disease. In Matrix Metalloproteinase Biology, Sagi/Matrix Metalloproteinase Biology; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2015; Volume 48, pp. 131–152. [Google Scholar]
- Cauwe, B.; Opdenakker, G. Intracellular substrate cleavage: A novel dimension in the biochemistry, biology and pathology of matrix metalloproteinases. Crit. Rev. Biochem. Mol. Biol. 2010, 45, 351–423. [Google Scholar] [CrossRef] [PubMed]
- Starr, A.E.; Dufour, A.; Maier, J.; Overall, C.M. Biochemical analysis of matrix metalloproteinase activation of chemokines CCL15 and CCL23 and increased glycosaminoglycan binding of CCL16. J. Biol. Chem. 2012, 287, 5848–5860. [Google Scholar] [CrossRef]
- Dufour, A.; Bellac, C.L.; Eckhard, U.; Solis, N.; Klein, T.; Kappelhoff, R.; Fortelny, N.; Jobin, P.; Rozmus, J.; Mark, J.; et al. C-terminal truncation of IFN-γ inhibits proinflammatory macrophage responses and is deficient in autoimmune disease. Nat. Commun. 2018, 9, 2416. [Google Scholar] [CrossRef] [PubMed]
- Marchant, D.J.; Bellac, C.L.; Moraes, T.J.; Wadsworth, S.J.; Dufour, A.; Butler, G.S.; Bilawchuk, L.M.; Hendry, R.G.; Robertson, A.G.; Cheung, C.T.; et al. A new transcriptional role for matrix metalloproteinase-12 in antiviral immunity. Nat. Med. 2014, 20, 493–502. [Google Scholar] [CrossRef]
- Dean, R.A.; Cox, J.H.; Bellac, C.L.; Doucet, A.; Starr, A.E.; Overall, C.M. Macrophage-specific metalloelastase (MMP-12) truncates and inactivates ELR+ CXC chemokines and generates CCL2, -7, -8, and -13 antagonists: Potential role of the macrophage in terminating polymorphonuclear leukocyte influx. Blood 2008, 112, 3455–3464. [Google Scholar] [CrossRef]
- Khokha, R.; Murthy, A.; Weiss, A. Metalloproteinases and their natural inhibitors in inflammation and immunity. Nat. Rev. Immunol. 2013, 13, 649–665. [Google Scholar] [CrossRef]
- Yang, Z.; Wu, B.; Jia, S.; Zhao, Y.; Hou, R.; Liu, X.; Wang, X.; Chen, L.; Yang, X.; Lei, D.; et al. The mechanically activated p38/MMP-2 signaling pathway promotes bone marrow mesenchymal stem cell migration in rats. Arch. Oral Biol. 2017, 76, 55–60. [Google Scholar] [CrossRef]
- Kulkarni, R.N.; Bakker, A.D.; Gruber, E.V.; Chae, T.D.; Veldkamp, J.B.B.; Klein-Nulend, J.; Everts, V. MT1-MMP modulates the mechanosensitivity of osteocytes. Biochem. Biophys. Res. Commun. 2012, 417, 824–829. [Google Scholar] [CrossRef] [PubMed]
- Gieseler, F.; Ungefroren, H.; Settmacher, U.; Hollenberg, M.D.; Kaufmann, R. Proteinase-activated receptors (PARs)-focus on receptor-receptor-interactions and their physiological and pathophysiological impact. Cell Commun. Signal. 2013, 11, 86. [Google Scholar] [CrossRef] [PubMed]
- Austin, K.M.; Covic, L.; Kuliopulos, A. Matrix metalloproteases and PAR1 activation. Blood 2013, 121, 431–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trivedi, V.; Boire, A.; Tchernychev, B.; Kaneider, N.C.; Leger, A.J.; O’Callaghan, K.; Covic, L.; Kuliopulos, A. Platelet matrix metalloprotease-1 mediates thrombogenesis by activating PAR1 at a cryptic ligand site. Cell 2009, 137, 332–343. [Google Scholar] [CrossRef]
- Wojtukiewicz, M.Z.; Hempel, D.; Sierko, E.; Tucker, S.C.; Honn, K.V. Protease-activated receptors (PARs)--biology and role in cancer invasion and metastasis. Cancer Metastasis Rev. 2015, 34, 775–796. [Google Scholar] [CrossRef] [PubMed]
- Boire, A.; Covic, L.; Agarwal, A.; Jacques, S.; Sherifi, S.; Kuliopulos, A. PAR1 is a matrix metalloprotease-1 receptor that promotes invasion and tumorigenesis of breast cancer cells. Cell 2005, 120, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Amour, A.; Knight, C.G.; English, W.R.; Webster, A.; Slocombe, P.M.; Knäuper, V.; Docherty, A.J.P.; Becherer, J.D.; Blobel, C.P.; Murphy, G. The enzymatic activity of ADAM8 and ADAM9 is not regulated by TIMPs. FEBS Lett. 2002, 524, 154–158. [Google Scholar] [CrossRef]
- Yan, P.; Hu, X.; Song, H.; Yin, K.; Bateman, R.J.; Cirrito, J.R.; Xiao, Q.; Hsu, F.F.; Turk, J.W.; Xu, J.; et al. Matrix metalloproteinase-9 degrades amyloid-beta fibrils in vitro and compact plaques in situ. J. Biol. Chem. 2006, 281, 24566–24574. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, M.; Takino, T.; Miyamori, H.; Yoshizaki, T.; Furukawa, M.; Sato, H. Cleavage of amyloid-beta precursor protein (APP) by membrane-type matrix metalloproteinases. J. Biochem. 2006, 139, 517–526. [Google Scholar] [CrossRef]
- LePage, R.N.; Fosang, A.J.; Fuller, S.J.; Murphy, G.; Evin, G.; Beyreuther, K.; Masters, C.L.; Small, D.H. Gelatinase A possesses a beta-secretase-like activity in cleaving the amyloid protein precursor of Alzheimer’s disease. FEBS Lett. 1995, 377, 267–270. [Google Scholar] [CrossRef]
- Kajita, M.; Itoh, Y.; Chiba, T.; Mori, H.; Okada, A.; Kinoh, H.; Seiki, M. Membrane-type 1 matrix metalloproteinase cleaves CD44 and promotes cell migration. J. Cell Biol. 2001, 153, 893–904. [Google Scholar] [CrossRef] [PubMed]
- Tsunezumi, J.; Higashi, S.; Miyazaki, K. Matrilysin (MMP-7) cleaves C-type lectin domain family 3 member A (CLEC3A) on tumor cell surface and modulates its cell adhesion activity. J. Cell. Biochem. 2009, 106, 693–702. [Google Scholar] [CrossRef] [PubMed]
- Dean, R.A.; Overall, C.M. Proteomics discovery of metalloproteinase substrates in the cellular context by iTRAQ labeling reveals a diverse MMP-2 substrate degradome. Mol. Cell Proteom. 2007, 6, 611–623. [Google Scholar] [CrossRef] [PubMed]
- Bozzi, M.; Inzitari, R.; Sbardell, D.; Monaco, S.; Pavoni, E.; Gioia, M.; Marini, S.; Morlacchi, S.; Sciandra, F.; Castagnola, M.; et al. Enzymatic processing of beta-dystroglycan recombinant ectodomain by MMP-9: Identification of the main cleavage site. IUBMB Life 2009, 61, 1143–1152. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Kesavan, P.; Nakada, M.T.; Yan, L. Tumor-stroma interaction: Positive feedback regulation of extracellular matrix metalloproteinase inducer (EMMPRIN) expression and matrix metalloproteinase-dependent generation of soluble EMMPRIN. Mol. Cancer Res. 2004, 2, 73–80. [Google Scholar] [PubMed]
- Haug, C.; Lenz, C.; Díaz, F.; Bachem, M.G. Oxidized low-density lipoproteins stimulate extracellular matrix metalloproteinase Inducer (EMMPRIN) release by coronary smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1823–1829. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.-T.; Sloniowski, S.; Ethell, D.W.; Ethell, I.M. Ephrin-B2-induced cleavage of EphB2 receptor is mediated by matrix metalloproteinases to trigger cell repulsion. J. Biol. Chem. 2008, 283, 28969–28979. [Google Scholar] [CrossRef]
- Strand, S.; Vollmer, P.; van den Abeelen, L.; Gottfried, D.; Alla, V.; Heid, H.; Kuball, J.; Theobald, M.; Galle, P.R.; Strand, D. Cleavage of CD95 by matrix metalloproteinase-7 induces apoptosis resistance in tumour cells. Oncogene 2004, 23, 3732–3736. [Google Scholar] [CrossRef] [Green Version]
- Almendro, V.; Ametller, E.; García-Recio, S.; Collazo, O.; Casas, I.; Augé, J.M.; Maurel, J.; Gascón, P. The role of MMP7 and its cross-talk with the FAS/FASL system during the acquisition of chemoresistance to oxaliplatin. PLoS ONE 2009, 4, e4728. [Google Scholar] [CrossRef]
- Levi, E.; Fridman, R.; Miao, H.Q.; Ma, Y.S.; Yayon, A.; Vlodavsky, I. Matrix metalloproteinase 2 releases active soluble ectodomain of fibroblast growth factor receptor 1. Proc. Natl. Acad. Sci. USA 1996, 93, 7069–7074. [Google Scholar] [CrossRef]
- Ratnikov, B.I.; Rozanov, D.V.; Postnova, T.I.; Baciu, P.G.; Zhang, H.; DiScipio, R.G.; Chestukhina, G.G.; Smith, J.W.; Deryugina, E.I.; Strongin, A.Y. An alternative processing of integrin alpha(v) subunit in tumor cells by membrane type-1 matrix metalloproteinase. J. Biol. Chem. 2002, 277, 7377–7385. [Google Scholar] [CrossRef]
- Vaisar, T.; Kassim, S.Y.; Gomez, I.G.; Green, P.S.; Hargarten, S.; Gough, P.J.; Parks, W.C.; Wilson, C.L.; Raines, E.W.; Heinecke, J.W. MMP-9 sheds the beta2 integrin subunit (CD18) from macrophages. Mol. Cell Proteom. 2009, 8, 1044–1060. [Google Scholar] [CrossRef]
- Maile, L.A.; Capps, B.E.; Miller, E.C.; Allen, L.B.; Veluvolu, U.; Aday, A.W.; Clemmons, D.R. Glucose regulation of integrin-associated protein cleavage controls the response of vascular smooth muscle cells to insulin-like growth factor-I. Mol. Endocrinol. 2008, 22, 1226–1237. [Google Scholar] [CrossRef]
- Sithu, S.D.; English, W.R.; Olson, P.; Krubasik, D.; Baker, A.H.; Murphy, G.; D’Souza, S.E. Membrane-type 1-matrix metalloproteinase regulates intracellular adhesion molecule-1 (ICAM-1)-mediated monocyte transmigration. J. Biol. Chem. 2007, 282, 25010–25019. [Google Scholar] [CrossRef]
- Tarín, C.; Gomez, M.; Calvo, E.; López, J.A.; Zaragoza, C. Endothelial nitric oxide deficiency reduces MMP-13-mediated cleavage of ICAM-1 in vascular endothelium: A role in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 27–32. [Google Scholar] [CrossRef]
- Essick, E.; Sithu, S.; Dean, W.; D’Souza, S. Pervanadate-induced shedding of the intercellular adhesion molecule (ICAM)-1 ectodomain is mediated by membrane type-1 matrix metalloproteinase (MT1-MMP). Mol. Cell. Biochem. 2008, 314, 151–159. [Google Scholar] [CrossRef]
- Amano, T.; Kwak, O.; Fu, L.; Marshak, A.; Shi, Y.-B. The matrix metalloproteinase stromelysin-3 cleaves laminin receptor at two distinct sites between the transmembrane domain and laminin binding sequence within the extracellular domain. Cell Res. 2005, 15, 150–159. [Google Scholar] [CrossRef]
- Szklarczyk, A.; Ewaleifoh, O.; Beique, J.-C.; Wang, Y.; Knorr, D.; Haughey, N.; Malpica, T.; Mattson, M.P.; Huganir, R.; Conant, K. MMP-7 cleaves the NR1 NMDA receptor subunit and modifies NMDA receptor function. FASEB J. 2008, 22, 3757–3767. [Google Scholar] [CrossRef]
- Milward, E.; Kim, K.J.; Szklarczyk, A.; Nguyen, T.; Melli, G.; Nayak, M.; Deshpande, D.; Fitzsimmons, C.; Hoke, A.; Kerr, D.; et al. Cleavage of myelin associated glycoprotein by matrix metalloproteinases. J. Neuroimmunol. 2008, 193, 140–148. [Google Scholar] [CrossRef] [Green Version]
- Chow, J.P.H.; Fujikawa, A.; Shimizu, H.; Suzuki, R.; Noda, M. Metalloproteinase- and gamma-secretase-mediated cleavage of protein-tyrosine phosphatase receptor type Z. J. Biol. Chem. 2008, 283, 30879–30889. [Google Scholar] [CrossRef]
- Lynch, C.C.; Hikosaka, A.; Acuff, H.B.; Martin, M.D.; Kawai, N.; Singh, R.K.; Vargo-Gogola, T.C.; Begtrup, J.L.; Peterson, T.E.; Fingleton, B.; et al. MMP-7 promotes prostate cancer-induced osteolysis via the solubilization of RANKL. Cancer Cell 2005, 7, 485–496. [Google Scholar] [CrossRef] [Green Version]
- Basile, J.R.; Holmbeck, K.; Bugge, T.H.; Gutkind, J.S. MT1-MMP controls tumor-induced angiogenesis through the release of semaphorin 4D. J. Biol. Chem. 2007, 282, 6899–6905. [Google Scholar] [CrossRef]
- Belkin, A.M.; Zemskov, E.A.; Hang, J.; Akimov, S.S.; Sikora, S.; Strongin, A.Y. Cell-surface-associated tissue transglutaminase is a target of MMP-2 proteolysis. Biochemistry 2004, 43, 11760–11769. [Google Scholar] [CrossRef]
- Belkin, A.M.; Akimov, S.S.; Zaritskaya, L.S.; Ratnikov, B.I.; Deryugina, E.I.; Strongin, A.Y. Matrix-dependent proteolysis of surface transglutaminase by membrane-type metalloproteinase regulates cancer cell adhesion and locomotion. J. Biol. Chem. 2001, 276, 18415–18422. [Google Scholar] [CrossRef]
- Schlöndorff, J.; Lum, L.; Blobel, C.P. Biochemical and pharmacological criteria define two shedding activities for TRANCE/OPGL that are distinct from the tumor necrosis factor alpha convertase. J. Biol. Chem. 2001, 276, 14665–14674. [Google Scholar] [CrossRef]
- Andolfo, A.; English, W.R.; Resnati, M.; Murphy, G.; Blasi, F.; Sidenius, N. Metalloproteases cleave the urokinase-type plasminogen activator receptor in the D1-D2 linker region and expose epitopes not present in the intact soluble receptor. Thromb. Haemost. 2002, 88, 298–306. [Google Scholar] [CrossRef]
- Peter, M.E.; Legembre, P.; Barnhart, B.C. Does CD95 have tumor promoting activities? Biochim. Biophys. Acta 2005, 1755, 25–36. [Google Scholar] [CrossRef]
- Shresta, S.; Pham, C.T.; Thomas, D.A.; Graubert, T.A.; Ley, T.J. How do cytotoxic lymphocytes kill their targets? Curr. Opin. Immunol. 1998, 10, 581–587. [Google Scholar] [CrossRef]
- Mollinedo, F.; Gajate, C. Fas/CD95 death receptor and lipid rafts: New targets for apoptosis-directed cancer therapy. Drug Resist. Updat. 2006, 9, 51–73. [Google Scholar] [CrossRef]
- Mitsiades, N.; Yu, W.H.; Poulaki, V.; Tsokos, M.; Stamenkovic, I. Matrix metalloproteinase-7-mediated cleavage of Fas ligand protects tumor cells from chemotherapeutic drug cytotoxicity. Cancer Res. 2001, 61, 577–581. [Google Scholar]
- Kayagaki, N.; Kawasaki, A.; Ebata, T.; Ohmoto, H.; Ikeda, S.; Inoue, S.; Yoshino, K.; Okumura, K.; Yagita, H. Metalloproteinase-mediated release of human Fas ligand. J. Exp. Med. 1995, 182, 1777–1783. [Google Scholar] [CrossRef]
- Pan, G.; Bauer, J.H.; Haridas, V.; Wang, S.; Liu, D.; Yu, G.; Vincenz, C.; Aggarwal, B.B.; Ni, J.; Dixit, V.M. Identification and functional characterization of DR6, a novel death domain-containing TNF receptor. FEBS Lett. 1998, 431, 351–356. [Google Scholar] [CrossRef] [Green Version]
- Benschop, R.; Wei, T.; Na, S. Tumor necrosis factor receptor superfamily member 21: TNFR-related death receptor-6, DR6. Adv. Exp. Med. Biol. 2009, 647, 186–194. [Google Scholar]
- Klíma, M.; Zájedová, J.; Doubravská, L.; Andera, L. Functional analysis of the posttranslational modifications of the death receptor 6. Biochim. Biophys. Acta 2009, 1793, 1579–1587. [Google Scholar] [CrossRef] [Green Version]
- Nikolaev, A.; McLaughlin, T.; O’Leary, D.D.M.; Tessier-Lavigne, M. APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature 2009, 457, 981–989. [Google Scholar] [CrossRef] [Green Version]
- DeRosa, D.C.; Ryan, P.J.; Okragly, A.; Witcher, D.R.; Benschop, R.J. Tumor-derived death receptor 6 modulates dendritic cell development. Cancer Immunol. Immunother. 2008, 57, 777–787. [Google Scholar] [CrossRef]
- Tam, E.M.; Morrison, C.J.; Wu, Y.I.; Stack, M.S.; Overall, C.M. Membrane protease proteomics: Isotope-coded affinity tag MS identification of undescribed MT1-matrix metalloproteinase substrates. Proc. Natl. Acad. Sci. USA 2004, 101, 6917–6922. [Google Scholar] [CrossRef]
- Brambilla, R.; Br uuml ckner, K.; Orioli, D.; Bergemann, A.; Flanagan, J.; Klein, R. Similarities and Differences in the Way Transmembrane-Type Ligands Interact with the Elk Subclass of Eph Receptors. Mol. Cell. Neurosci. 1996, 8, 199–209. [Google Scholar] [CrossRef]
- Flanagan, J.G.; Vanderhaeghen, P. The ephrins and Eph receptors in neural development. Annu. Rev. Neurosci. 1998, 21, 309–345. [Google Scholar] [CrossRef]
- Klein, R. Eph/ephrin signalling during development. Development 2012, 139, 4105–4109. [Google Scholar] [CrossRef] [Green Version]
- Zimmer, M.; Palmer, A.; Köhler, J.; Klein, R. EphB-ephrinB bi-directional endocytosis terminates adhesion allowing contact mediated repulsion. Nat. Cell Biol. 2003, 5, 869–878. [Google Scholar] [CrossRef]
- Marston, D.J.; Dickinson, S.; Nobes, C.D. Rac-dependent trans-endocytosis of ephrinBs regulates Eph-ephrin contact repulsion. Nat. Cell Biol. 2003, 5, 879–888. [Google Scholar] [CrossRef]
- Dufour, A.; Zucker, S.; Sampson, N.S.; Kuscu, C.; Cao, J. Role of matrix metalloproteinase-9 dimers in cell migration: Design of inhibitory peptides. J. Biol. Chem. 2010, 285, 35944–35956. [Google Scholar] [CrossRef]
- Dufour, A.; Sampson, N.S.; Li, J.; Kuscu, C.; Rizzo, R.C.; Deleon, J.L.; Zhi, J.; Jaber, N.; Liu, E.; Zucker, S.; et al. Small-molecule anticancer compounds selectively target the hemopexin domain of matrix metalloproteinase-9. Cancer Res. 2011, 71, 4977–4988. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Morath, I.; Hartmann, T.N.; Orian-Rousseau, V. CD44: More than a mere stem cell marker. Int. J. Biochem. Cell Biol. 2016, 81, 166–173. [Google Scholar] [CrossRef]
- Nakamura, H.; Suenaga, N.; Taniwaki, K.; Matsuki, H.; Yonezawa, K.; Fujii, M.; Okada, Y.; Seiki, M. Constitutive and induced CD44 shedding by ADAM-like proteases and membrane-type 1 matrix metalloproteinase. Cancer Res. 2004, 64, 876–882. [Google Scholar] [CrossRef]
- Zöller, M. CD44, Hyaluronan, the Hematopoietic Stem Cell, and Leukemia-Initiating Cells. Front. Immunol. 2015, 6, 235. [Google Scholar] [Green Version]
- Aruffo, A.; Stamenkovic, I.; Melnick, M.; Underhill, C.B.; Seed, B. CD44 is the principal cell surface receptor for hyaluronate. Cell 1990, 61, 1303–1313. [Google Scholar] [CrossRef]
- Naor, D.; Sionov, R.V.; Ish-Shalom, D. CD44: Structure, function, and association with the malignant process. Adv. Cancer Res. 1997, 71, 241–319. [Google Scholar]
- Zarrabi, K.; Dufour, A.; Li, J.; Kuscu, C.; Pulkoski-Gross, A.; Zhi, J.; Hu, Y.; Sampson, N.S.; Zucker, S.; Cao, J. Inhibition of matrix metalloproteinase 14 (MMP-14)-mediated cancer cell migration. J. Biol. Chem. 2011, 286, 33167–33177. [Google Scholar] [CrossRef]
- Hauser, A.S.; Attwood, M.M.; Rask-Andersen, M.; Schiöth, H.B.; Gloriam, D.E. Trends in GPCR drug discovery: New agents, targets and indications. Nat. Rev. Drug Discov. 2017, 16, 829–842. [Google Scholar] [CrossRef]
- Hilger, D.; Masureel, M.; Kobilka, B.K. Structure and dynamics of GPCR signaling complexes. Nat. Struct. Mol. Biol. 2018, 25, 4–12. [Google Scholar] [CrossRef]
- Latorraca, N.R.; Venkatakrishnan, A.J.; Dror, R.O. GPCR Dynamics: Structures in Motion. Chem. Rev. 2017, 117, 139–155. [Google Scholar] [CrossRef]
- Laguri, C.; Arenzana-Seisdedos, F.; Lortat-Jacob, H. Relationships between glycosaminoglycan and receptor binding sites in chemokines-the CXCL12 example. Carbohydr. Res. 2008, 343, 2018–2023. [Google Scholar] [CrossRef]
- Griffith, J.W.; Sokol, C.L.; Luster, A.D. Chemokines and chemokine receptors: Positioning cells for host defense and immunity. Annu. Rev. Immunol. 2014, 32, 659–702. [Google Scholar] [CrossRef]
- Eash, K.J.; Greenbaum, A.M.; Gopalan, P.K.; Link, D.C. CXCR2 and CXCR4 antagonistically regulate neutrophil trafficking from murine bone marrow. J. Clin. Invest. 2010, 120, 2423–2431. [Google Scholar] [CrossRef] [Green Version]
- Sabroe, I.; Hartnell, A.; Jopling, L.A.; Bel, S.; Ponath, P.D.; Pease, J.E.; Collins, P.D.; Williams, T.J. Differential regulation of eosinophil chemokine signaling via CCR3 and non-CCR3 pathways. J. Immunol. 1999, 162, 2946–2955. [Google Scholar]
- Serbina, N.V.; Pamer, E.G. Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat. Immunol. 2006, 7, 311–317. [Google Scholar] [CrossRef]
- Berkhout, T.A.; Sarau, H.M.; Moores, K.; White, J.R.; Elshourbagy, N.; Appelbaum, E.; Reape, R.J.; Brawner, M.; Makwana, J.; Foley, J.J.; et al. Cloning, in vitro expression, and functional characterization of a novel human CC chemokine of the monocyte chemotactic protein (MCP) family (MCP-4) that binds and signals through the CC chemokine receptor 2B. J. Biol. Chem. 1997, 272, 16404–16413. [Google Scholar] [CrossRef]
- Thompson, S.; Martínez-Burgo, B.; Sepuru, K.M.; Rajarathnam, K.; Kirby, J.A.; Sheerin, N.S.; Ali, S. Regulation of Chemokine Function: The Roles of GAG-Binding and Post-Translational Nitration. Int. J. Mol. Sci. 2017, 18, 1692. [Google Scholar] [CrossRef]
- McQuibban, G.A.; Gong, J.-H.; Wong, J.P.; Wallace, J.L.; Clark-Lewis, I.; Overall, C.M. Matrix metalloproteinase processing of monocyte chemoattractant proteins generates CC chemokine receptor antagonists with anti-inflammatory properties in vivo. Blood 2002, 100, 1160–1167. [Google Scholar]
- Starr, A.E.; Bellac, C.L.; Dufour, A.; Goebeler, V.; Overall, C.M. Biochemical characterization and N-terminomics analysis of leukolysin, the membrane-type 6 matrix metalloprotease (MMP25): Chemokine and vimentin cleavages enhance cell migration and macrophage phagocytic activities. J. Biol. Chem. 2012, 287, 13382–13395. [Google Scholar] [CrossRef]
- Prudova, A.; Auf dem Keller, U.; Butler, G.S.; Overall, C.M. Multiplex N-terminome analysis of MMP-2 and MMP-9 substrate degradomes by iTRAQ-TAILS quantitative proteomics. Mol. Cell Proteom. 2010, 9, 894–911. [Google Scholar] [CrossRef]
- Nelissen, I.; Martens, E.; Van den Steen, P.E.; Proost, P.; Ronsse, I.; Opdenakker, G. Gelatinase B/matrix metalloproteinase-9 cleaves interferon-beta and is a target for immunotherapy. Brain 2003, 126, 1371–1381. [Google Scholar] [CrossRef]
- Gearing, A.J.; Beckett, P.; Christodoulou, M.; Churchill, M.; Clements, J.; Davidson, A.H.; Drummond, A.H.; Galloway, W.A.; Gilbert, R.; Gordon, J.L. Processing of tumour necrosis factor-alpha precursor by metalloproteinases. Nature 1994, 370, 555–557. [Google Scholar] [CrossRef]
- Ito, A.; Mukaiyama, A.; Itoh, Y.; Nagase, H.; Thogersen, I.B.; Enghild, J.J.; Sasaguri, Y.; Mori, Y. Degradation of interleukin 1beta by matrix metalloproteinases. J. Biol. Chem. 1996, 271, 14657–14660. [Google Scholar] [CrossRef]
- D’Angelo, M.; Billings, P.C.; Pacifici, M.; Leboy, P.S.; Kirsch, T. Authentic matrix vesicles contain active metalloproteases (MMP). a role for matrix vesicle-associated MMP-13 in activation of transforming growth factor-beta. J. Biol. Chem. 2001, 276, 11347–11353. [Google Scholar]
- Yu, Q.; Stamenkovic, I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesis. Genes Dev. 2000, 14, 163–176. [Google Scholar]
- Shah, D.K.; Zúñiga-Pflücker, J.C. An overview of the intrathymic intricacies of T cell development. J. Immunol. 2014, 192, 4017–4023. [Google Scholar] [CrossRef]
- Koch, U.; Radtke, F. Mechanisms of T cell development and transformation. Annu. Rev. Cell Dev. Biol. 2011, 27, 539–562. [Google Scholar] [CrossRef]
- Uehara, S.; Grinberg, A.; Farber, J.M.; Love, P.E. A role for CCR9 in T lymphocyte development and migration. J. Immunol. 2002, 168, 2811–2819. [Google Scholar] [CrossRef]
- Ueno, T.; Hara, K.; Willis, M.S.; Malin, M.A.; Höpken, U.E.; Gray, D.H.D.; Matsushima, K.; Lipp, M.; Springer, T.A.; Boyd, R.L.; et al. Role for CCR7 ligands in the emigration of newly generated T lymphocytes from the neonatal thymus. Immunity 2002, 16, 205–218. [Google Scholar] [CrossRef]
- Al-Alwan, L.A.; Chang, Y.; Mogas, A.; Halayko, A.J.; Baglole, C.J.; Martin, J.G.; Rousseau, S.; Eidelman, D.H.; Hamid, Q. Differential roles of CXCL2 and CXCL3 and their receptors in regulating normal and asthmatic airway smooth muscle cell migration. J. Immunol. 2013, 191, 2731–2741. [Google Scholar] [CrossRef]
- Wuyts, A.; Govaerts, C.; Struyf, S.; Lenaerts, J.P.; Put, W.; Conings, R.; Proost, P.; van Damme, J. Isolation of the CXC chemokines ENA-78, GRO alpha and GRO gamma from tumor cells and leukocytes reveals NH2-terminal heterogeneity. Functional comparison of different natural isoforms. Eur. J. Biochem. 1999, 260, 421–429. [Google Scholar] [CrossRef]
- Van den Steen, P.E.; Wuyts, A.; Husson, S.J.; Proost, P.; Van Damme, J.; Opdenakker, G. Gelatinase B/MMP-9 and neutrophil collagenase/MMP-8 process the chemokines human GCP-2/CXCL6, ENA-78/CXCL5 and mouse GCP-2/LIX and modulate their physiological activities. Eur. J. Biochem. 2003, 270, 3739–3749. [Google Scholar] [CrossRef]
- Berahovich, R.D.; Miao, Z.; Wang, Y.; Premack, B.; Howard, M.C.; Schall, T.J. Proteolytic activation of alternative CCR1 ligands in inflammation. J. Immunol. 2005, 174, 7341–7351. [Google Scholar] [CrossRef]
- McQuibban, G.A.; Gong, J.H.; Tam, E.M.; McCulloch, C.A.; Clark-Lewis, I.; Overall, C.M. Inflammation dampened by gelatinase A cleavage of monocyte chemoattractant protein-3. Science 2000, 289, 1202–1206. [Google Scholar] [CrossRef]
- Altan-Bonnet, G.; Mukherjee, R. Cytokine-mediated communication: A quantitative appraisal of immune complexity. Nat. Rev. Immunol. 2019, 19, 205–217. [Google Scholar] [CrossRef]
- Van Lint, P.; Libert, C. Chemokine and cytokine processing by matrix metalloproteinases and its effect on leukocyte migration and inflammation. J. Leukoc. Biol. 2007, 82, 1375–1381. [Google Scholar] [CrossRef] [Green Version]
- Crow, M.K. Autoimmunity: Interferon α or β: Which is the culprit in autoimmune disease? Nat. Rev. Rheumatol. 2016, 12, 439–440. [Google Scholar] [CrossRef]
- Cheung, C.; Marchant, D.; Walker, E.K.-Y.; Luo, Z.; Zhang, J.; Yanagawa, B.; Rahmani, M.; Cox, J.; Overall, C.; Senior, R.M.; et al. Ablation of matrix metalloproteinase-9 increases severity of viral myocarditis in mice. Circulation 2008, 117, 1574–1582. [Google Scholar] [CrossRef]
- Wajant, H.; Pfizenmaier, K.; Scheurich, P. Tumor necrosis factor signaling. Cell Death Differ. 2003, 10, 45–65. [Google Scholar] [CrossRef] [Green Version]
- Le Gall, S.M.; Maretzky, T.; Issuree, P.D.A.; Niu, X.-D.; Reiss, K.; Saftig, P.; Khokha, R.; Lundell, D.; Blobel, C.P. ADAM17 is regulated by a rapid and reversible mechanism that controls access to its catalytic site. J. Cell. Sci. 2010, 123, 3913–3922. [Google Scholar] [CrossRef] [Green Version]
- Haro, H.; Crawford, H.C.; Fingleton, B.; Shinomiya, K.; Spengler, D.M.; Matrisian, L.M. Matrix metalloproteinase-7-dependent release of tumor necrosis factor-alpha in a model of herniated disc resorption. J. Clin. Investig. 2000, 105, 143–150. [Google Scholar] [CrossRef]
- English, W.R.; Puente, X.S.; Freije, J.M.; Knauper, V.; Amour, A.; Merryweather, A.; Lopez-Otin, C.; Murphy, G. Membrane type 4 matrix metalloproteinase (MMP17) has tumor necrosis factor-alpha convertase activity but does not activate pro-MMP2. J. Biol. Chem. 2000, 275, 14046–14055. [Google Scholar] [CrossRef]
- Chandler, S.; Cossins, J.; Lury, J.; Wells, G. Macrophage metalloelastase degrades matrix and myelin proteins and processes a tumour necrosis factor-alpha fusion protein. Biochem. Biophys. Res. Commun. 1996, 228, 421–429. [Google Scholar] [CrossRef]
- d’Ortho, M.P.; Will, H.; Atkinson, S.; Butler, G.; Messent, A.; Gavrilovic, J.; Smith, B.; Timpl, R.; Zardi, L.; Murphy, G. Membrane-type matrix metalloproteinases 1 and 2 exhibit broad-spectrum proteolytic capacities comparable to many matrix metalloproteinases. Eur. J. Biochem. 1997, 250, 751–757. [Google Scholar] [CrossRef]
- Brough, D.; Rothwell, N.J. Caspase-1-dependent processing of pro-interleukin-1beta is cytosolic and precedes cell death. J. Cell. Sci. 2007, 120, 772–781. [Google Scholar] [CrossRef]
- Thornberry, N.A.; Bull, H.G.; Calaycay, J.R.; Chapman, K.T.; Howard, A.D.; Kostura, M.J.; Miller, D.K.; Molineaux, S.M.; Weidner, J.R.; Aunins, J. A novel heterodimeric cysteine protease is required for interleukin-1 beta processing in monocytes. Nature 1992, 356, 768–774. [Google Scholar] [CrossRef]
- Schönbeck, U.; Mach, F.; Libby, P. Generation of biologically active IL-1 beta by matrix metalloproteinases: A novel caspase-1-independent pathway of IL-1 beta processing. J. Immunol. 1998, 161, 3340–3346. [Google Scholar]
- Bellehumeur, C.; Collette, T.; Maheux, R.; Mailloux, J.; Villeneuve, M.; Akoum, A. Increased soluble interleukin-1 receptor type II proteolysis in the endometrium of women with endometriosis. Hum. Reprod. 2005, 20, 1177–1184. [Google Scholar] [CrossRef] [Green Version]
- Orlando, S.; Sironi, M.; Bianchi, G.; Drummond, A.H.; Boraschi, D.; Yabes, D.; Mantovani, A. Role of metalloproteases in the release of the IL-1 type II decoy receptor. J. Biol. Chem. 1997, 272, 31764–31769. [Google Scholar] [CrossRef]
- Sheppard, D. Transforming growth factor beta: A central modulator of pulmonary and airway inflammation and fibrosis. Proc. Am. Thorac. Soc. 2006, 3, 413–417. [Google Scholar] [CrossRef]
- Maeda, S.; Dean, D.D.; Gomez, R.; Schwartz, Z.; Boyan, B.D. The first stage of transforming growth factor beta1 activation is release of the large latent complex from the extracellular matrix of growth plate chondrocytes by matrix vesicle stromelysin-1 (MMP-3). Calcif. Tissue Int. 2002, 70, 54–65. [Google Scholar] [CrossRef]
- Karsdal, M.A.; Larsen, L.; Engsig, M.T.; Lou, H.; Ferreras, M.; Lochter, A.; Delaissé, J.-M.; Foged, N.T. Matrix metalloproteinase-dependent activation of latent transforming growth factor-beta controls the conversion of osteoblasts into osteocytes by blocking osteoblast apoptosis. J. Biol. Chem. 2002, 277, 44061–44067. [Google Scholar] [CrossRef]
- Dallas, S.L.; Rosser, J.L.; Mundy, G.R.; Bonewald, L.F. Proteolysis of latent transforming growth factor-beta (TGF-beta )-binding protein-1 by osteoclasts. A cellular mechanism for release of TGF-beta from bone matrix. J. Biol. Chem. 2002, 277, 21352–21360. [Google Scholar] [CrossRef]
- Imai, K.; Hiramatsu, A.; Fukushima, D.; Pierschbacher, M.D.; Okada, Y. Degradation of decorin by matrix metalloproteinases: Identification of the cleavage sites, kinetic analyses and transforming growth factor-beta1 release. Biochem. J. 1997, 322 (Pt 3), 809–814. [Google Scholar] [CrossRef]
- Gordon, G.M.; Ledee, D.R.; Feuer, W.J.; Fini, M.E. Cytokines and signaling pathways regulating matrix metalloproteinase-9 (MMP-9) expression in corneal epithelial cells. J. Cell. Physiol. 2009, 221, 402–411. [Google Scholar] [CrossRef] [Green Version]
- Tseng, H.-C.; Lee, I.-T.; Lin, C.-C.; Chi, P.-L.; Cheng, S.-E.; Shih, R.-H.; Hsiao, L.-D.; Yang, C.-M. IL-1β promotes corneal epithelial cell migration by increasing MMP-9 expression through NF-κB- and AP-1-dependent pathways. PLoS ONE 2013, 8, e57955. [Google Scholar] [CrossRef]
- Dufour, A.; Sampson, N.S.; Zucker, S.; Cao, J. Role of the hemopexin domain of matrix metalloproteinases in cell migration. J. Cell. Physiol. 2008, 217, 643–651. [Google Scholar] [CrossRef] [Green Version]
- Zucker, S.; Schmidt, C.E.; Dufour, A.; Kaplan, R.C.; Park, H.I.; Jiang, W. ProMMP-2: TIMP-1 complexes identified in plasma of healthy individuals. Connect. Tissue Res. 2009, 50, 223–231. [Google Scholar] [CrossRef]
- Pavlaki, M.; Zucker, S.; Dufour, A.; Calabrese, N.; Bahou, W.; Cao, J. Furin Functions as a Nonproteolytic Chaperone for Matrix Metalloproteinase-28: MMP-28 Propeptide Sequence Requirement. Biochem. Res. Int. 2011, 2011, 630319. [Google Scholar] [CrossRef]
- Sela-Passwell, N.; Kikkeri, R.; Dym, O.; Rozenberg, H.; Margalit, R.; Arad-Yellin, R.; Eisenstein, M.; Brenner, O.; Shoham, T.; Danon, T.; et al. Antibodies targeting the catalytic zinc complex of activated matrix metalloproteinases show therapeutic potential. Nat. Med. 2011, 18, 143–147. [Google Scholar] [CrossRef]
- Talmi-Frank, D.; Altboum, Z.; Solomonov, I.; Udi, Y.; Jaitin, D.A.; Klepfish, M.; David, E.; Zhuravlev, A.; Keren-Shaul, H.; Winter, D.R.; et al. Extracellular Matrix Proteolysis by MT1-MMP Contributes to Influenza-Related Tissue Damage and Mortality. Cell Host Microbe 2016, 20, 458–470. [Google Scholar] [CrossRef] [Green Version]
- Devy, L.; Huang, L.; Naa, L.; Yanamandra, N.; Pieters, H.; Frans, N.; Chang, E.; Tao, Q.; Vanhove, M.; Lejeune, A.; et al. Selective inhibition of matrix metalloproteinase-14 blocks tumor growth, invasion, and angiogenesis. Cancer Res. 2009, 69, 1517–1526. [Google Scholar] [CrossRef]
- Ager, E.I.; Kozin, S.V.; Kirkpatrick, N.D.; Seano, G.; Kodack, D.P.; Askoxylakis, V.; Huang, Y.; Goel, S.; Snuderl, M.; Muzikansky, A.; et al. Blockade of MMP14 activity in murine breast carcinomas: Implications for macrophages, vessels, and radiotherapy. J. Natl. Cancer Inst. 2015, 107, 52. [Google Scholar] [CrossRef]
- Vandenbroucke, R.E.; Dejager, L.; Libert, C. The first MMP in sepsis. EMBO Mol. Med. 2011, 3, 367–369. [Google Scholar] [CrossRef]
- Vanlaere, I.; Libert, C. Matrix metalloproteinases as drug targets in infections caused by gram-negative bacteria and in septic shock. Clin. Microbiol. Rev. 2009, 22, 224–239. [Google Scholar] [CrossRef]
- Vandenbroucke, R.E.; Dejonckheere, E.; Van Hauwermeiren, F.; Lodens, S.; De Rycke, R.; Van Wonterghem, E.; Staes, A.; Gevaert, K.; López-Otín, C.; Libert, C. Matrix metalloproteinase 13 modulates intestinal epithelial barrier integrity in inflammatory diseases by activating TNF. EMBO Mol. Med. 2013, 5, 1000–1016. [Google Scholar] [CrossRef] [Green Version]
- Vandenbroucke, R.E.; Libert, C. Is there new hope for therapeutic matrix metalloproteinase inhibition? Nat. Rev. Drug Discov. 2014, 13, 904–927. [Google Scholar] [CrossRef]


{kind=link}
| Protein Name | MMP1 | MMP2 | MMP3 | MMP7 | MMP9 | MMP11 | MMP12 | MMP13 | MMP14/MT1–MMP | MMP16/MT3–MMP | MMP25/MT6–MMP | References |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Amyloid protein precursor (APP) | 687KL689 691FA692 694DV695 701AI702 | 579NM580 687KL689 | 463AM464 622HS623 579NM580 685HQ686 | [18,19,20,21] | ||||||||
| CD44 antigen (CD44) | 162RT163 186RS187 192GY193 | [22] | ||||||||||
| C-type lectin domain family 3 member A (CLEC3A) | 57AL58 63AL64 151FL152 | [23] | ||||||||||
| CX3CL1 (fractalkine) | 71AL72 4GM5 | [24] | ||||||||||
| β-dystroglycan | 715HL716 | [25] | ||||||||||
| EMMPRIN/CD147 | 209PM210 214NI215 | [26,27] | ||||||||||
| Ephrin B2 receptor | 394NI395 432DL433 | 394NI395 432DL433 | [28] | |||||||||
| Fas Receptor (FAS) | 19EL20 32NL33 | [29,30] | ||||||||||
| Fibroblast growth factor receptor 1 (FGFR1) | 368VM369 | [31] | ||||||||||
| Integrin αV (CD51) | 891DL892 | [32] | ||||||||||
| Integrin β2 (CD18) | 705AI706 | [33] | ||||||||||
| Integrin-associated protein (IAP/CD47) | [34] | |||||||||||
| Intercellular adhesion molecule (ICAM)-1 | 60IE61 97DG98 | [35,36,37] | ||||||||||
| Laminin receptor | 115AF116 133PI134 | [38] | ||||||||||
| Glutamate receptor ionotropic, NMDA 1 (NMDA receptor) | 516EK517 | [39] | ||||||||||
| Myelin-associated glycoprotein | 233SM234 508RL509 | 233SM234 508RL509 | 233SM234 508RL509 | [40] | ||||||||
| Protease-activated receptor-1 (PAR-1) | 41RS42 | [17] | ||||||||||
| Protein-tyrosine phosphatase receptor type Z (Ptprz) | 1625RI1626 1627GL1628 | [41] | ||||||||||
| Tumor necrosis factor ligand superfamily member 11(RANKL) | 145MM146 | [42] | ||||||||||
| Semaphorin 4D | [43] | |||||||||||
| Tissue transglutaminase | 375PV376 458RA459 461HL462 | 375PV376 458RA459 461HL462 | 375PV376 458RA459 461HL462 | [44,45] | ||||||||
| TRANCE/OPGL (TNF-related activation-induced cytokine/osteoprotegrin ligand) | 138RF139 145MM146 | [46] | ||||||||||
| Urokinase plasminogen activator surface receptor (uPAR/CD87) | 108TY109 | 108TY109 | 108TY109 109YS110 111RS112 | [47] |
| Chemokine Name | MMP1 | MMP2 | MMP3 | MMP7 | MMP8 | MMP9 | MMP12 | MMP13 | MMP 14/MT1–MMP | MMP 25/MT6–MMP | References |
|---|---|---|---|---|---|---|---|---|---|---|---|
| CCL2 | 4S↓A5 27A↓I28 | 4S↓A5 27A↓I28 | 4S↓A5 67K↓T68 | 4S↓A5 | 4S↓A5 | 4S↓A5 | 4S↓A5 | 4S↓A5 | [6,9,83,84] | ||
| CCL3 | 47I↓A48 | 8L↓V9 47I↓A48 63I↓F64 | 15M↓A16 | 15M↓A16 64F↓L65 | 47I↓A48 | [6] | |||||
| CCL4 | 15A↓A16 61P↓A62 | 5V↓T6 6T↓V7 44P↓R45 | 44P↓R45 | 5V↓T6 61P↓A62 | 6T↓V7 | 9L↓V14 61P↓A62 | 6T↓V7 | 5V↓T6 6T↓V7 44P↓R45 | 6T↓V7 | [6,84] | |
| CCL5 | 65V↓T66 | 4S↓A5 | [6] | ||||||||
| CCL7 | 4S↓A5 27G↓I28 | 4S↓A5 27G↓I28 | 4S↓A5 27G↓I28 | 4S↓A5 6A↓L7 | 4S↓A5 27G↓I28 | 4S↓A5 | 4S↓A5 27G↓I28 | 6A↓L7 8L↓C9 27G↓I28 | 4S↓A5 | [6,9,83,84,85] | |
| CCL8 | 4S↓A5 27G↓I28 | 4S↓A5 27G↓I28 | 4S↓A5 27G↓I28 | 4S↓A5 6A↓L7 | 4S↓A5 27G↓I28 | 4S↓A5 | 27G↓I28 | 6A↓L7 8L↓C9 27G↓I28 | 4S↓A5 | [6,9,83,84] | |
| CCL11 | 9W↓L10 | 9W↓L10 | 9W↓L10 | [6] | |||||||
| CCL13 | 4S↓A5 26D↓A27 A27↓L28 30V↓P31 72E↓I73 | 3V↓S4 4S↓A5 9C↓L10 74C↓A75 | 3V↓S4 4S↓A5 26D↓A27 A27↓L28 | 4S↓A5 9C↓L10 72E↓I73 74C↓A75 | 4S↓A5 72E↓I73 | 4S↓A5 72E↓I73 | 4S↓A5 72E↓I73 | 4S↓A5 72E↓I73 | 3V↓S4 | 4S↓A5 72E↓I73 | [6,9,84] |
| CCL14 | 66F↓I67 70R↓G71 | 3I↓S4 6A↓L7 71G↓H72 72H↓S73 | [6] | ||||||||
| CCL15 | 24I↓N25 | 13L↓V14 24I↓N25 26D↓A27 27A↓E28 | 16V↓L17 24I↓N25 27A↓E28 | 16V↓L17 24I↓N25 88K↓G89 | 24I↓N25 27A↓E28 42V↓V43 | 27A↓E28 | 24I↓N25 27A↓E28 | 13L↓V14 24I↓N25 26D↓A27 27A↓E28 | 13L↓V14 26D↓A27 | 24I↓N25 26D↓A27 | [6,84] |
| CCL16 | 7A↓L8 85Q↓E86 | 4S↓E5 7A↓L8 77T↓N78 | 7A↓L8 85Q↓E86 | 7A↓L885Q↓E86 | 4S↓E5 7A↓L8 85Q↓E86 | 7A↓L8 77T↓N78 85Q↓E86 | 7A↓L8 77T↓N78 85Q↓E86 | 7A↓L8 77T↓N78 | 4S↓E5 7A↓L8 85Q↓E86 | 4S↓E5 | [6,84] |
| CCL17 | 3P↓L4 69G↓R70 | 8A↓L9 | 3P↓L4 69G↓R70 | 3P↓L4 | [6] | ||||||
| CCL23 | 10C↓L11 16A↓L17 22R↓V23 25K↓D26 27A↓E28 29T↓E30 | 10C↓L11 16A↓L17 25K↓D26 27A↓E28 | 20Q↓A21 25K↓D26 90G↓R91 | 10C↓L11 16A↓L17 22R↓V23 25K↓D26 90G↓R91 | 10C↓L11 16A↓L17 25K↓D26 | 10C↓L11 27A↓E28 | 10C↓L11 16A↓L17 22R↓V23 25K↓D26 | 90G↓R91 | 13L↓V14 29T↓E30 | 10C↓L11 20Q↓A21 25K↓D26 | [6,84] |
| IFNα | 160L↓Q161 157N↓L158 | [8] | |||||||||
| IFNβ | 25N↓L26 29F↓L30 30L↓Q31 107N↓L108 114N↓L115 | [86] | |||||||||
| IFNγ | 136E↓L115 157M↓L158 | [7] | |||||||||
| Tumor necrosis factor (TNF) | ND | ND | ND | ND | ND | ND | 69L↓I70 72P↓L73 | [58,87] | |||
| IL-1β | 141E↓L142 | [88] | |||||||||
| TGFβ | ND | ND | ND | [89,90] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Young, D.; Das, N.; Anowai, A.; Dufour, A. Matrix Metalloproteases as Influencers of the Cells’ Social Media. Int. J. Mol. Sci. 2019, 20, 3847. https://doi.org/10.3390/ijms20163847
Young D, Das N, Anowai A, Dufour A. Matrix Metalloproteases as Influencers of the Cells’ Social Media. International Journal of Molecular Sciences. 2019; 20(16):3847. https://doi.org/10.3390/ijms20163847
Chicago/Turabian StyleYoung, Daniel, Nabangshu Das, Anthonia Anowai, and Antoine Dufour. 2019. "Matrix Metalloproteases as Influencers of the Cells’ Social Media" International Journal of Molecular Sciences 20, no. 16: 3847. https://doi.org/10.3390/ijms20163847
APA StyleYoung, D., Das, N., Anowai, A., & Dufour, A. (2019). Matrix Metalloproteases as Influencers of the Cells’ Social Media. International Journal of Molecular Sciences, 20(16), 3847. https://doi.org/10.3390/ijms20163847