Plakophilin-2 Haploinsufficiency Causes Calcium Handling Deficits and Modulates the Cardiac Response Towards Stress

 , , ,
, , ,
Abstract
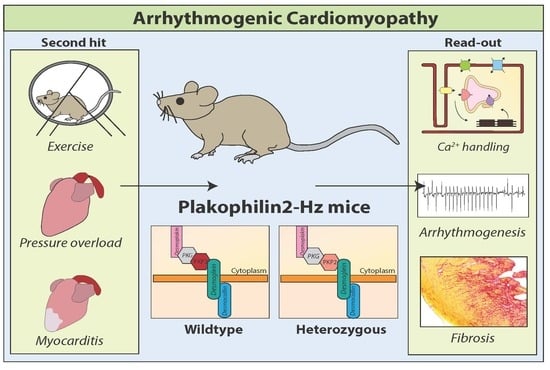
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. PKP2 Haploinsufficiency Impairs Expression of Calcium Handling-Related Proteins
2.2. Characterization of PKP2-Hz Hearts Over Time
2.3. Pro-Arrhythmic Cardiac Remodeling in PKP2-Hz Mice Exposed to Exercise
2.4. Cardiac Pressure Overload is Pro-Fibrotic in the PKP2 Happloinsufficient Heart
2.5. PKP2 Haploinsufficiency Alters the Cardiac Fibrotic and Inflammatory Response to Auto-Immune Myocarditis
3. Discussion
4. Study Limitations
5. Materials and Methods
5.1. Mouse Model
5.2. Transverse Aortic Constriction Procedure
5.3. Exercise Protocol
5.4. Experimental Autoimmune Myocarditis
5.5. Echocardiography
5.6. Electrocardiograms Recordings
5.7. Epicardial Activation Mapping in Langendorf-Perfused Hearts
5.8. Western Blot Analysis
5.9. PCR Validation of Gene Expression
5.10. Immunofluorescence Labeling
5.11. Immunohistochemistry
5.12. Picrosirius Red Staining
5.13. Hematoxylin and Eosin Staining
5.14. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACM | Arrhythmogenic Cardiomyopathy |
| AnkB | AnkyrinB |
| BCL | Basic cycle length |
| BW | Body weight |
| Ca2+ | Calcium |
| CaCL2 | Calcium chloride |
| Casq2 | Calsequestrin2 |
| Cav1.2 | L-type Ca2+ channel |
| cDNA | Complement DNA |
| CO2 | Carbon dioxide |
| Col1α1 | Collagen 1α1 |
| Col1α2 | Collagen 1α2 |
| CVL | Longitudinal conduction velocity |
| CVT | Transversal conduction velocity |
| Cx43 | Connexin43 |
| EAM | Experimental auto-immune myocarditis |
| ECG | Electrocardiogram |
| GAPDH | Glyceraldehyde 3-phosphate dehydrogenase |
| H&E | Hematoxylin and Eosin |
| HW | Heart weight |
| Hz | Heterozygous |
| IL | Interleukin |
| INa | Sodium current |
| KCL | Potassium chloride |
| LV | Left ventricle |
| LVFS | Left ventricular fractional shortening |
| MgSO4 | Magnesium sulfate |
| MMP9 | Matrix metallopeptidase 9 |
| NaCl | Sodium chloride |
| NaH2PO4 | Monosodium phosphate |
| NaHCO3 | Sodium bicarbonate |
| Nav1.5 | Sodium channel |
| Ncad | N-cadherin |
| NFκ-β | Nuclear Factor κ-β |
| O2 | Oxygen |
| PBS | Phosphate buffered saline |
| PKG | Plakoglobin |
| PKP2 | Plakophilin 2 |
| PKP2cKO | Plakophilin2 conditional knock out |
| RT-PCR | Reverse transcriptase polymerase chain reaction |
| RV | Right ventricle |
| RVEF | Right ventricular ejection fraction |
| TAC | Trans aortic constriction |
| TBP | TATA binding protein |
| Timp1 | Tissue inhibitor of metalloproteinase 1 |
| TNF | Tumor necrosis factor |
| WT | Wildtype |
References
- Müssigbrodt, A.; Knopp, H.; Czimbalmos, C.; Jahnke, C.; Richter, S.; Husser, D.; Gradistanac, T.; Hindricks, G. Exercise-related sudden cardiac death of an American football player with arrhythmogenic right ventricular dysplasia/cardiomyopathy AND sarcoidosis. Clin. Case Rep. 2019, 7, 686–688. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Huang, J.; He, S.; Feng, R.; Zhong, Z.; Liu, Y.; Ye, W.; Li, X.; Liao, H.; Fei, H.; et al. Case report of familial sudden cardiac death caused by a DSG2 p.F531C mutation as genetic background when carrying with heterozygous KCNE5 p.D92E/E93X mutation. Bmc Med. Genet. 2018, 19, 148. [Google Scholar] [CrossRef] [PubMed]
- Cadrin-Tourigny, J.; Bosman, L.P.; Nozza, A.; Wang, W.; Tadros, R.; Bhonsale, A.; Bourfiss, M.; Fortier, A.; Lie, H.; Saguner, A.M.; et al. A new prediction model for ventricular arrhythmias in arrhythmogenic right ventricular cardiomyopathy. Eur. Heart J. 2019, 40, 1850–1858. [Google Scholar] [CrossRef] [PubMed]
- Ohno, S. The genetic background of arrhythmogenic right ventricular cardiomyopathy. J. Arrhythmia 2016, 32, 398–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Austin, K.M.; Trembley, M.A.; Chandler, S.F.; Sanders, S.P.; Saffitz, J.E.; Abrams, D.J.; Pu, W.T. Molecular mechanisms of arrhythmogenic cardiomyopathy. Nat. Rev. Cardiol. 2019, 16, 519–537. [Google Scholar]
- Groeneweg, J.A.; Bhonsale, A.; James, C.A.; te Riele, A.S.; Dooijes, D.; Tichnell, C.; Murray, B.; Wiesfeld, A.C.; Sawant, A.C.; et al. Clinical Presentation, Long-Term Follow-Up, and Outcomes of 1001 Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy Patients and Family Members. Circ. Cardiovasc. Genet. 2015, 8, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Grossmann, K.S.; Grund, C.; Huelsken, J.; Behrend, M.; Erdmann, B.; Franke, W.W.; Birchmeier, W. Requirement of plakophilin 2 for heart morphogenesis and cardiac junction formation. J. Cell Biol. 2004, 167, 149–160. [Google Scholar] [CrossRef]
- Valente, M.; Calabrese, F.; Thiene, G.; Angelini, A.; Basso, C.; Nava, A.; Rossi, L. In vivo evidence of apoptosis in arrhythmogenic right ventricular cardiomyopathy. Am. J. Pathol. 1998, 152, 479–484. [Google Scholar] [PubMed]
- Basso, C.; Czarnowska, E.; Della Barbera, M.; Bauce, B.; Beffagna, G.; Wlodarska, E.K.; Pilichou, K.; Ramondo, A.; Lorenzon, A.; Wozniek, O.; et al. Ultrastructural evidence of intercalated disc remodelling in arrhythmogenic right ventricular cardiomyopathy: An electron microscopy investigation on endomyocardial biopsies. Eur. Heart, J. 2006, 27, 1847–1854. [Google Scholar] [CrossRef]
- d’Amati, G.; di Gioia, C.R.; Giordano, C.; Gallo, P. Myocyte transdifferentiation: A possible pathogenetic mechanism for arrhythmogenic right ventricular cardiomyopathy. Arch. Pathol. Lab. Med. 2000, 124, 287–290. [Google Scholar]
- Cerrone, M.; Montnach, J.; Lin, X.; Zhao, Y.-T.; Zhang, M.; Agullo-Pascual, E.; Leo-Marcias, A.; Alvardo, F.J.; Dolgalev, I.; Karathanos, T.V.; et al. Plakophilin-2 is required for transcription of genes that control calcium cycling and cardiac rhythm. Nat. Commun. 2017, 8, 106. [Google Scholar] [CrossRef]
- Tester, D.J.; Ackerman, J.P.; Giudicessi, J.R.; Ackerman, N.C.; Cerrone, M.; Delmar, M.; Ackerman, M.J. Plakophilin-2 Truncation Variants in Patients Clinically Diagnosed With Catecholaminergic Polymorphic Ventricular Tachycardia and Decedents With Exercise-Associated Autopsy Negative Sudden Unexplained Death in the Young. Jacc Clin. Electrophysiol 2019, 5, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Cerrone, M.; Lin, X.; Zhang, M.; Agullo-Pascual, E.; Pfenniger, A.; Chkourko Gusky, H.; Novelli, V.; Kim, C.; Tirasawadichai, T.; Judge, D.P.; et al. Missense mutations in plakophilin-2 cause sodium current deficit and associate with a Brugada syndrome phenotype. Circ. 2014, 129, 1092–1103. [Google Scholar] [CrossRef]
- Novelli, V.; Malkani, K.; Cerrone, M. Pleiotropic Phenotypes Associated With PKP2 Variants. Front. Cardiovasc. Med. 2018, 5, 184. [Google Scholar] [CrossRef]
- Cerrone, M.; Noorman, M.; Lin, X.; Chkourko, H.; Liang, F.-X.; van der Nagel, R.; Hund, T.; Birchmeier, W.; Mohler, P.; van Veen, T.A.; et al. Sodium current deficit and arrhythmogenesis in a murine model of plakophilin-2 haploinsufficiency. Cardiovasc. Res. 2012, 95, 460–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leo-Macías, A.; Liang, F.-X.; Delmar, M. Ultrastructure of the intercellular space in adult murine ventricle revealed by quantitative tomographic electron microscopy. Cardiovasc. Res. 2015, 107, 442–452. [Google Scholar] [CrossRef] [Green Version]
- Goff, Z.D.; Calkins, H. Sudden death related cardiomyopathies–Arrhythmogenic right ventricular cardiomyopathy, arrhythmogenic cardiomyopathy, and exercise-induced cardiomyopathy. Prog. Cardiovasc. Dis. 2019, 62, 217–226. [Google Scholar] [CrossRef]
- Cruz, F.M.; Sanz-Rosa, D.; Roche-Molina, M.; García-Prieto, J.; García-Ruiz, J.M.; Pizarro, G.; Jímenez-Borreguero, L.J.; Torres, M.; Bernad, A.; Ruiz-Cabello, J.; et al. Exercise Triggers ARVC Phenotype in Mice Expressing a Disease-Causing Mutated Version of Human Plakophilin-2. J. Am. Coll. Cardiol. 2015, 65, 1438–1450. [Google Scholar] [CrossRef]
- James, C.A.; Bhonsale, A.; Tichnell, C.; Murray, B.; Russell, S.D.; Tandri, H.; Tedford, R.J.; Judge, D.P.; Calkins, H. Exercise increases age-related penetrance and arrhythmic risk in arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated desmosomal mutation carriers. J. Am. Coll. Cardiol. 2013, 62, 1290–1297. [Google Scholar] [CrossRef] [PubMed]
- Benito, B.; Gay-Jordi, G.; Serrano-Mollar, A.; Guasch, E.; Shi, Y.; Tardif, J.C.; Brugada, J.; Nattel, S.; Mont, L. Cardiac Arrhythmogenic Remodeling in a Rat Model of Long-Term Intensive Exercise Training. Circ. 2011, 123, 13–22. [Google Scholar] [CrossRef] [Green Version]
- Merino, D.; Gil, A.; Gómez, J.; Ruiz, L.; Llano, M.; García, R.; Hurlé, M.A.; Nistal, J.F. Experimental modelling of cardiac pressure overload hypertrophy: Modified technique for precise, reproducible, safe and easy aortic arch banding-debanding in mice. Sci. Rep. 2018, 8, 3167. [Google Scholar] [CrossRef] [PubMed]
- Haugaa, K.H.; Haland, T.F.; Leren, I.S.; Saberniak, J.; Edvardsen, T. Arrhythmogenic right ventricular cardiomyopathy, clinical manifestations, and diagnosis. Europace. 2016, 18, 965–972. [Google Scholar] [CrossRef] [PubMed]
- Asimaki, A.; Tandri, H.; Duffy, E.R.; Winterfield, J.R.; Mackey-Bojack, S.; Picken, M.M.; Cooper, L.T.; Wilber, D.J.; Marcus, F.I.; Basso, C.; et al. Altered desmosomal proteins in granulomatous myocarditis and potential pathogenic links to arrhythmogenic right ventricular cardiomyopathy. Circ. Arrhythm Electrophysiol 2011, 4, 743–752. [Google Scholar] [CrossRef] [PubMed]
- Basso, C.; Thiene, G.; Corrado, D.; Angelini, A.; Nava, A.; Valente, M. Arrhythmogenic right ventricular cardiomyopathy. Dysplasia, dystrophy, or myocarditis? Circ. 1996, 94, 983–991. [Google Scholar] [CrossRef] [PubMed]
- Campuzano, O.; Alcalde, M.; Iglesias, A.; Barahona-Dussault, C.; Sarquella-Brugada, G.; Benito, B.; Arzamendi, D.; Flores, J.; Leung, T.K.; Talajic, M.; et al. Arrhythmogenic right ventricular cardiomyopathy: Severe structural alterations are associated with inflammation. J. Clin. Pathol. 2012, 65, 1077–1083. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, F.; Basso, C.; Carturan, E.; Valente, M.; Thiene, G. Arrhythmogenic right ventricular cardiomyopathy/dysplasia: Is there a role for viruses? Cardiovasc. Pathol. 2006, 15, 11–17. [Google Scholar] [CrossRef]
- Lopez-Ayala, J.M.; Pastor-Quirante, F.; Gonzalez-Carrillo, J.; Lopez-Cuenca, D.; Sanchez-Munoz, J.J.; Oliva-Sandoval, M.J.; Gimeno, J.R. Genetics of myocarditis in arrhythmogenic right ventricular dysplasia. Heart Rhythm. 2015, 12, 766–773. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J.D.; Murphy, N.P.; Hamilton, R.M.; Lubbers, E.R.; James, C.A.; Kline, C.F.; Gollob, M.H.; Krahn, A.D.; Sturm, A.C.; Musa, H.; et al. Ankyrin-B dysfunction predisposes to arrhythmogenic cardiomyopathy and is amenable to therapy. J. Clin. Invest. 2019, 129, 3171–3184. [Google Scholar] [CrossRef]
- van Veen, T.A.B.; Stein, M.; Royer, A.; Le Quang, K.; Charpentier, F.; Colledge, W.H.; Huang, C.L.; Wilders, R.; Grace, A.A.; Escande, D.; et al. Impaired Impulse Propagation in Scn5a -Knockout Mice. Circ. 2005, 112, 1927–1935. [Google Scholar] [CrossRef]
- Stein, M.; Noorman, M.; van Veen, T.A.B.; Herold, E.; Engelen, M.A.; Boulaksil, M.; Antoons, G.; Jansen, J.A.; van Oosterhout, M.F.; Hauer, R.N.; et al. Dominant arrhythmia vulnerability of the right ventricle in senescent mice. Heart Rhythm. 2008, 5, 438–448. [Google Scholar] [CrossRef]
- Corrado, D.; Basso, C.; Schiavon, M.; Thiene, G. Does sports activity enhance the risk of sudden cardiac death? J. Cardiovasc. Med. 2006, 7, 228–233. [Google Scholar] [CrossRef]
- Calore, M.; Lorenzon, A.; Vitiello, L.; Poloni, G.; Khan, M.A.F.; Beffagna, G.; Dazzo, E.; Sacchetto, C.; Polishchuk, R.; Sabetelli, P.; et al. A novel murine model for arrhythmogenic cardiomyopathy points to a pathogenic role of Wnt signalling and miRNA dysregulation. Cardiovasc. Res. 2019, 115, 739–751. [Google Scholar] [CrossRef] [PubMed]
- Basso, C.; Pilichou, K.; Bauce, B.; Corrado, D.; Thiene, G. Diagnostic Criteria, Genetics, and Molecular Basis of Arrhythmogenic Cardiomyopathy. Heart Fail. Clin. 2018, 14, 201–213. [Google Scholar] [CrossRef]
- Kim, J.-C.; Pérez-Hernández Duran, M.; Alvarado, F.J.; Maurya, S.R.; Montnach, J.; Yin, Y.; Zhang, M.; Lin, X.; Vasquez, C.; Heguy, A.; et al. Disruption of Ca2+i Homeostasis and Cx43 Hemichannel Function in the Right Ventricle Precedes Overt Arrhythmogenic Cardiomyopathy in PKP2-Deficient Mice. Circ. 2019. [Google Scholar] [CrossRef] [PubMed]
- Sen-Chowdhry, S.; Syrris, P.; McKenna, W.J. Genetics of right ventricular cardiomyopathy. J. Cardiovasc. Electrophysiol 2005, 16, 927–935. [Google Scholar] [CrossRef]
- Stein, M.; Boulaksil, M.; Jansen, J.A.; Herold, E.; Noorman, M.; Joles, J.A.; van Veen, T.A.; Houtman, M.J.; Engelen, M.A.; Hauer, R.N.; et al. Reduction of fibrosis-related arrhythmias by chronic renin-angiotensin-aldosterone system inhibitors in an aged mouse model. Am. J. Physiol. Circ. Physiol. 2010, 299, H310–H321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delmar, M.; McKenna, W.J. The Cardiac Desmosome and Arrhythmogenic Cardiomyopathies. Circ. Res. 2010, 107, 700–714. [Google Scholar] [CrossRef] [Green Version]
- Shaw, R.M.; Fay, A.J.; Puthenveedu, M.A.; von Zastrow, M.; Jan, Y.-N.; Jan, L.Y. Microtubule plus-end-tracking proteins target gap junctions directly from the cell interior to adherens junctions. Cell 2007, 128, 547–560. [Google Scholar] [CrossRef]
- Chatterjee, S.; Bavishi, C.; Sardar, P.; Agarwal, V.; Krishnamoorthy, P.; Grodzicki, T.; Messerli, F.H. Meta-analysis of left ventricular hypertrophy and sustained arrhythmias. Am. J. Cardiol. 2014, 114, 1049–1052. [Google Scholar] [CrossRef]
- Polidovitch, N.; Yang, S.; Sun, H.; Lakin, R.; Ahmad, F.; Gao, X.; Turnbull, P.C.; Chiarello, C.; Perry, C.G.R.; Manganiello, V.; et al. Phosphodiesterase type 3A (PDE3A), but not type 3B (PDE3B), contributes to the adverse cardiac remodeling induced by pressure overload. J. Mol. Cell. Cardiol. 2019, 132, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Velez Rueda, J.O.; Palomeque, J.; Mattiazzi, A. Early apoptosis in different models of cardiac hypertrophy induced by high renin-angiotensin system activity involves CaMKII. J. Appl. Physiol. 2012, 112, 2110–2120. [Google Scholar] [CrossRef] [PubMed]
- Lorenzon, A.; Calore, M.; Poloni, G.; De Windt, L.J.; Braghetta, P.; Rampazzo, A. Wnt/B-catenin pathway in arrhythmogenic cardiomyopathy. Oncotarget 2017, 8, 60640. [Google Scholar] [CrossRef] [PubMed]
- Niehrs, C. The complex world of WNT receptor signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 767–779. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Jiang, G.; Zhang, P.; Fan, J. Programmed cell death and its role in inflammation. Mil. Med. Res. 2015, 2, 12. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.L.; Najor, N.A.; Green, K.J. Desmosomes: Regulators of cellular signaling and adhesion in epidermal health and disease. Cold Spring Harb. Perspect. Med. 2014, 4, a015297. [Google Scholar] [CrossRef]
- Pieroni, M.; Dello Russo, A.; Marzo, F.; Pelargonio, G.; Casella, M.; Bellocci, F.; Crea, F. High Prevalence of Myocarditis Mimicking Arrhythmogenic Right Ventricular Cardiomyopathy: Differential Diagnosis by Electroanatomic Mapping-Guided Endomyocardial Biopsy. J. Am. Coll. Cardiol. 2009, 53, 681–689. [Google Scholar] [CrossRef] [PubMed]
- Sepehrkhouy, S.; Gho, J.M.I.H.; van Es, R.; Harakalova, M.; de Jonge, N.; Dooijes, D.; van der Smagt, J.J.; Buijsrogge, M.P.; Hauer, R.N.W.; Goldschmeding, R.; et al. Distinct fibrosis pattern in desmosomal and phospholamban mutation carriers in hereditary cardiomyopathies. Heart Rhythm 2017, 14, 1024–1032. [Google Scholar] [CrossRef]
- Dubash, A.D.; Kam, C.Y.; Aguado, B.A.; Patel, D.M.; Delmar, M.; Shea, L.D.; Green, K.J. Plakophilin-2 loss promotes TGF-β1/p38 MAPK-dependent fibrotic gene expression in cardiomyocytes. J. Cell Biol. 2016, 212, 425–438. [Google Scholar] [CrossRef]
- Grandy, S.A.; Brouilette, J.; Fiset, C. Reduction of Ventricular Sodium Current in a Mouse Model of HIV. J. Cardiovasc. Electrophysiol. 2010, 21, 916–922. [Google Scholar] [CrossRef]
- Boulaksil, M.; Winckels, S.K.G.; Engelen, M.A.; Stein, M.; van Veen, T.A.B.; Jansen, J.A.; Linnenbank, A.C.; Bierhuizen, M.F.; Groenewegen, W.A.; van Oosterhout, M.F.; et al. Heterogeneous Connexin43 distribution in heart failure is associated with dispersed conduction and enhanced susceptibility to ventricular arrhythmias. Eur. J. Heart Fail. 2010, 12, 913–921. [Google Scholar] [CrossRef] [Green Version]
- Smith, S.C. Autoimmune Myocarditis. In Curr. Protoc. Immunol.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2001; p. Unit 15.14. [Google Scholar]
- Ram, R.; Mickelsen, D.M.; Theodoropoulos, C.; Blaxall, B.C. New approaches in small animal echocardiography: Imaging the sounds of silence. Am. J. Physiol. Circ. Physiol. 2011, 301, 1765–1780. [Google Scholar] [CrossRef] [PubMed]
- Danik, S.; Cabo, C.; Chiello, C.; Kang, S.; Wit, A.L.; Coromilas, J. Correlation of repolarization of ventricular monophasic action potential with ECG in the murine heart. Am. J. Physiol. Circ. Physiol. 2002, 283, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, G.F.; Jeron, A.; Koren, G. Measurement of heart rate and Q-T interval in the conscious mouse. Am. J. Physiol. 1998, 274, 747–751. [Google Scholar] [CrossRef] [PubMed]
- Potse, M.; Linnenbank, A.C.; Grimbergen, C.A. Software design for analysis of multichannel intracardial and body surface electrocardiograms. Comput. Methods Programs Biomed 2002, 69, 225–236. [Google Scholar] [CrossRef] [Green Version]
- van Veen, T.A.B.; van Rijen, H.V.M.; Wiegerinck, R.F.; Opthof, T.; Colbert, M.C.; Clement, S.; de Bakker, J.M.; Jongsma, H.J. Remodeling of gap junctions in mouse hearts hypertrophied by forced retinoic acid signaling. J. Mol. Cell. Cardiol. 2002, 34, 1411–1423. [Google Scholar] [CrossRef]
- Fontes, M.S.C.; Raaijmakers, A.J.A.; van Doorn, T.; Kok, B.; Nieuwenhuis, S.; van der Nagel, R.; Vos, M.A.; de Boer, T.P.; van Rijen, H.V.; Bierhuizen, M.F. Changes in Cx43 and NaV1.5 expression precede the occurrence of substantial fibrosis in calcineurin-induced murine cardiac hypertrophy. PLoS ONE 2014, 9, e87226. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
van Opbergen, C.J.M.; Noorman, M.; Pfenniger, A.; Copier, J.S.; Vermij, S.H.; Li, Z.; van der Nagel, R.; Zhang, M.; de Bakker, J.M.T.; Glass, A.M.; et al. Plakophilin-2 Haploinsufficiency Causes Calcium Handling Deficits and Modulates the Cardiac Response Towards Stress. Int. J. Mol. Sci. 2019, 20, 4076. https://doi.org/10.3390/ijms20174076
van Opbergen CJM, Noorman M, Pfenniger A, Copier JS, Vermij SH, Li Z, van der Nagel R, Zhang M, de Bakker JMT, Glass AM, et al. Plakophilin-2 Haploinsufficiency Causes Calcium Handling Deficits and Modulates the Cardiac Response Towards Stress. International Journal of Molecular Sciences. 2019; 20(17):4076. https://doi.org/10.3390/ijms20174076
Chicago/Turabian Stylevan Opbergen, Chantal J.M., Maartje Noorman, Anna Pfenniger, Jaël S. Copier, Sarah H. Vermij, Zhen Li, Roel van der Nagel, Mingliang Zhang, Jacques M.T. de Bakker, Aaron M. Glass, and et al. 2019. "Plakophilin-2 Haploinsufficiency Causes Calcium Handling Deficits and Modulates the Cardiac Response Towards Stress" International Journal of Molecular Sciences 20, no. 17: 4076. https://doi.org/10.3390/ijms20174076
APA Stylevan Opbergen, C. J. M., Noorman, M., Pfenniger, A., Copier, J. S., Vermij, S. H., Li, Z., van der Nagel, R., Zhang, M., de Bakker, J. M. T., Glass, A. M., Mohler, P. J., Taffet, S. M., Vos, M. A., van Rijen, H. V. M., Delmar, M., & van Veen, T. A. B. (2019). Plakophilin-2 Haploinsufficiency Causes Calcium Handling Deficits and Modulates the Cardiac Response Towards Stress. International Journal of Molecular Sciences, 20(17), 4076. https://doi.org/10.3390/ijms20174076