Microtubules: A Key to Understand and Correct Neuronal Defects in CDKL5 Deficiency Disorder?

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
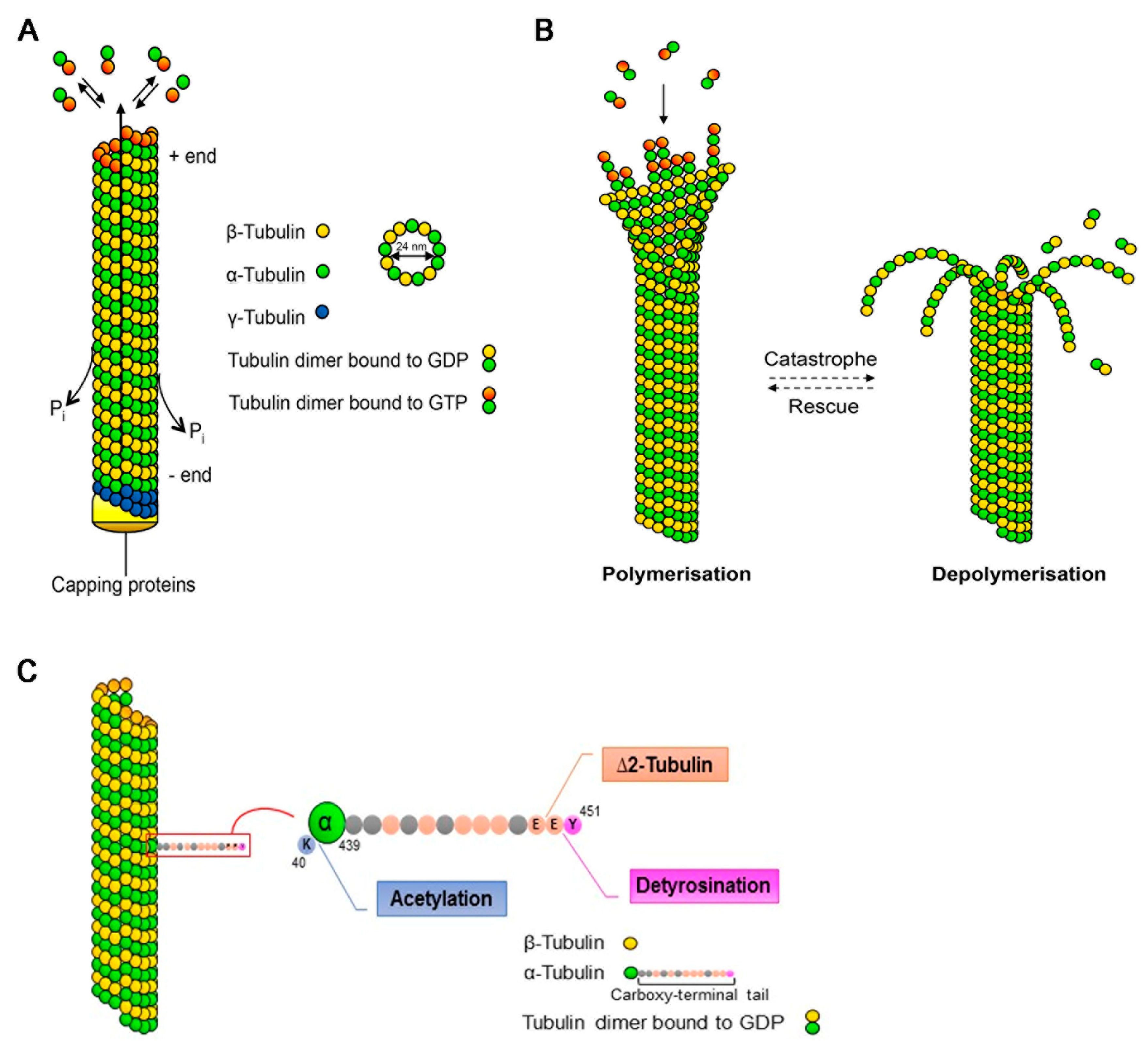
2. Microtubules
3. Neuronal CDKL5 Related Defects
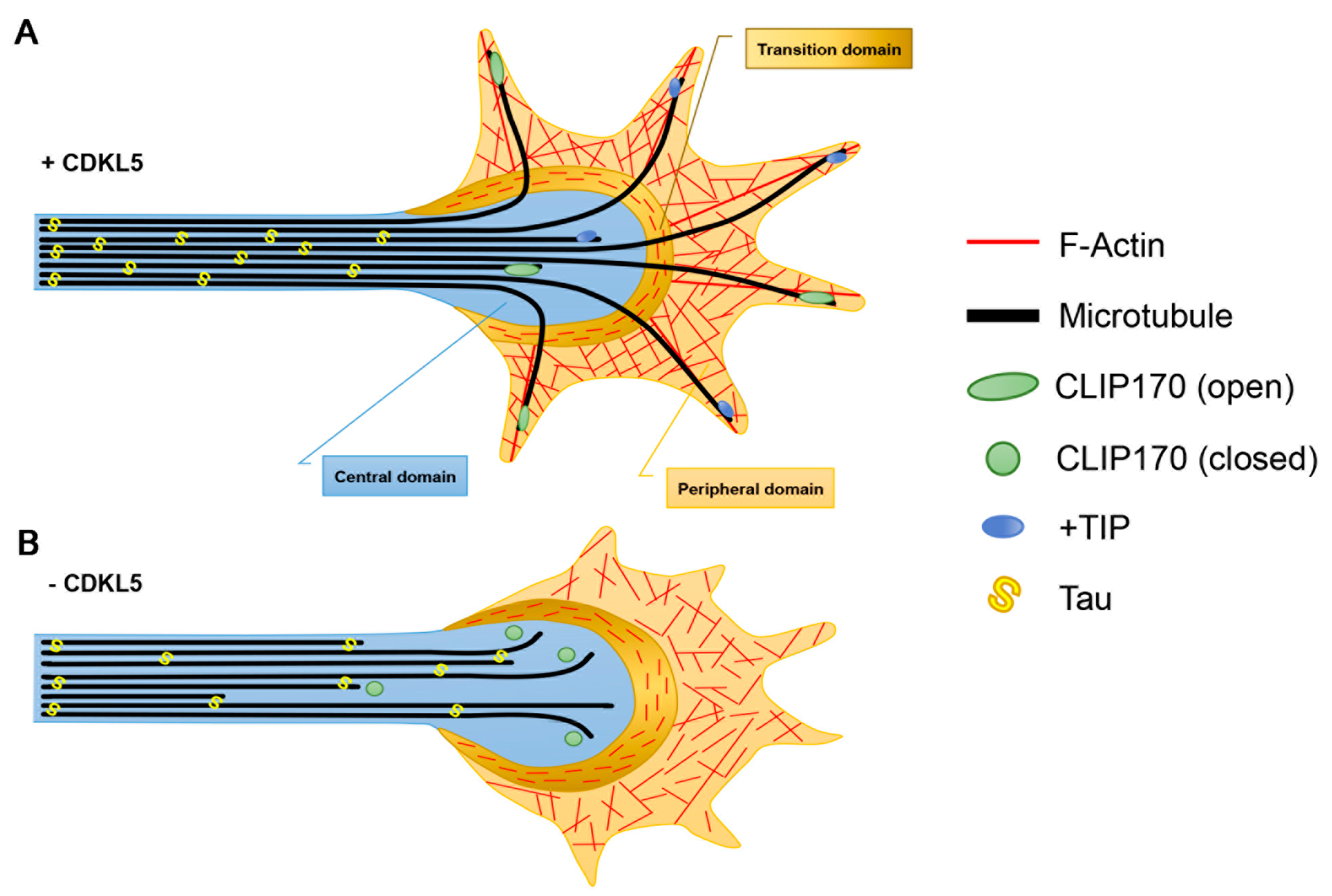
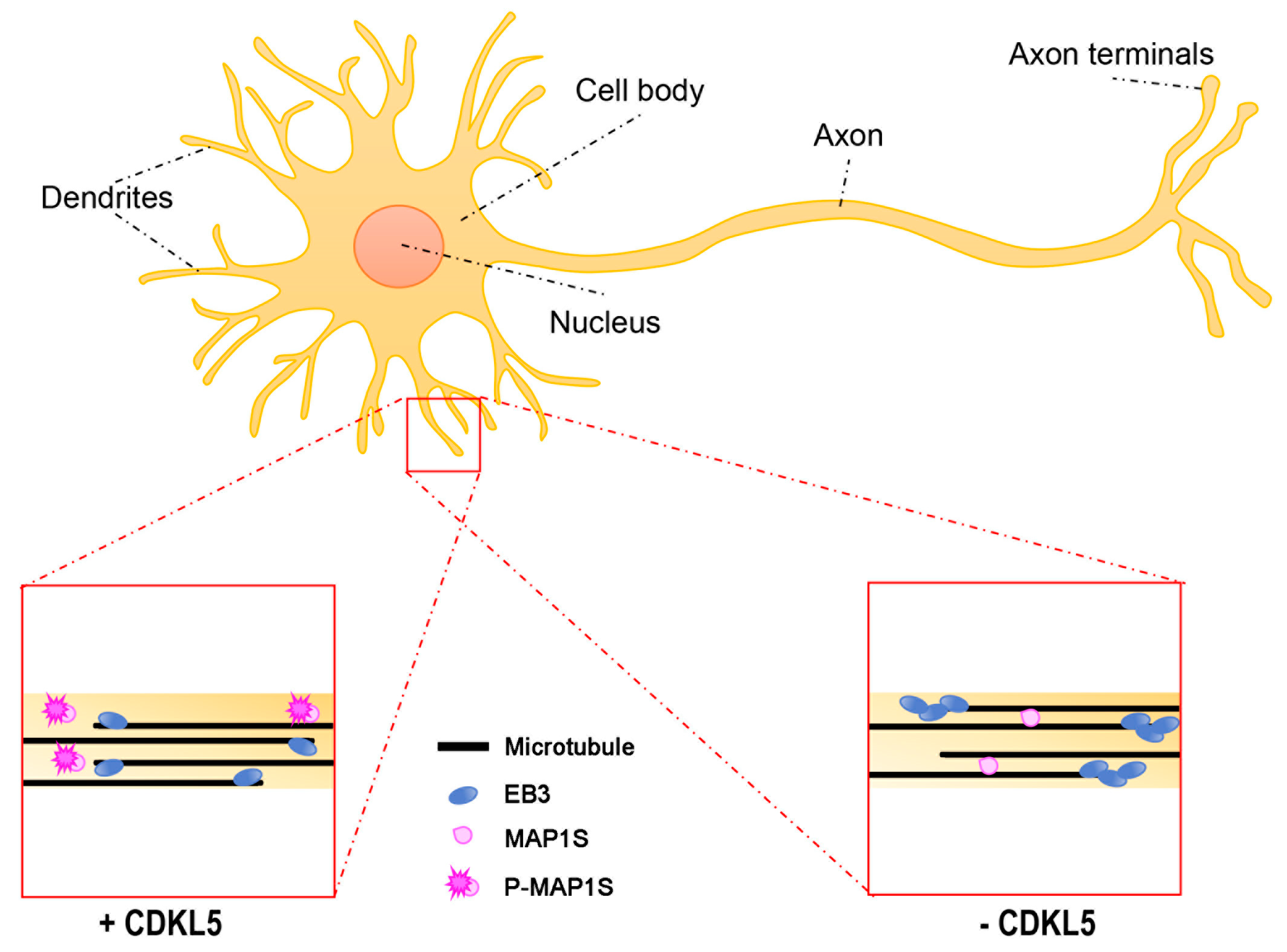
3.1. Axon Formation
3.2. Dendritic Arborisation
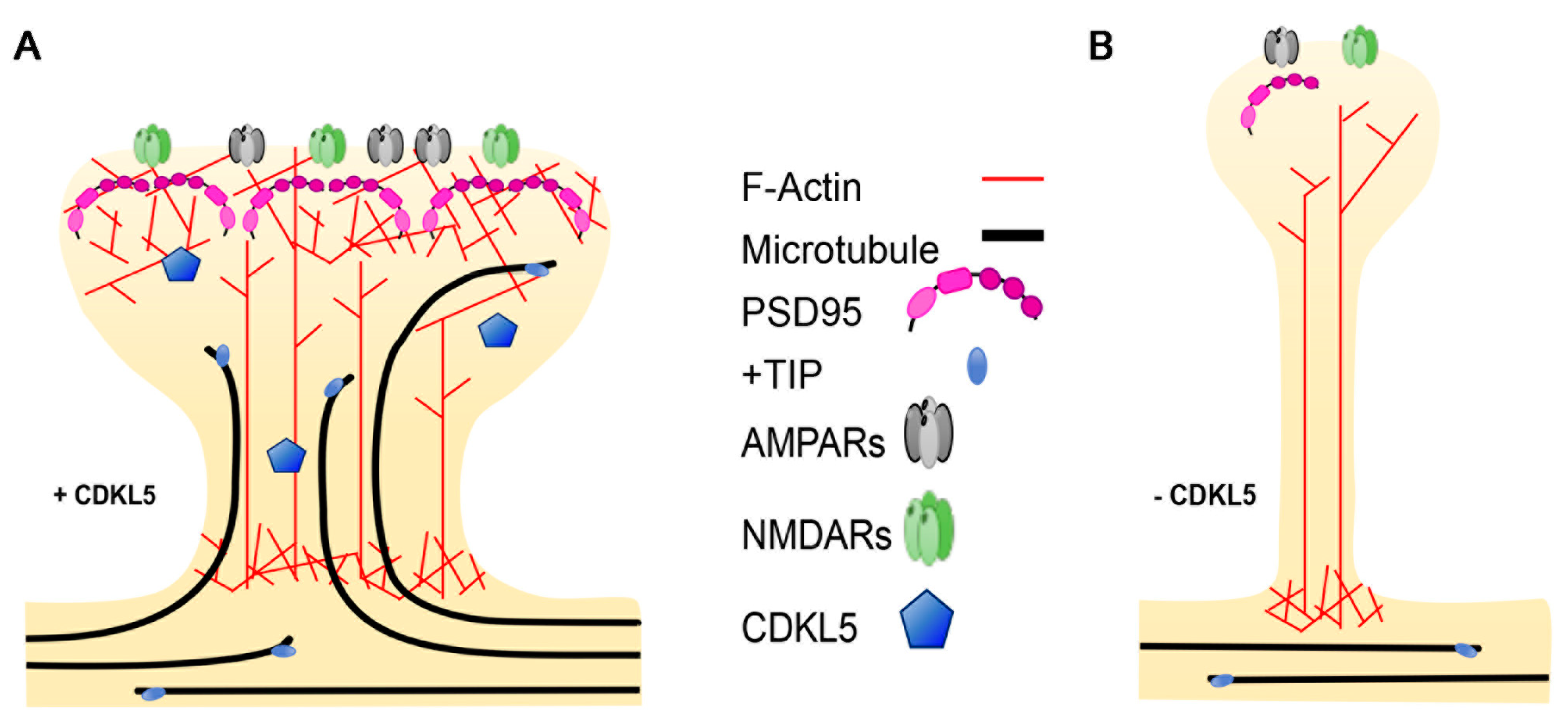
3.3. Dendritic Spines
4. Non-Neuronal CDKL5-Related Defects
5. Possible Functional Outcome of Specific Pathogenic CDKL5 Mutations
6. Therapeutic Relevance of CDKL5-Dependent MT Defects
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| +TIP | plus-end tracking protein |
| ADNP | activity-dependent neuroprotective protein |
| AMPA | α-amino-3-hydroxy-5-methylisozasole-4-propionic acid |
| APC | adenomatousis polyposis coli |
| Bdnf | brain-derived neurotrophic factor |
| CAP-Gly | cytoskeleton-associtated protein Gly-rich |
| CDD | CDKL5 deficiency disorder |
| CDKL5 | cyclin-dependent kinase-like 5 |
| CH | calponin homology |
| CLASP | CLIP-associated protein |
| CLIP | cytoplasmic linker protein |
| DLG5 | disks large homolog 5 |
| E | glutamate |
| EB | end-binding protein |
| EEY/F | glutamate-glutamate-tyrosine/phenylalanine |
| EpoD | Epothilone D |
| F-actin | filamentous actin |
| γ-TuRC | γ-tubulin ring complex |
| GFP | green fluorescent protein |
| GTP | guanine triphospate |
| GDP | guanine diphosphate |
| IQGAP1 | IQ motif containing GTPase activating protein 1 |
| iPSC | induced pluripotent stem cell |
| K | lysine |
| KIF | kinesin |
| LC | light chain |
| LTP | long-term potentiation |
| MAGUK | membrane-associated guanylate kinase |
| MAP | microtubule associated protein |
| MARK3 | MT affinity regulating kinase 3 |
| MT | microtubule |
| MTOC | microtubule organising centre |
| NMDA | N-methyl-D-aspartate |
| PCM | pericentriolar material |
| PREG | pregnenolone |
| PSD95 | post-synaptic density protein 95 |
| PTM | post-translational modification |
| RTT | Rett syndrome |
| SxIP | serine-any protein-isoleucine-proline |
| Y | tyrosine |
References
- Montini, E.; Andolfi, G.; Caruso, A.; Buchner, G.; Walpole, S.M.; Mariani, M.; Consalez, G.; Trump, D.; Ballabio, A.; Franco, B. Identification and characterization of a novel serine-threonine kinase gene from the Xp22 region. Genomics 1998, 51, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Kalscheuer, V.M.; Tao, J.; Donnelly, A.; Hollway, G.; Schwinger, E.; Kübart, S.; Menzel, C.; Hoeltzenbein, M.; Tommerup, N.; Eyre, H.; et al. Disruption of the serine/threonine kinase 9 gene causes severe X-linked infantile spasms and mental retardation. Am. J. Hum. Genet. 2003, 72, 1401–1411. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Van Esch, H.; Hagedorn-Greiwe, M.; Hoffmann, K.; Moser, B.; Raynaud, M.; Sperner, J.; Fryns, J.-P.; Schwinger, E.; Gécz, J.; et al. Mutations in the X-linked cyclin-dependent kinase-like 5 (CDKL5/STK9) gene are associated with severe neurodevelopmental retardation. Am. J. Hum. Genet. 2004, 75, 1149–1154. [Google Scholar] [CrossRef]
- Weaving, L.S.; Christodoulou, J.; Williamson, S.L.; Friend, K.L.; McKenzie, O.L.D.; Archer, H.; Evans, J.; Clarke, A.; Pelka, G.J.; Tam, P.P.L.; et al. Mutations of CDKL5 cause a severe neurodevelopmental disorder with infantile spasms and mental retardation. Am. J. Hum. Genet. 2004, 75, 1079–1093. [Google Scholar] [CrossRef]
- Scala, E.; Ariani, F.; Mari, F.; Caselli, R.; Pescucci, C.; Longo, I.; Meloni, I.; Giachino, D.; Bruttini, M.; Hayek, G.; et al. CDKL5/STK9 is mutated in Rett syndrome variant with infantile spasms. J. Med. Genet. 2005, 42, 103–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fehr, S.; Wilson, M.; Downs, J.; Williams, S.; Murgia, A.; Sartori, S.; Vecchi, M.; Ho, G.; Polli, R.; Psoni, S.; et al. The CDKL5 disorder is an independent clinical entity associated with early-onset encephalopathy. Eur. J. Hum. Genet. 2013, 21, 266–273. [Google Scholar] [CrossRef]
- Olson, H.E.; Demarest, S.T.; Pestana-Knight, E.M.; Swanson, L.C.; Iqbal, S.; Lal, D.; Leonard, H.; Cross, J.H.; Devinsky, O.; Benke, T.A. Cyclin-Dependent Kinase-Like 5 Deficiency Disorder: Clinical Review. Pediatr. Neurol. 2019, 97, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Neul, J.L.; Kaufmann, W.E.; Glaze, D.G.; Christodoulou, J.; Clarke, A.J.; Bahi-Buisson, N.; Leonard, H.; Bailey, M.E.S.; Schanen, N.C.; Zappella, M.; et al. Rett syndrome: Revised diagnostic criteria and nomenclature. Ann. Neurol. 2010, 68, 944–950. [Google Scholar] [CrossRef] [Green Version]
- Barbiero, I.; Peroni, D.; Tramarin, M.; Chandola, C.; Rusconi, L.; Landsberger, N.; Kilstrup-Nielsen, C. The neurosteroid pregnenolone reverts microtubule derangement induced by the loss of a functional CDKL5-IQGAP1 complex. Hum. Mol. Genet. 2017, 26, 3520–3530. [Google Scholar] [CrossRef] [PubMed]
- Barbiero, I.; Valente, D.; Chandola, C.; Magi, F.; Bergo, A.; Monteonofrio, L.; Tramarin, M.; Fazzari, M.; Soddu, S.; Landsberger, N.; et al. CDKL5 localizes at the centrosome and midbody and is required for faithful cell division. Sci. Rep. 2017, 7, 6228. [Google Scholar] [CrossRef]
- Baltussen, L.L.; Negraes, P.D.; Silvestre, M.; Claxton, S.; Moeskops, M.; Christodoulou, E.; Flynn, H.R.; Snijders, A.P.; Muotri, A.R.; Ultanir, S.K. Chemical genetic identification of CDKL5 substrates reveals its role in neuronal microtubule dynamics. EMBO J. 2018, 37, e99763. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, I.M.; Morgan, M.E.; Peltier, J.; Weiland, F.; Gregorczyk, M.; Brown, F.C.; Macartney, T.; Toth, R.; Trost, M.; Rouse, J. Phosphoproteomic screening identifies physiological substrates of the CDKL5 kinase. EMBO J. 2018, 37, e99559. [Google Scholar] [CrossRef] [PubMed]
- Kapitein, L.C.; Hoogenraad, C.C. Building the Neuronal Microtubule Cytoskeleton. Neuron 2015, 87, 492–506. [Google Scholar] [PubMed] [Green Version]
- Varidaki, A.; Hong, Y.; Coffey, E.T. Repositioning Microtubule Stabilizing Drugs for Brain Disorders. Front. Cell. Neurosci. 2018, 12, 226. [Google Scholar] [CrossRef] [PubMed]
- Bonini, S.A.; Mastinu, A.; Ferrari-Toninelli, G.; Memo, M. Potential Role of Microtubule Stabilizing Agents in Neurodevelopmental Disorders. Int. J. Mol. Sci. 2017, 18, 1627. [Google Scholar] [Green Version]
- Stiess, M.; Maghelli, N.; Kapitein, L.C.; Gomis-Rüth, S.; Wilsch-Bräuninger, M.; Hoogenraad, C.C.; Tolić-Nørrelykke, I.M.; Bradke, F. Axon extension occurs independently of centrosomal microtubule nucleation. Science 2010, 327, 704–707. [Google Scholar] [CrossRef]
- van de Willige, D.; Hoogenraad, C.C.; Akhmanova, A. Microtubule plus-end tracking proteins in neuronal development. Cell. Mol. Life Sci. 2016, 73, 2053–2077. [Google Scholar]
- Yu, I.; Garnham, C.P.; Roll-Mecak, A. Writing and Reading the Tubulin Code. J. Biol. Chem. 2015, 290, 17163–17172. [Google Scholar] [CrossRef] [Green Version]
- Erck, C.; Peris, L.; Andrieux, A.; Meissirel, C.; Gruber, A.D.; Vernet, M.; Schweitzer, A.; Saoudi, Y.; Pointu, H.; Bosc, C.; et al. A vital role of tubulin-tyrosine-ligase for neuronal organization. Proc. Natl. Acad. Sci. USA 2005, 102, 7853–7858. [Google Scholar] [Green Version]
- Halpain, S.; Dehmelt, L. The MAP1 family of microtubule-associated proteins. Genome Biol. 2006, 7, 224. [Google Scholar] [CrossRef]
- Dehmelt, L.; Halpain, S. Actin and microtubules in neurite initiation: Are MAPs the missing link? J. Neurobiol. 2004, 58, 18–33. [Google Scholar] [CrossRef]
- Akhmanova, A.; Steinmetz, M.O. Tracking the ends: A dynamic protein network controls the fate of microtubule tips. Nat. Rev. Mol. Cell Biol. 2008, 9, 309–322. [Google Scholar] [CrossRef] [PubMed]
- Perez, F.; Diamantopoulos, G.S.; Stalder, R.; Kreis, T.E. CLIP-170 highlights growing microtubule ends in vivo. Cell 1999, 96, 517–527. [Google Scholar] [CrossRef]
- Mustyatsa, V.V.; Boyakhchyan, A.V.; Ataullakhanov, F.I.; Gudimchuk, N.B. EB-Family Proteins: Functions and Microtubule Interaction Mechanisms. Biochem. Mosc. 2017, 82, 791–802. [Google Scholar] [CrossRef] [PubMed]
- Peris, L.; Thery, M.; Fauré, J.; Saoudi, Y.; Lafanechère, L.; Chilton, J.K.; Gordon-Weeks, P.; Galjart, N.; Bornens, M.; Wordeman, L.; et al. Tubulin tyrosination is a major factor affecting the recruitment of CAP-Gly proteins at microtubule plus ends. J. Cell Biol. 2006, 174, 839–849. [Google Scholar] [CrossRef] [PubMed]
- Bieling, P.; Kandels-Lewis, S.; Telley, I.A.; van Dijk, J.; Janke, C.; Surrey, T. CLIP-170 tracks growing microtubule ends by dynamically recognizing composite EB1/tubulin-binding sites. J. Cell Biol. 2008, 183, 1223–1233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamura, N.; Draviam, V.M. Microtubule plus-ends within a mitotic cell are “moving platforms” with anchoring, signalling and force-coupling roles. Open Biol. 2012, 2, 120132. [Google Scholar] [CrossRef]
- Witte, H.; Neukirchen, D.; Bradke, F. Microtubule stabilization specifies initial neuronal polarization. J. Cell Biol. 2008, 180, 619–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharp, D.J.; Yu, W.; Baas, P.W. Transport of dendritic microtubules establishes their nonuniform polarity orientation. J. Cell Biol. 1995, 130, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Jaworski, J.; Kapitein, L.C.; Gouveia, S.M.; Dortland, B.R.; Wulf, P.S.; Grigoriev, I.; Camera, P.; Spangler, S.A.; Di Stefano, P.; Demmers, J.; et al. Dynamic microtubules regulate dendritic spine morphology and synaptic plasticity. Neuron 2009, 61, 85–100. [Google Scholar] [CrossRef] [PubMed]
- Lasser, M.; Tiber, J.; Lowery, L.A. The Role of the Microtubule Cytoskeleton in Neurodevelopmental Disorders. Front. Cell. Neurosci. 2018, 12, 165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukata, M.; Watanabe, T.; Noritake, J.; Nakagawa, M.; Yamaga, M.; Kuroda, S.; Matsuura, Y.; Iwamatsu, A.; Perez, F.; Kaibuchi, K. Rac1 and Cdc42 capture microtubules through IQGAP1 and CLIP-170. Cell 2002, 109, 873–885. [Google Scholar] [CrossRef]
- Hedman, D.; Reza Barzegar, H.; Rosén, A.; Wågberg, T.; Andreas Larsson, J. On the Stability and Abundance of Single Walled Carbon Nanotubes. Sci. Rep. 2015, 5, 16850. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.-C.; Xiong, Z.-Q. Molecular and Synaptic Bases of CDKL5 Disorder. Dev Neurobiol 2019, 79, 8–19. [Google Scholar] [CrossRef] [PubMed]
- Nawaz, M.S.; Giarda, E.; Bedogni, F.; La Montanara, P.; Ricciardi, S.; Ciceri, D.; Alberio, T.; Landsberger, N.; Rusconi, L.; Kilstrup-Nielsen, C. CDKL5 and Shootin1 Interact and Concur in Regulating Neuronal Polarization. PLoS ONE 2016, 11, e0148634. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, C.; Medici, G.; Trazzi, S.; Gennaccaro, L.; Galvani, G.; Berteotti, C.; Ren, E.; Loi, M.; Ciani, E. CDKL5 deficiency predisposes neurons to cell death through the deregulation of SMAD3 signaling. Brain Pathol. 2019. [Google Scholar] [CrossRef]
- Conde, C.; Cáceres, A. Microtubule assembly, organization and dynamics in axons and dendrites. Nat. Rev. Neurosci. 2009, 10, 319–332. [Google Scholar] [CrossRef] [PubMed]
- Nirschl, J.J.; Magiera, M.M.; Lazarus, J.E.; Janke, C.; Holzbaur, E.L.F. α-Tubulin Tyrosination and CLIP-170 Phosphorylation Regulate the Initiation of Dynein-Driven Transport in Neurons. Cell Rep. 2016, 14, 2637–2652. [Google Scholar] [CrossRef] [PubMed]
- Neukirchen, D.; Bradke, F. Cytoplasmic linker proteins regulate neuronal polarization through microtubule and growth cone dynamics. J. Neurosci. 2011, 31, 1528–1538. [Google Scholar] [CrossRef] [PubMed]
- Barbiero, I.; Peroni, D.; Siniscalchi, P.; Rusconi, L.; Tramarin, M.; De Rosa, R.; Motta, P.; Bianchi, M.; Kilstrup-Nielsen, C. Pregnenolone and pregnenolone-methyl-ether rescue neuronal defects caused by dysfunctional CLIP170 in a neuronal model of CDKL5 Deficiency Disorder. Neuropharmacology. under review.
- Lansbergen, G.; Komarova, Y.; Modesti, M.; Wyman, C.; Hoogenraad, C.C.; Goodson, H.V.; Lemaitre, R.P.; Drechsel, D.N.; van Munster, E.; Gadella, T.W.J.; et al. Conformational changes in CLIP-170 regulate its binding to microtubules and dynactin localization. J. Cell Biol. 2004, 166, 1003–1014. [Google Scholar] [CrossRef] [PubMed]
- Weng, J.-H.; Liang, M.-R.; Chen, C.-H.; Tong, S.-K.; Huang, T.-C.; Lee, S.-P.; Chen, Y.-R.; Chen, C.-T.; Chung, B.-C. Pregnenolone activates CLIP-170 to promote microtubule growth and cell migration. Nat. Chem. Biol. 2013, 9, 636–642. [Google Scholar] [CrossRef]
- Toriyama, M.; Shimada, T.; Kim, K.B.; Mitsuba, M.; Nomura, E.; Katsuta, K.; Sakumura, Y.; Roepstorff, P.; Inagaki, N. Shootin1: A protein involved in the organization of an asymmetric signal for neuronal polarization. J. Cell Biol. 2006, 175, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Kubo, Y.; Baba, K.; Toriyama, M.; Minegishi, T.; Sugiura, T.; Kozawa, S.; Ikeda, K.; Inagaki, N. Shootin1-cortactin interaction mediates signal-force transduction for axon outgrowth. J. Cell Biol. 2015, 210, 663–676. [Google Scholar] [CrossRef]
- Chen, Q.; Zhu, Y.-C.; Yu, J.; Miao, S.; Zheng, J.; Xu, L.; Zhou, Y.; Li, D.; Zhang, C.; Tao, J.; et al. CDKL5, a protein associated with rett syndrome, regulates neuronal morphogenesis via Rac1 signaling. J. Neurosci. 2010, 30, 12777–12786. [Google Scholar] [CrossRef] [PubMed]
- Amendola, E.; Zhan, Y.; Mattucci, C.; Castroflorio, E.; Calcagno, E.; Fuchs, C.; Lonetti, G.; Silingardi, D.; Vyssotski, A.L.; Farley, D.; et al. Mapping pathological phenotypes in a mouse model of CDKL5 disorder. PLoS ONE 2014, 9, e91613. [Google Scholar] [CrossRef]
- Tang, S.; Wang, I.-T.J.; Yue, C.; Takano, H.; Terzic, B.; Pance, K.; Lee, J.Y.; Cui, Y.; Coulter, D.A.; Zhou, Z. Loss of CDKL5 in Glutamatergic Neurons Disrupts Hippocampal Microcircuitry and Leads to Memory Impairment in Mice. J. Neurosci. 2017, 37, 7420–7437. [Google Scholar] [CrossRef] [Green Version]
- Okuda, K.; Takao, K.; Watanabe, A.; Miyakawa, T.; Mizuguchi, M.; Tanaka, T. Comprehensive behavioral analysis of the Cdkl5 knockout mice revealed significant enhancement in anxiety- and fear-related behaviors and impairment in both acquisition and long-term retention of spatial reference memory. PLoS ONE 2018, 13, e0196587. [Google Scholar] [CrossRef]
- Jacquemet, G.; Humphries, M.J. IQGAP1 is a key node within the small GTPase network. Small GTPases 2013, 4, 199–207. [Google Scholar] [CrossRef] [Green Version]
- Swiech, L.; Blazejczyk, M.; Urbanska, M.; Pietruszka, P.; Dortland, B.R.; Malik, A.R.; Wulf, P.S.; Hoogenraad, C.C.; Jaworski, J. CLIP-170 and IQGAP1 cooperatively regulate dendrite morphology. J. Neurosci. 2011, 31, 4555–4568. [Google Scholar] [CrossRef]
- Ramkumar, A.; Jong, B.Y.; Ori-McKenney, K.M. ReMAPping the microtubule landscape: How phosphorylation dictates the activities of microtubule-associated proteins. Dev. Dyn. 2018, 247, 138–155. [Google Scholar] [PubMed]
- Ricciardi, S.; Ungaro, F.; Hambrock, M.; Rademacher, N.; Stefanelli, G.; Brambilla, D.; Sessa, A.; Magagnotti, C.; Bachi, A.; Giarda, E.; et al. CDKL5 ensures excitatory synapse stability by reinforcing NGL-1-PSD95 interaction in the postsynaptic compartment and is impaired in patient iPSC-derived neurons. Nat. Cell Biol. 2012, 14, 911–923. [Google Scholar] [CrossRef] [PubMed]
- Della Sala, G.; Putignano, E.; Chelini, G.; Melani, R.; Calcagno, E.; Michele Ratto, G.; Amendola, E.; Gross, C.T.; Giustetto, M.; Pizzorusso, T. Dendritic Spine Instability in a Mouse Model of CDKL5 Disorder Is Rescued by Insulin-like Growth Factor 1. Biol. Psychiatry 2016, 80, 302–311. [Google Scholar] [CrossRef] [PubMed]
- Lamprecht, R.; LeDoux, J. Structural plasticity and memory. Nat. Rev. Neurosci. 2004, 5, 45–54. [Google Scholar] [CrossRef] [PubMed]
- La Montanara, P.; Rusconi, L.; Locarno, A.; Forti, L.; Barbiero, I.; Tramarin, M.; Chandola, C.; Kilstrup-Nielsen, C.; Landsberger, N. Synaptic synthesis, dephosphorylation, and degradation: A novel paradigm for an activity-dependent neuronal control of CDKL5. J. Biol. Chem. 2015, 290, 4512–4527. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.-C.; Li, D.; Wang, L.; Lu, B.; Zheng, J.; Zhao, S.-L.; Zeng, R.; Xiong, Z.-Q. Palmitoylation-dependent CDKL5-PSD-95 interaction regulates synaptic targeting of CDKL5 and dendritic spine development. Proc. Natl. Acad. Sci. USA 2013, 110, 9118–9123. [Google Scholar] [CrossRef] [PubMed]
- Pizzo, R.; Gurgone, A.; Castroflorio, E.; Amendola, E.; Gross, C.; Sassoè-Pognetto, M.; Giustetto, M. Lack of Cdkl5 Disrupts the Organization of Excitatory and Inhibitory Synapses and Parvalbumin Interneurons in the Primary Visual Cortex. Front. Cell. Neurosci. 2016, 10, 261. [Google Scholar] [CrossRef]
- Tramarin, M.; Rusconi, L.; Pizzamiglio, L.; Barbiero, I.; Peroni, D.; Scaramuzza, L.; Guilliams, T.; Cavalla, D.; Antonucci, F.; Kilstrup-Nielsen, C. The antidepressant tianeptine reverts synaptic AMPA receptor defects caused by deficiency of CDKL5. Hum. Mol. Genet. 2018, 27, 2052–2063. [Google Scholar] [CrossRef] [PubMed]
- Ren, E.; Roncacé, V.; Trazzi, S.; Fuchs, C.; Medici, G.; Gennaccaro, L.; Loi, M.; Galvani, G.; Ye, K.; Rimondini, R.; et al. Functional and Structural Impairments in the Perirhinal Cortex of a Mouse Model of CDKL5 Deficiency Disorder Are Rescued by a TrkB Agonist. Front. Cell. Neurosci. 2019, 13, 169. [Google Scholar] [CrossRef] [PubMed]
- Yennawar, M.; White, R.S.; Jensen, F.E. AMPA Receptor Dysregulation and Therapeutic Interventions in a Mouse Model of CDKL5 Deficiency Disorder. J. Neurosci. 2019, 39, 4814–4828. [Google Scholar] [CrossRef] [Green Version]
- Ethell, I.M.; Pasquale, E.B. Molecular mechanisms of dendritic spine development and remodeling. Prog. Neurobiol. 2005, 75, 161–205. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Viesselmann, C.; Nam, S.; Merriam, E.; Dent, E.W. Activity-dependent dynamic microtubule invasion of dendritic spines. J. Neurosci. 2008, 28, 13094–13105. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Firestein, B.L.; Zheng, J.Q. Microtubules in dendritic spine development. J. Neurosci. 2008, 28, 12120–12124. [Google Scholar] [CrossRef] [PubMed]
- Schätzle, P.; Esteves da Silva, M.; Tas, R.P.; Katrukha, E.A.; Hu, H.Y.; Wierenga, C.J.; Kapitein, L.C.; Hoogenraad, C.C. Activity-Dependent Actin Remodeling at the Base of Dendritic Spines Promotes Microtubule Entry. Curr. Biol. 2018, 28, 2081–2093. [Google Scholar] [CrossRef] [PubMed]
- Pchitskaya, E.; Kraskovskaya, N.; Chernyuk, D.; Popugaeva, E.; Zhang, H.; Vlasova, O.; Bezprozvanny, I. Stim2-Eb3 Association and Morphology of Dendritic Spines in Hippocampal Neurons. Sci. Rep. 2017, 7, 17625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrick, C.; Fischer, A.; Srivastava, D.P.; Tronson, N.C.; Penzes, P.; Radulovic, J. N-cadherin regulates cytoskeletally associated IQGAP1/ERK signaling and memory formation. Neuron 2007, 55, 786–798. [Google Scholar] [CrossRef] [PubMed]
- Jausoro, I.; Mestres, I.; Quassollo, G.; Masseroni, L.; Heredia, F.; Caceres, A. Regulation of spine density and morphology by IQGAP1 protein domains. PLoS ONE 2013, 8, e56574. [Google Scholar] [CrossRef]
- Gao, C.; Frausto, S.F.; Guedea, A.L.; Tronson, N.C.; Jovasevic, V.; Leaderbrand, K.; Corcoran, K.A.; Guzmán, Y.F.; Swanson, G.T.; Radulovic, J. IQGAP1 regulates NR2A signaling, spine density, and cognitive processes. J. Neurosci. 2011, 31, 8533–8542. [Google Scholar] [CrossRef]
- Nuriya, M.; Oh, S.; Huganir, R.L. Phosphorylation-dependent interactions of alpha-Actinin-1/IQGAP1 with the AMPA receptor subunit GluR4. J. Neurochem. 2005, 95, 544–552. [Google Scholar] [CrossRef]
- Wang, S.-H.J.; Celic, I.; Choi, S.-Y.; Riccomagno, M.; Wang, Q.; Sun, L.O.; Mitchell, S.P.; Vasioukhin, V.; Huganir, R.L.; Kolodkin, A.L. Dlg5 regulates dendritic spine formation and synaptogenesis by controlling subcellular N-cadherin localization. J. Neurosci. 2014, 34, 12745–12761. [Google Scholar] [CrossRef]
- Sandí, M.-J.; Marshall, C.B.; Balan, M.; Coyaud, É.; Zhou, M.; Monson, D.M.; Ishiyama, N.; Chandrakumar, A.A.; La Rose, J.; Couzens, A.L.; et al. MARK3-mediated phosphorylation of ARHGEF2 couples microtubules to the actin cytoskeleton to establish cell polarity. Sci. Signal. 2017, 10, eaan3286. [Google Scholar] [CrossRef] [PubMed]
- McVicker, D.P.; Awe, A.M.; Richters, K.E.; Wilson, R.L.; Cowdrey, D.A.; Hu, X.; Chapman, E.R.; Dent, E.W. Transport of a kinesin-cargo pair along microtubules into dendritic spines undergoing synaptic plasticity. Nat. Commun. 2016, 7, 12741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schatten, H. The mammalian centrosome and its functional significance. Histochem. Cell Biol. 2008, 129, 667–686. [Google Scholar] [PubMed] [Green Version]
- Bettencourt-Dias, M.; Glover, D.M. Centrosome biogenesis and function: Centrosomics brings new understanding. Nat. Rev. Mol. Cell Biol. 2007, 8, 451–463. [Google Scholar] [CrossRef] [PubMed]
- Nigg, E.A.; Raff, J.W. Centrioles, centrosomes, and cilia in health and disease. Cell 2009, 139, 663–678. [Google Scholar] [CrossRef]
- Glotzer, M. Cytokinesis: GAP gap. Curr. Biol. 2009, 19, R162–R165. [Google Scholar] [CrossRef]
- Lim, H.H.; Zhang, T.; Surana, U. Regulation of centrosome separation in yeast and vertebrates: Common threads. Trends Cell Biol. 2009, 19, 325–333. [Google Scholar] [CrossRef]
- Staples, C.J.; Myers, K.N.; Beveridge, R.D.D.; Patil, A.A.; Lee, A.J.X.; Swanton, C.; Howell, M.; Boulton, S.J.; Collis, S.J. The centriolar satellite protein Cep131 is important for genome stability. J. Cell. Sci. 2012, 125, 4770–4779. [Google Scholar] [CrossRef]
- Canning, P.; Park, K.; Gonçalves, J.; Li, C.; Howard, C.J.; Sharpe, T.D.; Holt, L.J.; Pelletier, L.; Bullock, A.N.; Leroux, M.R. CDKL Family Kinases Have Evolved Distinct Structural Features and Ciliary Function. Cell Rep. 2018, 22, 885–894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graser, S.; Stierhof, Y.-D.; Lavoie, S.B.; Gassner, O.S.; Lamla, S.; Le Clech, M.; Nigg, E.A. Cep164, a novel centriole appendage protein required for primary cilium formation. J. Cell Biol. 2007, 179, 321–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malicki, J.J.; Johnson, C.A. The Cilium: Cellular Antenna and Central Processing Unit. Trends Cell Biol. 2017, 27, 126–140. [Google Scholar] [CrossRef] [PubMed]
- Valente, E.M.; Rosti, R.O.; Gibbs, E.; Gleeson, J.G. Primary cilia in neurodevelopmental disorders. Nat. Rev. Neurol. 2014, 10, 27–36. [Google Scholar] [CrossRef]
- Tam, L.-W.; Ranum, P.T.; Lefebvre, P.A. CDKL5 regulates flagellar length and localizes to the base of the flagella in Chlamydomonas. Mol. Biol. Cell. 2013, 24, 588–600. [Google Scholar] [CrossRef] [PubMed]
- Hector, R.D.; Kalscheuer, V.M.; Hennig, F.; Leonard, H.; Downs, J.; Clarke, A.; Benke, T.A.; Armstrong, J.; Pineda, M.; Bailey, M.E.S.; et al. CDKL5 variants: Improving our understanding of a rare neurologic disorder. Neurol. Genet. 2017, 3, e200. [Google Scholar] [CrossRef] [PubMed]
- Bertani, I.; Rusconi, L.; Bolognese, F.; Forlani, G.; Conca, B.; De Monte, L.; Badaracco, G.; Landsberger, N.; Kilstrup-Nielsen, C. Functional consequences of mutations in CDKL5, an X-linked gene involved in infantile spasms and mental retardation. J. Biol. Chem. 2006, 281, 32048–32056. [Google Scholar] [CrossRef]
- Rusconi, L.; Salvatoni, L.; Giudici, L.; Bertani, I.; Kilstrup-Nielsen, C.; Broccoli, V.; Landsberger, N. CDKL5 expression is modulated during neuronal development and its subcellular distribution is tightly regulated by the C-terminal tail. J. Biol. Chem. 2008, 283, 30101–30111. [Google Scholar] [CrossRef] [PubMed]
- Rosas-Vargas, H.; Bahi-Buisson, N.; Philippe, C.; Nectoux, J.; Girard, B.; N’Guyen Morel, M.A.; Gitiaux, C.; Lazaro, L.; Odent, S.; Jonveaux, P.; et al. Impairment of CDKL5 nuclear localisation as a cause for severe infantile encephalopathy. J. Med. Genet. 2008, 45, 172–178. [Google Scholar] [CrossRef]
- Nectoux, J.; Heron, D.; Tallot, M.; Chelly, J.; Bienvenu, T. Maternal origin of a novel C-terminal truncation mutation in CDKL5 causing a severe atypical form of Rett syndrome. Clin. Genet. 2006, 70, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Russo, S.; Marchi, M.; Cogliati, F.; Bonati, M.T.; Pintaudi, M.; Veneselli, E.; Saletti, V.; Balestrini, M.; Ben-Zeev, B.; Larizza, L. Novel mutations in the CDKL5 gene, predicted effects and associated phenotypes. Neurogenetics 2009, 10, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Wang, I.-T.J.; Allen, M.; Goffin, D.; Zhu, X.; Fairless, A.H.; Brodkin, E.S.; Siegel, S.J.; Marsh, E.D.; Blendy, J.A.; Zhou, Z. Loss of CDKL5 disrupts kinome profile and event-related potentials leading to autistic-like phenotypes in mice. Proc. Natl. Acad. Sci. USA 2012, 109, 21516–21521. [Google Scholar] [CrossRef] [Green Version]
- Tang, S.; Terzic, B.; Wang, I.-T.J.; Sarmiento, N.; Sizov, K.; Cui, Y.; Takano, H.; Marsh, E.D.; Zhou, Z.; Coulter, D.A. Altered NMDAR signaling underlies autistic-like features in mouse models of CDKL5 deficiency disorder. Nat. Commun. 2019, 10, 2655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szafranski, P.; Golla, S.; Jin, W.; Fang, P.; Hixson, P.; Matalon, R.; Kinney, D.; Bock, H.-G.; Craigen, W.; Smith, J.L.; et al. Neurodevelopmental and neurobehavioral characteristics in males and females with CDKL5 duplications. Eur. J. Hum. Genet. 2015, 23, 915–921. [Google Scholar] [CrossRef] [PubMed]
- Marchisella, F.; Coffey, E.T.; Hollos, P. Microtubule and microtubule associated protein anomalies in psychiatric disease. Cytoskeleton (Hoboken) 2016, 73, 596–611. [Google Scholar] [CrossRef] [PubMed]
- Brunden, K.R.; Lee, V.M.-Y.; Smith, A.B.; Trojanowski, J.Q.; Ballatore, C. Altered microtubule dynamics in neurodegenerative disease: Therapeutic potential of microtubule-stabilizing drugs. Neurobiol. Dis. 2017, 105, 328–335. [Google Scholar] [CrossRef] [PubMed]
- Amos, L.A. Microtubule structure and its stabilisation. Org. Biomol. Chem. 2004, 2, 2153–2160. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Maiti, A.; Shively, S.; Lakhani, F.; McDonald-Jones, G.; Bruce, J.; Lee, E.B.; Xie, S.X.; Joyce, S.; Li, C.; et al. Microtubule-binding drugs offset tau sequestration by stabilizing microtubules and reversing fast axonal transport deficits in a tauopathy model. Proc. Natl. Acad. Sci. USA 2005, 102, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Brunden, K.R.; Ballatore, C.; Lee, V.M.-Y.; Smith, A.B.; Trojanowski, J.Q. Brain-penetrant microtubule-stabilizing compounds as potential therapeutic agents for tauopathies. Biochem. Soc. Trans. 2012, 40, 661–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Carroll, J.; Trojanowski, J.Q.; Yao, Y.; Iba, M.; Potuzak, J.S.; Hogan, A.-M.L.; Xie, S.X.; Ballatore, C.; Smith, A.B.; et al. The microtubule-stabilizing agent, epothilone D, reduces axonal dysfunction, neurotoxicity, cognitive deficits, and Alzheimer-like pathology in an interventional study with aged tau transgenic mice. J. Neurosci. 2012, 32, 3601–3611. [Google Scholar] [CrossRef] [PubMed]
- Brunden, K.R.; Zhang, B.; Carroll, J.; Yao, Y.; Potuzak, J.S.; Hogan, A.-M.L.; Iba, M.; James, M.J.; Xie, S.X.; Ballatore, C.; et al. Epothilone D improves microtubule density, axonal integrity, and cognition in a transgenic mouse model of tauopathy. J. Neurosci. 2010, 30, 13861–13866. [Google Scholar] [CrossRef] [PubMed]
- Gozes, I. NAP (davunetide) provides functional and structural neuroprotection. Curr. Pharm. Des. 2011, 17, 1040–1044. [Google Scholar] [CrossRef] [PubMed]
- Helsmoortel, C.; Vulto-van Silfhout, A.T.; Coe, B.P.; Vandeweyer, G.; Rooms, L.; van den Ende, J.; Schuurs-Hoeijmakers, J.H.M.; Marcelis, C.L.; Willemsen, M.H.; Vissers, L.E.L.M.; et al. A SWI/SNF-related autism syndrome caused by de novo mutations in ADNP. Nat. Genet. 2014, 46, 380–384. [Google Scholar] [CrossRef] [Green Version]
- Oz, S.; Kapitansky, O.; Ivashco-Pachima, Y.; Malishkevich, A.; Giladi, E.; Skalka, N.; Rosin-Arbesfeld, R.; Mittelman, L.; Segev, O.; Hirsch, J.A.; et al. The NAP motif of activity-dependent neuroprotective protein (ADNP) regulates dendritic spines through microtubule end binding proteins. Mol. Psychiatry 2014, 19, 1115–1124. [Google Scholar] [CrossRef] [Green Version]
- Gozes, I. ADNP Regulates Cognition: A Multitasking Protein. Front. Neurosci. 2018, 12, 873. [Google Scholar] [CrossRef] [Green Version]
- Oz, S.; Ivashko-Pachima, Y.; Gozes, I. The ADNP derived peptide, NAP modulates the tubulin pool: Implication for neurotrophic and neuroprotective activities. PLoS ONE 2012, 7, e51458. [Google Scholar]
- Gozes, I. The cytoskeleton as a drug target for neuroprotection: The case of the autism- mutated ADNP. Biol. Chem. 2016, 397, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.alzforum.org/ (accessed on 21 July 2019).
- Javitt, D.C.; Buchanan, R.W.; Keefe, R.S.E.; Kern, R.; McMahon, R.P.; Green, M.F.; Lieberman, J.; Goff, D.C.; Csernansky, J.G.; McEvoy, J.P.; et al. Effect of the neuroprotective peptide davunetide (AL-108) on cognition and functional capacity in schizophrenia. Schizophr. Res. 2012, 136, 25–31. [Google Scholar] [CrossRef]
- Weng, J.-H.; Chung, B.-C. Nongenomic actions of neurosteroid pregnenolone and its metabolites. Steroids 2016, 111, 54–59. [Google Scholar] [CrossRef]
- Flood, J.F.; Morley, J.E.; Roberts, E. Memory-enhancing effects in male mice of pregnenolone and steroids metabolically derived from it. Proc. Natl. Acad. Sci. USA 1992, 89, 1567–1571. [Google Scholar] [CrossRef] [PubMed]
- Brown, E.S.; Park, J.; Marx, C.E.; Hynan, L.S.; Gardner, C.; Davila, D.; Nakamura, A.; Sunderajan, P.; Lo, A.; Holmes, T. A randomized, double-blind, placebo-controlled trial of pregnenolone for bipolar depression. Neuropsychopharmacology 2014, 39, 2867–2873. [Google Scholar] [CrossRef]
- Osuji, I.J.; Vera-Bolaños, E.; Carmody, T.J.; Brown, E.S. Pregnenolone for cognition and mood in dual diagnosis patients. Psychiatry Res. 2010, 178, 309–312. [Google Scholar] [CrossRef]
- Bianchi, M.; Baulieu, E.-E. 3β-Methoxy-pregnenolone (MAP4343) as an innovative therapeutic approach for depressive disorders. Proc. Natl. Acad. Sci. USA 2012, 109, 1713–1718. [Google Scholar] [CrossRef] [PubMed]
- Parésys, L.; Hoffmann, K.; Froger, N.; Bianchi, M.; Villey, I.; Baulieu, E.-E.; Fuchs, E. Effects of the Synthetic Neurosteroid: 3β-Methoxypregnenolone (MAP4343) on Behavioral and Physiological Alterations Provoked by Chronic Psychosocial Stress in Tree Shrews. Int. J. Neuropsychopharmacol. 2016, 19, pyv119. [Google Scholar] [CrossRef] [PubMed]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barbiero, I.; De Rosa, R.; Kilstrup-Nielsen, C. Microtubules: A Key to Understand and Correct Neuronal Defects in CDKL5 Deficiency Disorder? Int. J. Mol. Sci. 2019, 20, 4075. https://doi.org/10.3390/ijms20174075
Barbiero I, De Rosa R, Kilstrup-Nielsen C. Microtubules: A Key to Understand and Correct Neuronal Defects in CDKL5 Deficiency Disorder? International Journal of Molecular Sciences. 2019; 20(17):4075. https://doi.org/10.3390/ijms20174075
Chicago/Turabian StyleBarbiero, Isabella, Roberta De Rosa, and Charlotte Kilstrup-Nielsen. 2019. "Microtubules: A Key to Understand and Correct Neuronal Defects in CDKL5 Deficiency Disorder?" International Journal of Molecular Sciences 20, no. 17: 4075. https://doi.org/10.3390/ijms20174075
APA StyleBarbiero, I., De Rosa, R., & Kilstrup-Nielsen, C. (2019). Microtubules: A Key to Understand and Correct Neuronal Defects in CDKL5 Deficiency Disorder? International Journal of Molecular Sciences, 20(17), 4075. https://doi.org/10.3390/ijms20174075