The Role of Fibroblast Growth Factor 23 in Inflammation and Anemia


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
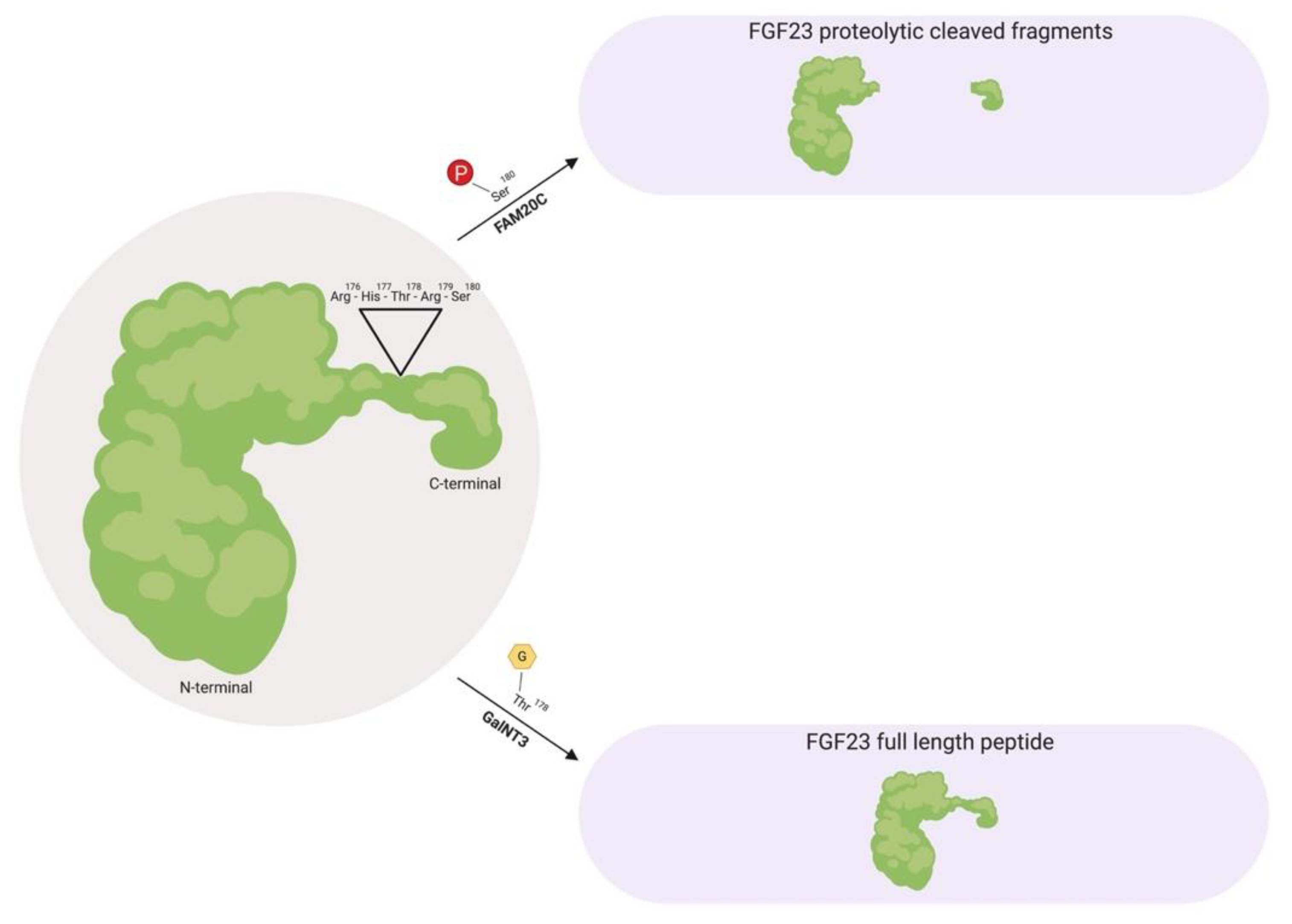
2. The Evolution and Biology of FGF23
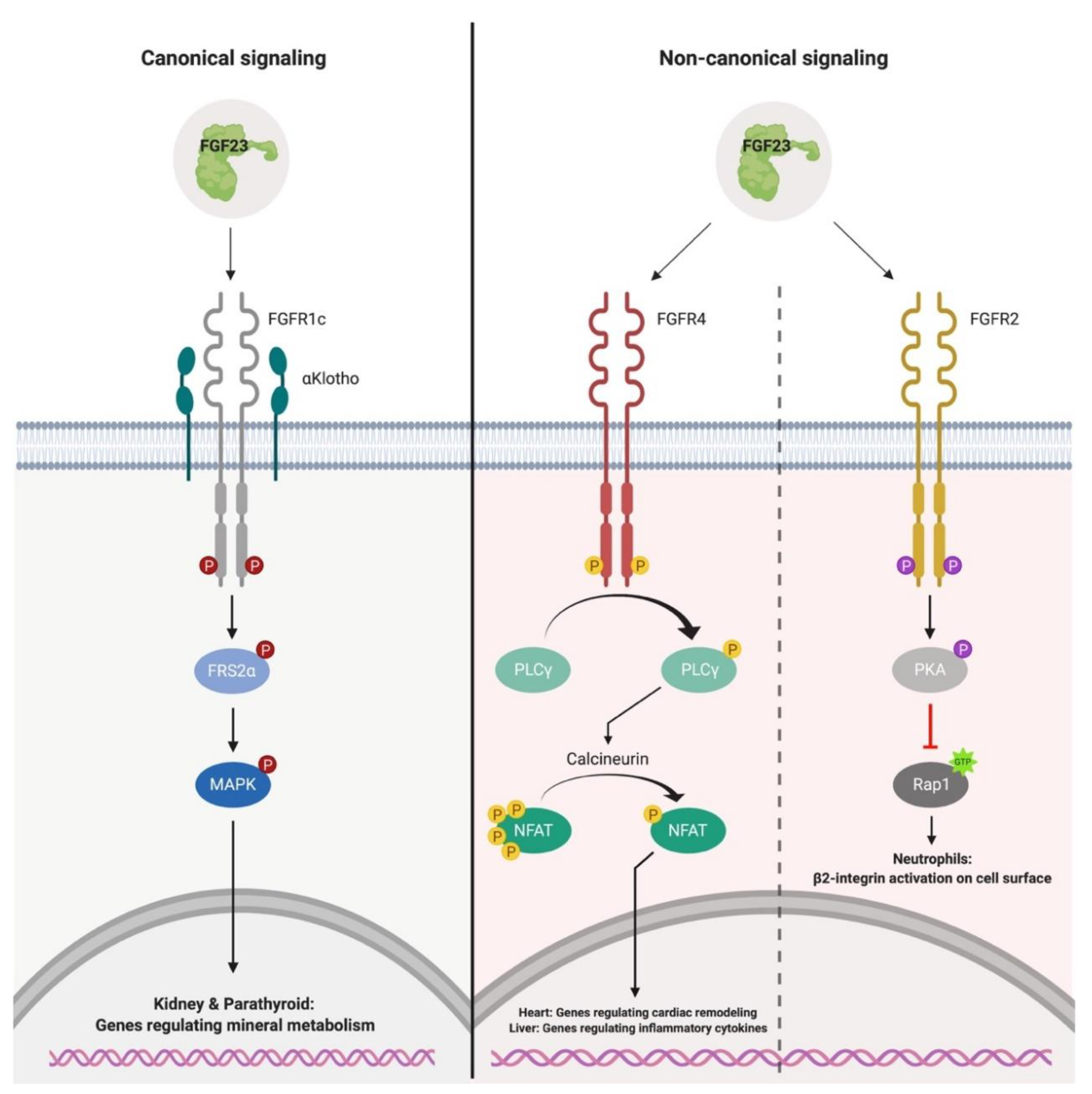
3. Canonical and Non-Canonical FGF23-Mediated Signaling
4. FGF23 in Chronic Kidney Disease
4.1. Inflammation and Iron Deficiency, Two Novel Determinants of FGF23 Production
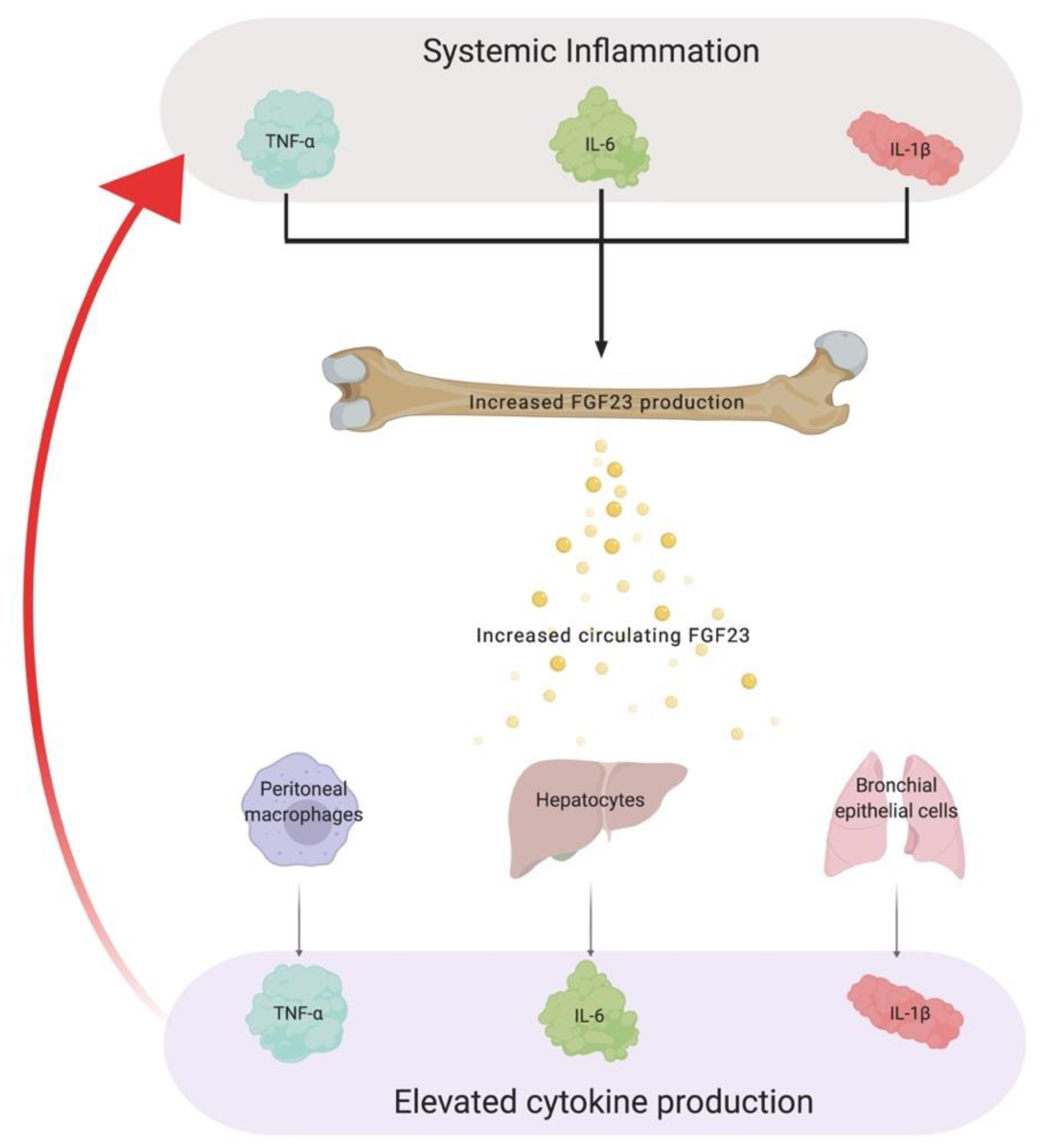
4.2. The Role of FGF23 in Chronic Inflammation
5. Chronic Inflammation, a Silent Culprit of Chronic Kidney Disease
6. Mechanisms Underlying Iron Dysregulation in Chronic Kidney Disease
6.1. Absolute Iron Deficiency
6.2. Functional Iron Deficiency
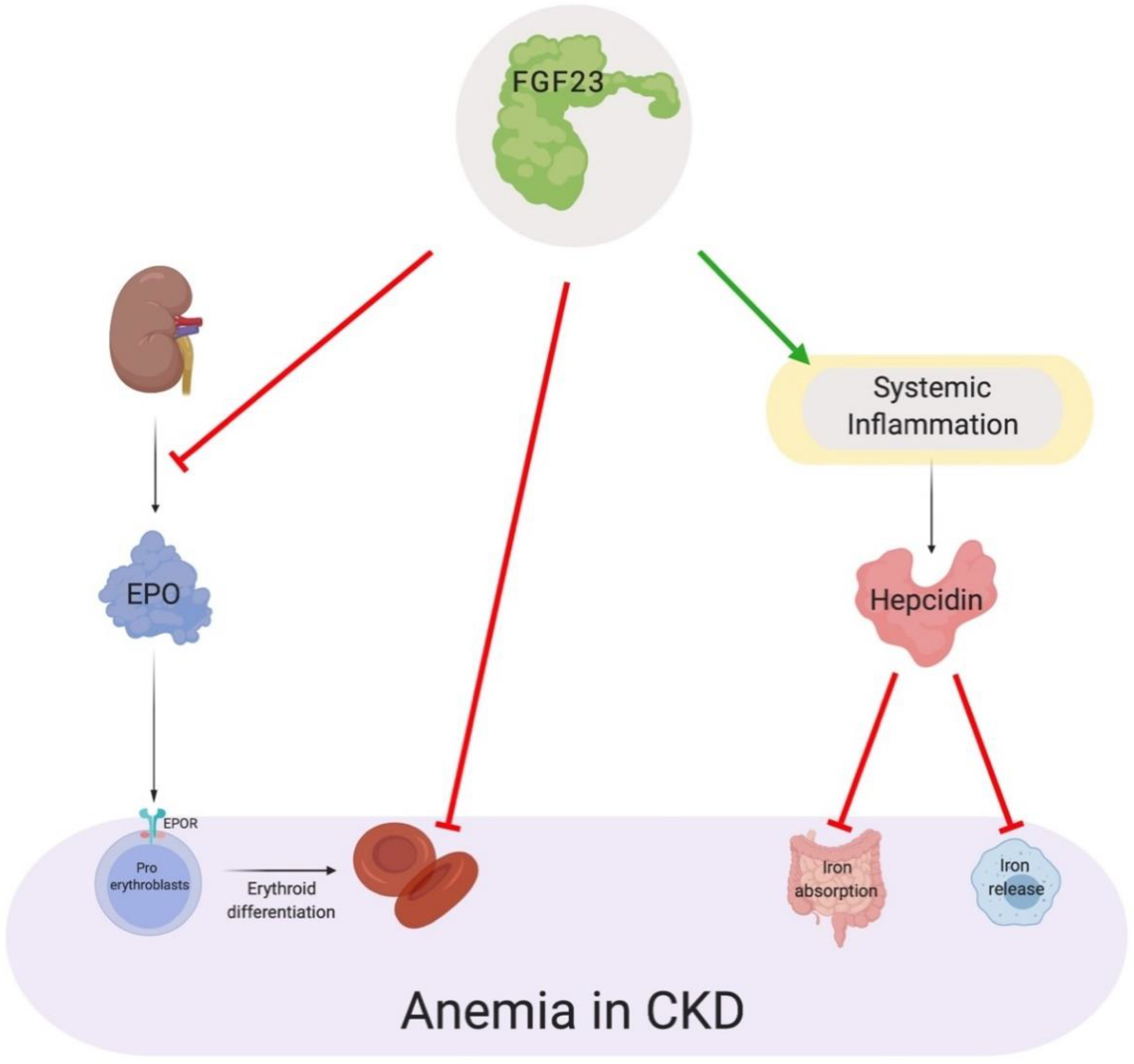
7. Direct and Indirect Actions of FGF23 in Anemia
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Go, A.S.; Chertow, G.M. Chronic Kidney Disease and the Risks of Death, Cardiovascular Events, and Hospitalization. N. Engl. J. Med. 2004, 351, 1296–1305. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez, O.M.; Mannstadt, M. Fibroblast Growth Factor 23 and Mortality among Patients Undergoing Hemodialysis. N. Engl. J. Med. 2008, 359, 584–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willerson, J.T. Inflammation as a Cardiovascular Risk Factor. Circulation 2004, 109, II2–II10. [Google Scholar] [CrossRef] [PubMed]
- Vlagopoulos, P.T.; Tighiouart, H.; Weiner, D.E.; Griffith, J.; Pettitt, D.; Salem, D.N.; Levey, A.S.; Sarnak, M.J. Anemia as a Risk Factor for Cardiovascular Disease and All-Cause Mortality in Diabetes: The Impact of Chronic Kidney Disease. J. Am. Soc. Nephrol. 2005, 16, 3403–3410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nam, K.; Lee, K.W.; Chung, O.; Yim, H.-S.; Cha, S.-S.; Lee, S.-W.; Jun, J.; Cho, Y.S.; Bhak, J.; de Magalhães, J.P.; et al. Analysis of the FGF gene family provides insights into aquatic adaptation in cetaceans. Sci. Rep. 2016, 7, 40233. [Google Scholar] [CrossRef] [PubMed]
- Thewissen, J.G.M.; Cooper, L.N.; Clementz, M.T.; Bajpai, S.; Tiwari, B.N. Whales originated from aquatic artiodactyls in the Eocene epoch of India. Nature 2007, 450, 1190–1194. [Google Scholar] [CrossRef]
- Thewissen, J.G.M.; Williams, E.M. The Early Radiations of Cetacea (Mammalia): Evolutionary Pattern and Developmental Correlations. Annu. Rev. Ecol. Syst. 2002, 33, 73–90. [Google Scholar] [CrossRef]
- Masato, N.; Alejandro, R.; Okada, N. Phylogenetic relationships among cetartiodactyls based on insertions of short and long interspersed elements: Hippopotamuses are the closest extant relatives of whales. Proc. Natl. Acad. Sci. USA 1999, 96, 10261–10266. [Google Scholar]
- Gatsey, J.; OLeary, M. Deciphering whale origins with molecules and fossils. Trends Ecol. Evol. 2001, 16, 562–570. [Google Scholar] [CrossRef]
- Itoh, N.; Ornitz, D.M. Evolution of the Fgf and Fgfr gene families. Trends Genet. 2004, 20, 563–569. [Google Scholar] [CrossRef]
- Sidow, A. Gen(om)e duplications in the evolution of early vertebrates. Curr. Opin. Genet. Dev. 1996, 6, 715–722. [Google Scholar] [CrossRef]
- Horton, A.; Mahadevan, N. Phylogenetic Analyses Alone Are Insufficient to Determine Whether Genome Duplication(s) Occurred During Early Vertebrate Evolution. J. Exp. Zool. B Mol. Dev. Evol. 2003, 299, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Itoh, N.; Ornitz, D.M. Fibroblast growth factors: From molecular evolution to roles in development, metabolism and disease. J. Biochem. 2011, 149, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Itoh, N. The Fgf Families in Humans, Mice, and Zebrafish: Their Evolutional Processes and Roles in Development, Metabolism, and Disease. Biol. Pharm. Bull. 2007, 30, 1819–1825. [Google Scholar] [CrossRef] [Green Version]
- Fon Tacer, K.; Bookout, A.L.; Ding, X.; Kurosu, H.; John, G.B.; Wang, L.; Goetz, R.; Mohammadi, M.; Kuro-o, M.; Mangelsdorf, D.J.; et al. Research Resource: Comprehensive Expression Atlas of the Fibroblast Growth Factor System in Adult Mouse. Mol. Endocrinol. 2010, 24, 2050–2064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itoh, N.; Ornitz, D.M. Functional evolutionary history of the mouse Fgf gene family. Dev. Dyn. 2007, 237, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Cousens, L.; Barr, P.; Sprang, S. Three-dimensional structure of human basic fibroblast growth factor a structural homolog of interleukin. Proc. Natl. Acad. Sci. 1991, 88, 3446–3450. [Google Scholar] [CrossRef]
- Eriksson, A.E.; Cousens, L.; Mastthews, B. Three-dimensional structure of human basic fibroblast growth factor. Proc. Natl. Acad. Sci. USA 1991, 88, 3441–3445. [Google Scholar] [CrossRef]
- Eswarakumar, V.P.; Lax, I.; Schlessinger, J. Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev. 2005, 16, 139–149. [Google Scholar] [CrossRef]
- Turner, N.; Grose, R. Fibroblast growth factor signaling: From development to cancer. Nat. Rev. Cancer 2010, 10, 116–129. [Google Scholar] [CrossRef]
- Goldfarb, M.; Schoorlemmer, J.; Williams, A.; Diwakar, S.; Wang, Q.; Huang, X.; Giza, J.; Tchetchik, D.; Kelley, K.; Vega, A.; et al. Fibroblast Growth Factor Homologous Factors Control Neuronal Excitability through Modulation of Voltage-Gated Sodium Channels. Neuron 2007, 55, 449–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoorlemmer, J.; Goldfarb, M. Fibroblast Growth Factor Homologous Factors and the Islet Brain-2 Scaffold Protein Regulate Activation of a Stress-activated Protein Kinase. J. Biol. Chem. 2002, 277, 49111–49119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, M.; Xu, L.; Laezza, F.; Yamada, K.; Feng, S.; Ornitz, D.M. Impaired hippocampal synaptic transmission and plasticity in mice lacking fibroblast growth factor 14. Mol. Cell. Neurosci. 2007, 34, 366–377. [Google Scholar] [CrossRef] [PubMed]
- Shakkottai, V.G.; Xiao, M.; Xu, L.; Wong, M.; Nerbonne, J.M.; Ornitz, D.M.; Yamada, K.A. FGF14 regulates the intrinsic excitability of cerebellar Purkinje neurons. Neurobiol. Dis. 2009, 33, 81–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dover, K.; Solinas, S.; D’Angelo, E.; Goldfarb, M. Long-term inactivation particle for voltage-gated sodium channels. J. Physiol. 2010, 588, 3695–3711. [Google Scholar] [CrossRef] [PubMed]
- Ornitz, D.M.; Itoh, N. Fibroblast growth factors. Genome Biol. 2001, 2, REVIEWS3005. [Google Scholar] [CrossRef] [PubMed]
- Goetz, R.; Beenken, A.; Ibrahimi, O.A.; Kalinina, J.; Olsen, S.K.; Eliseenkova, A.V.; Xu, C.; Neubert, T.A.; Zhang, F.; Linhardt, R.J.; et al. Molecular Insights into the Klotho-Dependent, Endocrine Mode of Action of Fibroblast Growth Factor 19 Subfamily Members. Mol. Cell. Biol. 2007, 27, 3417–3428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harmer, N.J.; Pellegrini, L.; Chirgadze, D.; Fernandez-Recio, J.; Blundell, T.L. The Crystal Structure of Fibroblast Growth Factor (FGF) 19 Reveals Novel Features of the FGF Family and Offers a Structural Basis for Its Unusual Receptor Affinity. Biochemistry 2004, 43, 629–640. [Google Scholar] [CrossRef]
- Mohammadi, M.; Olsen, S.K.; Ibrahimi, O.A. Structural basis for fibroblast growth factor receptor activation. Cytokine Growth Factor Rev. 2005, 16, 107–137. [Google Scholar] [CrossRef]
- Angelin, B.; Larsson, T.E.; Rudling, M. Circulating Fibroblast Growth Factors as Metabolic Regulators—A Critical Appraisal. Cell Metab. 2012, 16, 693–705. [Google Scholar] [CrossRef]
- Goetz, R.; Mohammadi, M. Exploring mechanisms of FGF signaling through the lens of structural biology. Nat. Rev. Mol. Cell Biol. 2013, 14, 166–180. [Google Scholar] [CrossRef] [PubMed]
- Quarles, L.D. Endocrine functions of bone in mineral metabolism regulation. J. Clin. Investig. 2008, 118, 3820–3828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erben, R.G.; Andrukhova, O. FGF23 regulation of renal tubular solute transport. Curr. Opin. Nephrol. Hypertens. 2015, 24, 450–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimada, T.; Hasegawa, H.; Yamazaki, Y.; Muto, T.; Hino, R.; Takeuchi, Y.; Fujita, T.; Nakahara, K.; Fukumoto, S.; Yamashita, T. FGF-23 is a Potent Regulator of Vitamin D Metabolism and Phosphate Homeostasis. J. Bone Miner. Res. 2003, 19, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Shimada, T.; Kakitani, M.; Yamazaki, Y.; Hasegawa, H.; Takeuchi, Y.; Fujita, T.; Fukumoto, S.; Tomizuka, K.; Yamashita, T. Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. J. Clin. Investig. 2004, 113, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.C.; Shiizaki, K.; Kuro-o, M.; Moe, O.W. Fibroblast Growth Factor 23 and Klotho: Physiology and Pathophysiology of an Endocrine Network of Mineral Metabolism. Annu. Rev. Physiol. 2013, 75, 503–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamashita, T.; Yoshioka, M.; Itoh, N. Identification of a Novel Fibroblast Growth Factor, FGF-23, Preferentially Expressed in the Ventrolateral Thalamic Nucleus of the Brain. Biochem. Biophys. Res. Commun. 2000, 277, 494–498. [Google Scholar] [CrossRef] [PubMed]
- White, K.E.; Carn, G.; Lorenz-Depiereux, B.; Benet-Pagès, A.; Strom, T.M.; Econs, M.J. Autosomal-dominant hypophosphatemic rickets (ADHR) mutations stabilize FGF-23. Kidney Int. 2001, 60, 2079–2086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimada, T.; Yamazaki, Y.; Takahashi, M.; Hasegawa, H.; Urakawa, I.; Oshima, T.; Ono, K.; Kakitani, M.; Tomizuka, K.; Fujita, T.; et al. Vitamin D receptor-independent FGF23 actions in regulating phosphate and vitamin D metabolism. Am. J. Ren. Physiol. 2005, 289, F1088–F1095. [Google Scholar] [CrossRef]
- Martin, A.; David, V.; Quarles, L.D. Regulation and Function of the FGF23/Klotho Endocrine Pathways. Physiol. Rev. 2012, 92, 131–155. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Tamada, T.; Kasai, N.; Urakawa, I.; Aono, Y.; Hasegawa, H.; Fujita, T.; Kuroki, R.; Yamashita, T.; Fukumoto, S.; et al. Anti-FGF23 Neutralizing Antibodies Show the Physiological Role and Structural Features of FGF23. J. Bone Miner. Res. 2008, 23, 1509–1518. [Google Scholar] [CrossRef]
- Goetz, R.; Nakada, Y.; Hu, M.C.; Kurosu, H.; Wang, L.; Nakatani, T.; Shi, M.; Eliseenkova, A.V.; Razzaque, M.S.; Moe, O.W.; et al. Isolated C-terminal tail of FGF23 alleviates hypophosphatemia by inhibiting FGF23-FGFR-Klotho complex formation. Proc. Natl. Acad. Sci. USA 2010, 107, 407–412. [Google Scholar] [CrossRef]
- Xu, Y.; Sun, Z. Molecular Basis of Klotho: From Gene to Function in Aging. Endocr. Rev. 2015, 36, 174–193. [Google Scholar] [CrossRef]
- Shimada, T.; Muto, T.; Urakawa, I. Mutant FGF-23 responsible for autosomal dominant hypophsophatemic rickets is resistant to proteolytic cleavage and causes hypophosphatemia in vivo. Endocrinolgy 2002, 143, 3179–3182. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Okazaki, R.; Hasegawa, Y.; Satoh, K.; Tajima, T.; Takeuchi, Y.; Fujita, T.; Nakahara, K.; Yamashita, T.; Fukumoto, S. Increased circulatory level of biologically active full-length FGF-23 in patients with hypophosphatemic rickets/osteomalacia. J. Clin. Endocrinol. Metab. 2002, 87, 4957–4960. [Google Scholar] [CrossRef]
- Benet-Pagès, A.; Lorenz-Depiereux, B.; Zischka, H.; White, K.E.; Econs, M.J.; Strom, T.M. FGF23 is processed by proprotein convertases but not by PHEX. Bone 2004, 35, 455–462. [Google Scholar] [CrossRef]
- Frishberg, Y.; Ito, N.; Rinat, C.; Yamazaki, Y.; Feinstein, S.; Urakawa, I.; Navon-Elkan, P.; Becker-Cohen, R.; Yamashita, T.; Araya, K.; et al. Hyperostosis-Hyperphosphatemia Syndrome: A Congenital Disorder of O-Glycosylation Associated with Augmented Processing of Fibroblast Growth Factor 23. J. Bone Miner. Res. 2006, 22, 235–242. [Google Scholar] [CrossRef]
- Bergwitz, C.; Banerjee, S.; Abu-Zahra, H.; Kaji, H.; Miyauchi, A.; Sugimoto, T.; Jüppner, H. Defective O-Glycosylation due to a Novel Homozygous S129P Mutation Is Associated with Lack of Fibroblast Growth Factor 23 Secretion and Tumoral Calcinosis. J. Clin. Endocrinol. Metab. 2009, 94, 4267–4274. [Google Scholar] [CrossRef] [Green Version]
- Kato, K.; Jeanneau, C.; Tarp, M. Polypeptide GalNAc-transferase T3 and Familial Tumoral Calcinosis. J. Biol. Chem. 2006, 281, 18370–18377. [Google Scholar] [CrossRef] [Green Version]
- Khosravi, A.; Cutler, C.M.; Kelly, M.H.; Chang, R.; Royal, R.E.; Sherry, R.M.; Wodajo, F.M.; Fedarko, N.S.; Collins, M.T. Determination of the Elimination Half-Life of Fibroblast Growth Factor-23. J. Clin. Endocrinol. Metab. 2007, 92, 2374–2377. [Google Scholar] [CrossRef] [Green Version]
- Christov, M.; Waikar, S.S.; Pereira, R.C.; Havasi, A.; Leaf, D.E.; Goltzman, D.; Pajevic, P.D.; Wolf, M.; Jüppner, H. Plasma FGF23 levels increase rapidly after acute kidney injury. Kidney Int. 2013, 84, 776–785. [Google Scholar] [CrossRef] [Green Version]
- Mace, M.L.; Gravesen, E.; Hofman-Bang, J.; Olgaard, K.; Lewin, E. Key role of the kidney in the regulation of fibroblast growth factor 23. Kidney Int. 2015, 88, 1304–1313. [Google Scholar] [CrossRef] [Green Version]
- Lindberg, I.; Pang, H.W.; Stains, J.P.; Clark, D.; Yang, A.J.; Bonewald, L.; Li, K.Z. FGF23 is endogenously phosphorylated in bone cells. J. Bone Miner. Res. 2015, 30, 449–454. [Google Scholar] [CrossRef]
- Tagliabracci, V.S.; Engel, J.L.; Wiley, S.E.; Xiao, J.; Gonzalez, D.J.; Nidumanda Appaiah, H.; Koller, A.; Nizet, V.; White, K.E.; Dixon, J.E. Dynamic regulation of FGF23 by Fam20C phosphorylation, GalNAc-T3 glycosylation, and furin proteolysis. Proc. Natl. Acad. Sci. USA 2014, 111, 5520–5525. [Google Scholar] [CrossRef] [Green Version]
- ADHR Consortium. Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. Nat. Genet. 2000, 26, 345–348. [Google Scholar] [CrossRef]
- Wang, X.; Wang, S.; Li, C.; Gao, T.; Liu, Y.; Rangiani, A.; Sun, Y.; Hao, J.; George, A.; Lu, Y.; et al. Inactivation of a Novel FGF23 Regulator, FAM20C, Leads to Hypophosphatemic Rickets in Mice. PLoS Genet. 2012, 8, e1002708. [Google Scholar] [CrossRef]
- Topaz, O.; Shurman, D.L.; Bergman, R.; Indelman, M.; Ratajczak, P.; Mizrachi, M.; Khamaysi, Z.; Behar, D.; Petronius, D.; Friedman, V.; et al. Mutations in GALNT3, encoding a protein involved in O-linked glycosylation, cause familial tumoral calcinosis. Nat. Genet. 2004, 36, 579–581. [Google Scholar] [CrossRef] [Green Version]
- Larsson, T.; Davis, S.I.; Garringer, H.J.; Mooney, S.D.; Draman, M.S.; Cullen, M.J.; White, K.E. Fibroblast Growth Factor-23 Mutants Causing Familial Tumoral Calcinosis Are Differentially Processed. Endocrinology 2005, 146, 3883–3891. [Google Scholar] [CrossRef] [Green Version]
- Ichikawa, S.; Sorenson, A.H.; Austin, A.M.; Mackenzie, D.S.; Fritz, T.A.; Moh, A.; Hui, S.L.; Econs, M.J. Ablation of the GalnT3 Gene Leads to Low-Circulating Intact Fibroblast Growth Factor 23 (Fgf23) Concentrations and Hyperphosphatemia Despite Increased Fgf23 Expression. Endocrinology 2009, 150, 2543–2550. [Google Scholar] [CrossRef]
- Leaf, D.E.; Jacob, K.A.; Srivastava, A.; Chen, M.E.; Christov, M.; Jüppner, H.; Sabbisetti, V.S.; Martin, A.; Wolf, M.; Waikar, S.S. Fibroblast Growth Factor 23 Levels Associate with AKI and Death in Critical Illness. J. Am. Soc. Nephrol. 2017, 28, 1877–1885. [Google Scholar] [CrossRef]
- Lemmon, M.A.; Schlessinger, J. Cell Signaling by Receptor Tyrosine Kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [Green Version]
- Tiong, K.H.; Mah, L.Y.; Leong, C.-O. Functional roles of fibroblast growth factor receptors (FGFRs) signaling in human cancers. Apoptosis 2013, 18, 1447–1468. [Google Scholar] [CrossRef] [Green Version]
- Schlessinger, J.; Plotnikov, A.N.; Ibrahimi, O.A. Crystal Structure of a Ternary FGF-FGFR-Heparin Complex Reveals a Dual Role for Heparin in FGFR Binding and Dimerization. Cell 2000, 6, 743–750. [Google Scholar] [CrossRef]
- Harmer, N.J.; Robinson, C.J.; Adam, L.E.; Ilag, L.L.; Robinson, C.V.; Gallagher, J.T.; Blundell, T.L. Multimers of the fibroblast growth factor (FGF)-FGF receptor-saccharide complex are formed on long oligomers of heparin. Biochem. J. 2006, 393, 741–748. [Google Scholar] [CrossRef]
- Richter, B.; Faul, C. FGF23 Actions on Target Tissues—With and Without Klotho. Front. Endocrinol. 2018, 9, 15–21. [Google Scholar] [CrossRef]
- Faul, C. Cardiac actions of fibroblast growth factor 23. Bone 2017, 100, 69–79. [Google Scholar] [CrossRef]
- Katoh, M. Therapeutics Targeting FGF Signaling Network in Human Diseases. Trends Pharmacol. Sci. 2016, 37, 1081–1096. [Google Scholar] [CrossRef]
- Mohammadi, M.; Honegger, A.M.; Rotin, D. A Tyrosine-Phosphorylated Carboxy-Terminal Peptide of the Fibroblast Growth Factor Receptor (Flg) is a novel binding site for the SH2 Domain of Phospholipase C-y1. Mol. Cell. Biol. 1991, 11, 5068–5078. [Google Scholar] [CrossRef]
- Vainikka, S.; Joukov, V.; Wennström, S. Signal Transduction by Fibroblast Growth Factor Receptor-4 (FGFR-4). J. Biol. Chem. 1994, 269, 18320–18326. [Google Scholar]
- Peters, K.; Marie, J.; Wilson, E. Point mutation of an FGF receptor abolishes phosphatidylinositol turnover and calcium flux but not mitogenesis. Nature 1992, 358, 678–681. [Google Scholar] [CrossRef]
- Klint, P.; Claesson-Welsh, L. Signal transduction by fibroblast growth factor receptors. Front. Biosci. 1999, 4, D165–D177. [Google Scholar] [CrossRef]
- Burgess, W.; Dionne, C.; Kaplow, J. Characterization and cDNA Cloning of Phospholipase C-y, a major substrate for heparin-binding growth factor 1—activated tyrosine kinase. Mol. Cell. Biol. 1990, 10, 4770–4777. [Google Scholar] [CrossRef]
- Vervloet, M. Renal and extrarenal effects of fibroblast growth factor 23. Nat. Rev. Nephrol. 2019, 15, 109–120. [Google Scholar] [CrossRef]
- Kouhara, H.; Hadari, Y.R.; Spivak-Kroizman, T. A Lipid-Anchored Grb2-Binding Protein That Links FGF-Receptor Activation to the Ras/MAPK Signaling Pathway. Cell 1997, 89, 693–702. [Google Scholar] [CrossRef] [Green Version]
- Ben-Dov, I.Z.; Galitzer, H.; Lavi-Moshayoff, V.; Goetz, R.; Kuro-o, M.; Mohammadi, M.; Sirkis, R.; Naveh-Many, T.; Silver, J. The parathyroid is a target organ for FGF23 in rats. J. Clin. Investig. 2007, 117, 4003–4008. [Google Scholar] [CrossRef] [Green Version]
- Krajisnik, T.; Bjorklund, P.; Marsell, R.; Ljunggren, O.; Akerstrom, G.; Jonsson, K.B.; Westin, G.; Larsson, T.E. Fibroblast growth factor-23 regulates parathyroid hormone and 1-hydroxylase expression in cultured bovine parathyroid cells. J. Endocrinol. 2007, 195, 125–131. [Google Scholar] [CrossRef]
- Chen, G.; Liu, Y.; Goetz, R.; Fu, L.; Jayaraman, S.; Hu, M.C.; Moe, O.W.; Liang, G.; Li, X.; Mohammadi, M. α-Klotho is a non-enzymatic molecular scaffold for FGF23 hormone signaling. Nature 2018, 553, 461–466. [Google Scholar] [CrossRef]
- Shiraki-Iida, T.; Aizawa, H.; Matsumura, Y. Structure of the mouse klotho gene and its two transcripts encoding membrane and secreted protein. FEBJ Lett. 1998, 424, 6–10. [Google Scholar] [CrossRef]
- Urakawa, I.; Yamazaki, Y.; Shimada, T.; Iijima, K.; Hasegawa, H.; Okawa, K.; Fujita, T.; Fukumoto, S.; Yamashita, T. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature 2006, 444, 770–774. [Google Scholar] [CrossRef]
- Kurosu, H.; Ogawa, Y.; Miyoshi, M.; Yamamoto, M.; Nandi, A.; Rosenblatt, K.P.; Baum, M.G.; Schiavi, S.; Hu, M.C.; Moe, O.W.; et al. Regulation of Fibroblast Growth Factor-23 Signaling by Klotho. J. Biol. Chem. 2006, 281, 6120–6123. [Google Scholar] [CrossRef] [Green Version]
- Goetz, R.; Ohnishi, M.; Kir, S.; Kurosu, H.; Wang, L.; Pastor, J.; Ma, J.; Gai, W.; Kuro-o, M.; Razzaque, M.S.; et al. Conversion of a paracrine fibroblast growth factor into an endocrine fibroblast growth factor. J. Biol. Chem. 2012, 287, 29134–29146. [Google Scholar] [CrossRef]
- Kuro-o, M.; Matsumura, Y.; Kawaguchi, H. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature 1997, 390, 45–51. [Google Scholar] [CrossRef]
- Olauson, H.; Mencke, R.; Hillebrands, J.-L.; Larsson, T.E. Tissue expression and source of circulating αKlotho. Bone 2017, 100, 19–35. [Google Scholar] [CrossRef]
- Gattineni, J.; Bates, C.; Twombley, K.; Dwarakanath, V.; Robinson, M.L.; Goetz, R.; Mohammadi, M.; Baum, M. FGF23 decreases renal NaPi-2a and NaPi-2c expression and induces hypophosphatemia in vivo predominantly via FGF receptor 1. Am. J. Physiol. Ren. Physiol. 2009, 297, F282–F291. [Google Scholar] [CrossRef] [Green Version]
- Vervloet, M.G.; Sezer, S.; Massy, Z.A.; Johansson, L.; Cozzolino, M.; Fouque, D. The role of phosphate in kidney disease. Nat. Rev. Nephrol. 2016, 13, 27–38. [Google Scholar] [CrossRef]
- Shimada, T.; Mizutani, S.; Muto, T. Cloning and characterization of FGF23 as a causative factor of tumor-induced osteomalacia. Proc. Natl. Acad. Sci. USA 2001, 98, 6500–6505. [Google Scholar] [CrossRef] [Green Version]
- Olauson, H.; Lindberg, K.; Amin, R.; Sato, T.; Jia, T.; Goetz, R.; Mohammadi, M.; Andersson, G.; Lanske, B.; Larsson, T.E. Parathyroid-Specific Deletion of Klotho Unravels a Novel Calcineurin-Dependent FGF23 Signaling Pathway That Regulates PTH Secretion. PLoS Genet. 2013, 9, e1003975. [Google Scholar] [CrossRef]
- Lanske, B.; Razzaque, M.S. Molecular interactions of FGF23 and PTH in phosphate regulation. Kidney Int. 2014, 86, 1072–1074. [Google Scholar] [CrossRef] [Green Version]
- Kawakami, K.; Takeshita, A.; Furushima, K.; Miyajima, M.; Hatamura, I.; Kuro-o, M.; Furuta, Y.; Sakaguchi, K. Persistent fibroblast growth factor 23 signaling in the parathyroid glands for secondary hyperparathyroidism in mice with chronic kidney disease. Sci. Rep. 2017, 7, 40534. [Google Scholar] [CrossRef]
- Grabner, A.; Amaral, A.P.; Schramm, K.; Singh, S.; Sloan, A.; Yanucil, C.; Li, J.; Shehadeh, L.A.; Hare, J.M.; David, V.; et al. Activation of Cardiac Fibroblast Growth Factor Receptor 4 Causes Left Ventricular Hypertrophy. Cell Metab. 2015, 22, 1020–1032. [Google Scholar] [CrossRef] [Green Version]
- Grabner, A.; Schramm, K.; Silswal, N.; Hendrix, M.; Yanucil, C.; Czaya, B.; Singh, S.; Wolf, M.; Hermann, S.; Stypmann, J.X.R.; et al. FGF23/FGFR4-mediated left ventricular hypertrophy is reversible. Sci. Rep. 2017, 7, 1993. [Google Scholar] [CrossRef]
- Singh, S.; Grabner, A.; Yanucil, C.; Schramm, K.; Czaya, B.; Krick, S.; Czaja, M.J.; Bartz, R.; Abraham, R.; Di Marco, G.S.; et al. Fibroblast growth factor 23 directly targets hepatocytes to promote inflammation in chronic kidney disease. Kidney Int. 2016, 90, 985–996. [Google Scholar] [CrossRef] [Green Version]
- Smith, E.R.; Tan, S.J.; Holt, S.G.; Hewitson, T.D. FGF23 is synthesized locally by renal tubules and activates injury-primed fibroblasts. Sci. Rep. 2017, 7, 3345. [Google Scholar] [CrossRef]
- Smith, E.R.; Holt, S.G.; Hewitson, T.D. FGF23 activates injury-primed renal fibroblasts via FGFR4-dependent signaling and enhancement of TGF-β autoinduction. Int. J. Biochem. Cell Biol. 2017, 7, 40534. [Google Scholar]
- Rossaint, J.; Oehmichen, J.; Van Aken, H.; Reuter, S.; Pavenstädt, H.J.; Meersch, M.; Unruh, M.; Zarbock, A. FGF23 signaling impairs neutrophil recruitment and host defense during CKD. J. Clin. Investig. 2016, 126, 962–974. [Google Scholar] [CrossRef] [Green Version]
- Leifheit-Nestler, M.; Haffner, D. Paracrine Effects of FGF23 on the Heart. Front. Endocrinol. 2018, 9, 278. [Google Scholar] [CrossRef] [Green Version]
- Hanudel, M.; Jüppner, H.; Salusky, I.B. Fibroblast growth factor 23: Fueling the fire. Kidney Int. 2016, 90, 928–930. [Google Scholar] [CrossRef]
- Faul, C.; Amaral, A.P.; Oskouei, B.; Hu, M.C.; Sloan, A.; Isakova, T.; Gutiérrez, O.M.; Aguillon-Prada, R.; Lincoln, J.; Hare, J.M.; et al. FGF23 induces left ventricular hypertrophy. J. Clin. Investig. 2011, 121, 4393–4408. [Google Scholar] [CrossRef] [Green Version]
- Erben, R.G. Pleiotropic Actions of FGF23. Toxicol. Pathol. 2017, 45, 904–910. [Google Scholar] [CrossRef]
- Hanudel, M.R.; Chua, K.; Rappaport, M.; Gabayan, V.; Valore, E.; Goltzman, D.; Ganz, T.; Nemeth, E.; Salusky, I.B. Effects of dietary iron intake and chronic kidney disease on fibroblast growth factor 23 metabolism in wild-type and hepcidin knockout mice. Am. J. Physiol. Ren. Physiol. 2016, 311, F1369–F1377. [Google Scholar] [CrossRef]
- Clinkenbeard, E.L.; Noonan, M.L.; Thomas, J.C.; Ni, P.; Hum, J.M.; Aref, M.; Swallow, E.A.; Moe, S.M.; Allen, M.R.; White, K.E. Increased FGF23 protects against detrimental cardio-renal consequences during elevated blood phosphate in CKD. JCI Insight 2019, 4, 123817. [Google Scholar] [CrossRef]
- Nakatani, T.; Sarraj, B.; Ohnishi, M.; Densmore, M.J.; Taguchi, T.; Goetz, R.; Mohammadi, M.; Lanske, B.; Razzaque, M.S. In vivo genetic evidence for klotho-dependent, fibroblast growth factor 23 (Fgf23)-mediated regulation of systemic phosphate homeostasis. FASEB J. 2009, 23, 433–441. [Google Scholar] [CrossRef]
- Nakatani, T.; Ohnishi, M.; Razzaque, M.S. Inactivation of klotho function induces hyperphosphatemia even in presence of high serum fibroblast growth factor 23 levels in a genetically engineered hypophosphatemic (Hyp) mouse model. FASEB J. 2009, 23, 3702–3711. [Google Scholar] [CrossRef]
- Cavallaro, U.; Niedermeyer, J.; Fuxa, M.; Christofori, G. N-CAM modulates tumor-cell adhesion to matrix by inducing FGF-receptor signaling. Nat. Cell Biol. 2018, 3, 650–657. [Google Scholar] [CrossRef]
- Francavilla, C.; Loeffler, S.; Piccini, D.; Kren, A.; Christofori, G.; Cavallaro, U. Neural cell adhesion molecule regulates the cellular response to fibroblast growth factor. J. Cell Sci. 2007, 120, 4388–4394. [Google Scholar] [CrossRef] [Green Version]
- Francavilla, C.; Cattaneo, P.; Berezin, V.; Bock, E.; Ami, D.; de Marco, A.; Christofori, G.; Cavallaro, U. The binding of NCAM to FGFR1 induces a specific cellular response mediated by receptor trafficking. J. Cell Biol. 2009, 187, 1101–1116. [Google Scholar] [CrossRef]
- Williams, E.; Furness, J.; Walsh, F.; Doherty, P. Activation of the FGF Receptor Underlies Neurite Outgrowth Stimulated by 11, N-CAM, and- N-Cadherin. Neuron 1994, 13, 583–594. [Google Scholar] [CrossRef]
- Sanchez-Heras, E.; Howell, F.; Williams, G.; Doherty, P. The Fibroblast Growth Factor Receptor Acid Box Is Essential for Interactions with N-Cadherin and All of the Major Isoforms of Neural Cell Adhesion Molecule. J. Biol. Chem. 2006, 281, 35208–35216. [Google Scholar] [CrossRef] [Green Version]
- Christensen, C.; Lauridsen, J.B.; Berezin, V.; Bock, E.; Kiselyov, V.V. The neural cell adhesion molecule binds to fibroblast growth factor receptor 2. FEBS Lett. 2006, 580, 3386–3390. [Google Scholar] [CrossRef] [Green Version]
- Johnson, K.; Levine, K.; Sergi, J.; Chamoun, J.; Roach, R.; Vekich, J.; Favis, M.; Horn, M.; Cao, X.; Miller, B.; et al. Therapeutic Effects of FGF23 c-tail Fc in a Murine Preclinical Model of X-Linked Hypophosphatemia via the Selective Modulation of Phosphate Reabsorption. J. Bone Miner. Res. 2017, 32, 2062–2073. [Google Scholar] [CrossRef]
- Berndt, T.J.; Craig, T.A.; McCormick, D.J.; Lanske, B.; Sitara, D.; Razzaque, M.S.; Pragnell, M.; Bowe, A.E.; O’Brien, S.P.; Schiavi, S.C.; et al. Biological activity of FGF-23 fragments. Pflugers Arch. 2007, 454, 615–623. [Google Scholar] [CrossRef] [Green Version]
- Agoro, R.; Montagna, A.; Goetz, R.; Aligbe, O.; Singh, G.; Coe, L.M.; Mohammadi, M.; Rivella, S.; Sitara, D. Inhibition of fibroblast growth factor 23 (FGF23) signaling rescues renal anemia. FASEB J. 2018, 32, 3752–3764. [Google Scholar] [CrossRef] [Green Version]
- Sneddon, W.B.; Ruiz, G.W.; Gallo, L.I.; Xiao, K.; Zhang, Q.; Rbaibi, Y.; Weisz, O.A.; Apodaca, G.L.; Friedman, P.A. Convergent Signaling Pathways Regulate Parathyroid Hormone and Fibroblast Growth Factor-23 Action on NPT2A-mediated Phosphate Transport. J. Biol. Chem. 2016, 291, 18632–18642. [Google Scholar] [CrossRef] [Green Version]
- Naveh-Many, T.; Silver, J. The Pas de Trois of Vitamin D, FGF23, and PTH. J. Am. Soc. Nephrol. 2017, 28, 393–395. [Google Scholar] [CrossRef]
- Hruska, K.A.; Mathew, S.; Lund, R.; Qiu, P.; Pratt, R. Hyperphosphatemia of chronic kidney disease. Kidney Int. 2008, 74, 148–157. [Google Scholar] [CrossRef] [Green Version]
- Komaba, H.; Fukagawa, M. Phosphate—A poison for humans? Kidney Int. 2016, 90, 753–763. [Google Scholar] [CrossRef]
- Vervloet, M.G.; van Ballegooijen, A.J. Prevention and treatment of hyperphosphatemia in chronic kidney disease. Kidney Int. 2018, 93, 1060–1072. [Google Scholar] [CrossRef]
- Fukagawa, M.; Komaba, H.; Miyamoto, K.-I. Source matters: From phosphorus load to bioavailability. Clin. J. Am. Soc. Nephrol. 2011, 6, 239–240. [Google Scholar] [CrossRef]
- Kalantar-Zadeh, K.; Gutekunst, L.; Mehrotra, R.; Kovesdy, C.P.; Bross, R.; Shinaberger, C.S.; Noori, N.; Hirschberg, R.; Benner, D.; Nissenson, A.R.; et al. Understanding sources of dietary phosphorus in the treatment of patients with chronic kidney disease. Clin. J. Am. Soc. Nephrol. 2010, 5, 519–530. [Google Scholar] [CrossRef]
- Boaz, M.; Smetana, S. Regression Equation Predicts Dietary Phosphorus Intake from Estimate of Dietary Protein Intake. J. Am. Diet. Assoc. 1996, 96, 1268–1270. [Google Scholar] [CrossRef]
- Kayne, L.H.; D’Argenio, D.Z.; Meyer, J.H.; Hu, M.S.; Jamgotchian, N.; Lee, D.B. Analysis of segmental phosphate absorption in intact rats. A compartmental analysis approach. J. Clin. Investig. 1993, 91, 915–922. [Google Scholar] [CrossRef]
- Uribarri, J. Phosphorus Metabolism and Management in Chronic Kidney Disease: Phosphorus Homeostasis in Normal Health and in Chronic Kidney Disease Patients with Special Emphasis on Dietary Phosphorus Intake. Semin. Dial. 2007, 20, 295–301. [Google Scholar] [CrossRef]
- Schlemmer, U.; Frølich, W. Phytate in foods and significance for humans: Food sources, intake, processing, bioavailability, protective role and analysis. Mol. Nutr. 2009, 53, S330–S375. [Google Scholar] [CrossRef]
- Sandberg, A.S.; Andersson, H.; Kivistö, B.; Sandström, B. Extrusion cooking of a high-fiber cereal product. Br. J. Nutr. 1986, 55, 245–254. [Google Scholar] [CrossRef]
- Waldroup, P.W.; Kersey, J.H.; Saleh, E.A. Nonphytate Phosphorus Requirement and Phosphorus Excretion of Broiler Chicks Fed Diets Composed of Normal or High Available Phosphate Corn with and Without Microbial Phytase. Poult. Sci. 2000, 79, 1451–1459. [Google Scholar] [CrossRef]
- Lei, X.G.; Porres, J.M. Phytase enzymology, applications, and biotechnology. Biotechnol. Lett. 2003, 25, 1787–1794. [Google Scholar] [CrossRef]
- Ritz, E.; Hahn, K.; Ketteler, M.; Kuhlmann, M.K.; Mann, J. Phosphate Additives in Food. Deutsch. Aerztebl. Int. 2012, 109, 49–55. [Google Scholar] [CrossRef]
- Sullivan, C.; Sayre, S.S.; Leon, J.B.; Machekano, R.; Love, T.E.; Porter, D.; Marbury, M.; Sehgal, A.R. Effect of Food Additives on Hyperphosphatemia Among Patients with End-stage Renal Disease. JAMA 2009, 301, 629–635. [Google Scholar] [CrossRef]
- DSc, O.B.; RD, C.D.; VS, D.G.; PhD, A.C.M. Extra-Phosphate Load From Food Additives in Commonly Eaten Foods: A Real and Insidious Danger for Renal Patients. J. Ren. Nutr. 2011, 21, 303–308. [Google Scholar]
- Karalis, M.; Murphy-Gutekunst, L. Enhanced Foods: Hidden Phosphorus and Sodium in Foods Commonly Eaten. J. Ren. Nutr. 2006, 16, 79–81. [Google Scholar] [CrossRef]
- Kemi, V.E.; Rita, H.J.; Kärkkäinen, M.U.; Viljakainen, H.T.; Laaksonen, M.M.; Outila, T.A.; Lamberg-Allardt, C.J. Habitual high phosphorus intakes and foods with phosphate additives negatively affect serum parathyroid hormone concentration: A cross-sectional study on healthy premenopausal women. Public Health Nutr. 2009, 12, 1885–1892. [Google Scholar] [CrossRef]
- Uribarri, J. Hidden Sources of Phosphorus in the Typical American Diet: Does it Matter in Nephrology? Semin. Dial. 2003, 16, 186–188. [Google Scholar] [CrossRef]
- McCutcheon, J.; Campbell, K.; Ferguson, M.; Day, S.; Rossi, M. Prevalence of Phosphorus-Based Additives in the Australian Food Supply: A Challenge for Dietary Education? J. Ren. Nutr. 2015, 25, 440–444. [Google Scholar] [CrossRef]
- Calvo, M. Dietary Considerations to Prevent Loss of Bone and Renal Function. Nutrition 2000, 16, 564–566. [Google Scholar] [CrossRef]
- León, J.B.; Sullivan, C.M.; Sehgal, A.R. The Prevalence of Phosphorus-Containing Food Additives in Top-Selling Foods in Grocery Stores. J. Ren. Nutr. 2013, 23, 265–270. [Google Scholar] [CrossRef]
- Moser, M.; White, K.; Henry, B.; Oh, S.; Miller, E.R.; Anderson, C.A.; Benjamin, J.; Charleston, J.; Appel, L.J.; Chang, A.R. Phosphorus Content of Popular Beverages. Am. J. Kidney Dis. 2015, 65, 969–971. [Google Scholar] [CrossRef]
- Bell, R.R.; Draper, H.H.; Tzeng, D.Y.M.; Shin, H.K.; Schmidt, G.R. Physiological Responses of Human Adults to Foods Containing Phosphate Additives. J. Nutr. 1977, 107, 42–50. [Google Scholar] [CrossRef]
- Shafey, T.M.; McDonald, M.W.; Pym, R.A.E. Effects of dietary calcium, available phosphorus and vitamin D on growth rate, food utilization, plasma and bone constituents and calcium and phosphorus retention of commercial broiler strains. Br. Poult. Sci. 1990, 31, 587–602. [Google Scholar] [CrossRef]
- Wolf, M. Update on fibroblast growth factor 23 in chronic kidney disease. Kidney Int. 2012, 82, 737–747. [Google Scholar] [CrossRef] [Green Version]
- Jüppner, H. Phosphate and FGF-23. Kidney Int. 2011, 79, S24–S27. [Google Scholar] [CrossRef] [Green Version]
- Graciolli, F.G.; Neves, K.R.; Barreto, F.; Barreto, D.V.; Reis, dos, L.M.; Canziani, M.E.; Sabbagh, Y.; Carvalho, A.B.; Jorgetti, V.; Elias, R.M.; et al. The complexity of chronic kidney disease-mineral and bone disorder across stages of chronic kidney disease. Kidney Int. 2017, 91, 1436–1446. [Google Scholar] [CrossRef]
- Drew, D.A.; Katz, R.; Kritchevsky, S.; Ix, J.; Shlipak, M.; Gutiérrez, O.M.; Newman, A.; Hoofnagle, A.; Fried, L.; Semba, R.D.; et al. Association between Soluble Klotho and Change in Kidney Function: The Health Aging and Body Composition Study. J. Am. Soc. Nephrol. 2017, 28, 1859–1866. [Google Scholar] [CrossRef] [Green Version]
- Hu, M.C.; Kuro-o, M.; Moe, O.W. Klotho and Chronic Kidney Disease. Contrib. Nephrol. 2013, 180, 47–63. [Google Scholar] [Green Version]
- Di Zou; Wu, W.; He, Y.; Ma, S.; Gao, J. The role of klotho in chronic kidney disease. BMC Nephrol. 2018, 19, 285. [Google Scholar]
- Hasegawa, H.; Nagano, N.; Urakawa, I.; Yamazaki, Y.; Iijima, K.; Fujita, T.; Yamashita, T.; Fukumoto, S.; Shimada, T. Direct evidence for a causative role of FGF23 in the abnormal renal phosphate handling and vitamin D metabolism in rats with early-stage chronic kidney disease. Kidney Int. 2010, 78, 975–980. [Google Scholar] [CrossRef] [Green Version]
- Isakova, T.; Gutiérrez, O.M.; Wolf, M. A blueprint for randomized trials targeting phosphorus metabolism in chronic kidney disease. Kidney Int. 2009, 76, 705–716. [Google Scholar] [CrossRef] [Green Version]
- Isakova, T.; Wahl, P.; Vargas, G.S.; Gutiérrez, O.M.; Scialla, J.; Xie, H.; Appleby, D.; Nessel, L.; Bellovich, K.; Chen, J.; et al. Fibroblast growth factor 23 is elevated before parathyroid hormone and phosphate in chronic kidney disease. Kidney Int. 2011, 79, 1370–1378. [Google Scholar] [CrossRef] [Green Version]
- Kato, S.; Chmielewski, M.; Honda, H.; Pecoits-Filho, R.; Matsuo, S.; Yuzawa, Y.; Tranaeus, A.; Stenvinkel, P.; Lindholm, B. Aspects of Immune Dysfunction in End-stage Renal Disease. Clin. J. Am. Soc. Nephrol. 2008, 3, 1526–1533. [Google Scholar] [CrossRef] [Green Version]
- Meuwese, C.L.; Stevinkel, P.; Carrero, J.J. Monitoring of inflammation in patients on dialysis: Forewarned is forearmed. Nat. Rev. Nephrol. 2011, 7, 166–176. [Google Scholar] [CrossRef]
- Fishbane, S.; Spinowitz, B. Update on Anemia in ESRD and Earlier Stages of CKD: Core Curriculum 2018. Am. J. Kidney Dis. 2018, 71, 423–435. [Google Scholar] [CrossRef]
- Moe, S.M.; Chen, N.X. Mechanisms of Vascular Calcification in Chronic Kidney Disease. J. Am. Soc. Nephrol. 2008, 19, 213–216. [Google Scholar] [CrossRef]
- Wang, X.H.; Mitch, W.E. Mechanisms of muscle wasting in chronic kidney disease. Nat. Rev. Nephrol. 2014, 10, 504–516. [Google Scholar] [CrossRef] [Green Version]
- Mathew, J.; Katz, R.; St Sutton, M.J.; Dixit, S.; Gerstenfeld, E.P.; Ghio, S.; Gold, M.R.; Linde, C.; Shlipak, M.G.; Deo, R. Chronic kidney disease and cardiac remodeling in patients with mild heart failure: Results from the REsynchronization reVErses Remodeling in Systolic Left vEntricular Dysfunction (REVERSE) study. Eur. J. Heart Fail. 2014, 14, 1420–1428. [Google Scholar] [CrossRef]
- Tonelli, M. Chronic Kidney Disease and Mortality Risk: A Systematic Review. J. Am. Soc. Nephrol. 2006, 17, 2034–2047. [Google Scholar] [CrossRef] [Green Version]
- Isakova, T.; Wolf, M. Fibroblast Growth Factor 23 and Risks of Mortality and End-Stage Renal Disease in Patients with Chronic Kidney Disease. JAMA 2011, 305, 2432–2439. [Google Scholar] [CrossRef]
- Kendrick, J.; Cheung, A.K.; Kaufman, J.S.; Greene, T.; Roberts, W.L.; Smits, G.; Chonchol, M.; HOST Investigators. FGF-23 Associates with Death, Cardiovascular Events, and Initiation of Chronic Dialysis. J. Am. Soc. Nephrol. 2011, 22, 1913–1922. [Google Scholar] [CrossRef]
- Mendoza, J.M.; Isakova, T.; Cai, X.; Bayes, L.Y.; Faul, C.; Scialla, J.J.; Lash, J.P.; Chen, J.; He, J.; Navaneethan, S.; et al. Inflammation and elevated levels of fibroblast growth factor 23 are independent risk factors for death in chronic kidney disease. Kidney Int. 2016, 91, 711–719. [Google Scholar] [CrossRef] [Green Version]
- Mehta, R.; Cai, X.; Hodakowski, A.; Lee, J.; Leonard, M.; Ricardo, A.; Chen, J.; Hamm, L.; Sondheimer, J.; Dobre, M.; et al. Fibroblast Growth Factor 23 and Anemia in the Chronic Renal Insufficiency Cohort Study. Clin. J. Am. Soc. Nephrol. 2017, 12, 1795–1803. [Google Scholar] [CrossRef]
- Weiss, G.; Ganz, T.; Goodnough, L. Anemia of inflammation. Blood 2019, 133, 40–50. [Google Scholar] [CrossRef] [Green Version]
- Nemeth, E.; Ganz, T. Anemia of Inflammation. Hematol. Oncol. Clin. N. Am. 2014, 28, 671–681. [Google Scholar] [CrossRef] [Green Version]
- Francis, C.; David, V. Inflammation regulates fibroblast growth factor 23 production. Curr. Opin. Nephrol. Hypertens. 2016, 25, 325–332. [Google Scholar] [CrossRef] [Green Version]
- Wrighting, D.M.; Andrews, N.C. Interleukin-6 induces hepcidin expression through STAT3. Blood 2006, 108, 3204–3209. [Google Scholar] [CrossRef]
- Kanamori, Y.; Murakami, M.; Sugiyama, M.; Hashimoto, O.; Matsui, T.; Funaba, M. Interleukin-1β (IL-1β) transcriptionally activates hepcidin by inducing CCAAT enhancer-binding protein δ (C/EBPδ) expression in hepatocytes. J. Biol. Chem. 2017, 292, 10275–10287. [Google Scholar] [CrossRef]
- Chung, B.; Verdier, F.; Matak, P.; Deschemin, J.-C.; Mayeux, P.; Vaulont, S. Oncostatin M is a potent inducer of hepcidin, the iron regulatory hormone. FASEB J. 2010, 24, 2093–2103. [Google Scholar] [CrossRef]
- Nemeth, E.; Rivera, S.; Gabayan, V.; Keller, C.; Taudorf, S.; Pedersen, B.K.; Ganz, T. IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J. Clin. Investig. 2004, 113, 1271–1276. [Google Scholar] [CrossRef] [Green Version]
- Pietrangelo, A.; Dierssen, U.; Valli, L.; Garuti, C.; Rump, A.; Corradini, E.; Ernst, M.; Klein, C.; Trautwein, C. STAT3 Is Required for IL-6-gp130–Dependent Activation of Hepcidin In Vivo. Gastroenterologty 2007, 132, 294–300. [Google Scholar] [CrossRef]
- Andrews, N.C. Anemia of inflammation: The cytokine-hepcidin link. J. Clin. Invest. 2004, 113, 1251–1253. [Google Scholar] [CrossRef]
- Lee, P.; Gelbart, T.; Wang, L.; Beutler, E. Regulation of hepcidin transcription by interleukin-1 and interleukin-6. Proc. Natl. Acad. Sci. USA 2005, 102, 1906–1910. [Google Scholar] [CrossRef] [Green Version]
- Fatih, N.; Camberlein, E.; Island, M.L.; Corlu, A.; Abgueguen, E.; Détivaud, L.; Leroyer, P.; Brissot, P.; Loréal, O. Natural and synthetic STAT3 inhibitors reduce hepcidin expression in differentiated mouse hepatocytes expressing the active phosphorylated STAT3 form. J. Mol. Med. 2010, 88, 477–486. [Google Scholar] [CrossRef]
- Ganz, T. Hepcidin and iron regulation, 10 years later. Blood 2011, 117, 4425–4433. [Google Scholar] [CrossRef] [Green Version]
- Young, B.; Zaritsky, J. Hepcidin for clinicians. Clin. J. Am. Soc. Nephrol. 2009, 4, 1384–1387. [Google Scholar] [CrossRef]
- Ganz, T.; Nemeth, E. Hepcidin and iron homeostasis. BBA Mol. Cell Res. 2012, 1823, 1434–1443. [Google Scholar] [CrossRef] [Green Version]
- Akchurin, O.; Sureshbabu, A.; Doty, S.B.; Zhu, Y.S.; Patino, E.; Cunningham-Rundles, S.; Choi, M.E.; Boskey, A.; Rivella, S. Lack of hepcidin ameliorates anemia and improves growth in an adenine-induced mouse model of chronic kidney disease. Am. J. Physiol. Ren. Physiol. 2016, 311, F877–F889. [Google Scholar] [CrossRef] [Green Version]
- David, V.; Francis, C.; Babitt, J.L. Ironing out the crosstalk between FGF23 and inflammation. Am. J. Physiol. Ren. Physiol. 2017, 312, F1–F8. [Google Scholar] [CrossRef]
- Mendoza, J.M.; Isakova, T.; Ricardo, A. Fibroblast Growth Factor 23 and Inflammation in CKD. Clin. J. Am. Soc. Nephrol. 2012, 7, 1155–1162. [Google Scholar] [CrossRef] [Green Version]
- Wallquist, C.; Mansouri, L.; Norrbäck, M.; Hylander, B.; Jacobson, S.H.; Larsson, T.E.; Lundahl, J. Associations of Fibroblast Growth Factor 23 with Markers of Inflammation and Leukocyte Transmigration in Chronic Kidney Disease. Nephron 2018, 138, 287–295. [Google Scholar] [CrossRef] [Green Version]
- Hanks, L.J.; Casazza, K.; Judd, S.E.; Jenny, N.S.; Gutiérrez, O.M. Associations of Fibroblast Growth Factor-23 with Markers of Inflammation, Insulin Resistance and Obesity in Adults. PLoS ONE 2015, 10, e0122885. [Google Scholar] [CrossRef]
- El-Hodhod, M.; Hamdy, A.; Abbas, A. Fibroblast growth factor 23 contributes to diminished bone mineral density in childhood inflammatory bowel disease. Gastroenterology 2012, 12, 44. [Google Scholar] [CrossRef]
- David, V.; Martin, A.; Isakova, T.; Spaulding, C.; Qi, L.; Ramirez, V.; Zumbrennen-Bullough, K.B.; Sun, C.C.; Lin, H.Y.; Babitt, J.L.; et al. Inflammation and functional iron deficiency regulate fibroblast growth factor 23 production. Kidney Int. 2016, 89, 135–146. [Google Scholar] [CrossRef] [Green Version]
- Durlacher-Betzer, K.; Hassan, A.; Levi, R.; Axelrod, J.; Silver, J.; Naveh-Many, T. Interleukin-6 contributes to the increase in fibroblast growth factor 23 expression in acute and chronic kidney disease. Kidney Int. 2018, 94, 315–325. [Google Scholar] [CrossRef]
- Glosse, P.; Fajol, A.; Hirche, F.; Feger, M.; Voelkl, J.; Lang, F.; Stangl, G.I.; Föller, M. A high-fat diet stimulates fibroblast growth factor 23 formation in mice through TNFα upregulation. Nutr. Diabetes 2018, 8, 36. [Google Scholar] [CrossRef]
- Egli-Spichtig, D.; Silva, P.H.I.; Glaudemans, B.; Gehring, N.; Bettoni, C.; Zhang, M.Y.H.; Pastor-Arroyo, E.M.; Schönenberger, D.; Rajski, M.; Hoogewijs, D.; et al. Tumor necrosis factor stimulates fibroblast growth factor 23 levels in chronic kidney disease and non-renal inflammation. Kidney Int. 2019. [Google Scholar] [CrossRef]
- Richter, M.; Lautze, H.-J.; Walther, T.; Braun, T.; Kostin, S.; Kubin, T. The failing heart is a major source of circulating FGF23 via oncostatin M receptor activation. J. Heart Lung Transplant. 2015, 34, 1211–1214. [Google Scholar] [CrossRef]
- Onal, M.; Carlson, A.; Thostenson, J. A Novel Distal Enhancer Mediates Inflammation-, PTH-, and Early Onset Murine Kidney Disease-Induced Expression of the Mouse Fgf23 Gene. J. Bone Mineral. Metab. 2018, 2, 32–47. [Google Scholar] [CrossRef]
- Farrow, E.G.; Summers, L.J.; Davis, S.I. Iron deficiency drives an autosomal dominant hypophosphatemic rickets (ADHR) phenotype in fibroblast growth factor-23 (Fgf23) knock-in mice. Proc. Natl. Acad. Sci. USA 2011, 108, E1146–E1155. [Google Scholar] [CrossRef] [Green Version]
- Fishbane, S.; Block, G.A.; Loram, L.; Neylan, J.; Pergola, P.E.; Uhlig, K.; Chertow, G.M. Effects of Ferric Citrate in Patients with Nondialysis-Dependent CKD and Iron Deficiency Anemia. J. Am. Soc. Nephrol. 2017, 28, 1851–1858. [Google Scholar] [CrossRef] [Green Version]
- Fukao, W.; Hasuike, Y.; Yamakawa, T.; Toyoda, K.; Aichi, M.; Masachika, S.; Kantou, M.; Takahishi, S.I.; Iwasaki, T.; Yahiro, M.; et al. Versus Intravenous Iron Supplementation for the Treatment of Iron Deficiency Anemia in Patients on Maintenance Hemodialysis—Effect on Fibroblast Growth Factor-23 Metabolism. J. Ren. Nutr. 2018, 28, 270–277. [Google Scholar] [CrossRef]
- Iguchi, A.; Yamamoto, S.; Yamazaki, M.; Tasaki, K.; Suzuki, Y.; Kazama, J.J.; Narita, I. Effect of ferric citrate hydrate on FGF23 and PTH levels in patients with non-dialysis-dependent chronic kidney disease with normophosphatemia and iron deficiency. Clin. Exp. Nephrol. 2017, 22, 789–796. [Google Scholar] [CrossRef]
- Maruyama, N.; Otsuki, T.; Yoshida, Y.; Nagura, C.; Kitai, M.; Shibahara, N.; Tomita, H.; Maruyama, T.; Abe, M. Ferric Citrate Decreases Fibroblast Growth Factor 23 and Improves Erythropoietin Responsiveness in Hemodialysis Patients. Am. J. Nephrol. 2018, 47, 406–414. [Google Scholar] [CrossRef]
- Rabadi, S.; Udo, I.; Leaf, D.E.; Waikar, S.; Christov, M. Acute blood loss stimulates fibroblast growth factor 23 production. Am. J. Physiol. Ren. Physiol. 2017, 314, F132–F139. [Google Scholar] [CrossRef]
- Hanudel, M.R.; Eisenga, M.F.; Rappaport, M.; Chua, K.; Qiao, B.; Jung, G.; Gabayan, V.; Gales, B.; Ramos, G.; de Jong, M.A.; et al. Effects of erythropoietin on fibroblast growth factor 23 in mice and humans. Nephrol. Dial. Transplant. 2018, 13, 504–509. [Google Scholar] [CrossRef]
- Toro, L.; Barrientos, V.; León, P.; Rojas, M.; Gonzalez, M.; González-Ibáñez, A.; Illanes, S.; Sugikawa, K.; Abarzúa, N.; Bascuñán, C.; et al. Erythropoietin induces bone marrow and plasma fibroblast growth factor 23 during acute kidney injury. Kidney Int. 2018, 93, 1131–1141. [Google Scholar] [CrossRef]
- Mihai, S.; Codrici, E.; Popescu, I.D.; Enciu, A.-M.; Necula, L.G.; Anton, G.; Tanase, C. Inflammation and Chronic Kidney Disease: Current Approaches and Recent Advances. Chronic Kidney Dis. Pathophysiol. Clin. Improv. 2018, 2018, 2180373. [Google Scholar]
- Amdur, R.; Feldman, H.; Gupta, J. Inflammation and Progression of CKD: The CRIC Study. Clin. J. Am. Soc. Nephrol. 2016, 11, 1546–1556. [Google Scholar] [CrossRef] [Green Version]
- Gupta, J.; Mitra, N.; Kanetsky, P.A.; Devaney, J.; Wing, M.R.; Reilly, M.; Shah, V.O.; Balakrishnan, V.S.; Guzman, N.J.; Girndt, M.; et al. CRIC Study Investigators Association between albuminuria, kidney function, and inflammatory biomarker profile in CKD in CRIC. Clin. J. Am. Soc. Nephrol. 2012, 7, 1938–1946. [Google Scholar] [CrossRef]
- Mihai, S.; Codrici, E.; Popescu, I.D.; Enciu, A.-M.; Albulescu, L.; Necula, L.G.; Mambet, C.; Anton, G.; Tanase, C. Inflammation-Related Mechanisms in Chronic Kidney Disease Prediction, Progression, and Outcome. J. Immunol. Res. 2018, 2018, 1–16. [Google Scholar] [CrossRef]
- Lin, B.; Wang, M.; Blackmore, C.; Desnoyers, L. Liver-specific Activities of FGF19 Require Klotho beta. J. Biol. Chem. 2007, 282, 27277–27284. [Google Scholar] [CrossRef] [Green Version]
- Kan, M.; Wu, X.; Wang, F.; Mckeehan, W. Specificity for Fibroblast Growth Factors Determined by Heparan Sulfate in a Binary Complex with the Receptor Kinase. J. Biol. Chem. 1999, 274, 15947–15952. [Google Scholar] [CrossRef] [Green Version]
- Sandhu, D.S.; Baichoo, E.; Roberts, L.R. Fibroblast growth factor signaling in liver carcinogenesis. Hepatology 2013, 59, 1166–1173. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Yang, C.; Jin, C.; Luo, Y.; Wang, F.; McKeehan, W.L. Resident hepatocyte fibroblast growth factor receptor 4 limits hepatocarcinogenesis. Mol. Carcinog. 2009, 48, 553–562. [Google Scholar] [CrossRef]
- Gulati, S.; Wells, J.M.; Urdaneta, G.P.; Balestrini, K.; Vital, I.; Tovar, K.; Barnes, J.W.; Bhatt, S.P.; Campos, M.; Krick, S. Fibroblast Growth Factor 23 is Associated with a Frequent Exacerbator Phenotype in COPD: A Cross-Sectional Pilot Study. Int. J. Med. Sci. 2019, 20, 2292. [Google Scholar] [CrossRef]
- Elsammak, M.; Attia, A.; Suleman, M. Fibroblast Growth Factor-23 and Hypophosphatemia in Chronic Obstructive Pulmonary Disease Patients. J. Med. Biochem. 2011, 31, 12–18. [Google Scholar] [CrossRef]
- Krick, S.; Grabner, A.; Baumlin, N.; Yanucil, C.; Helton, S.; Grosche, A.; Sailland, J.; Geraghty, P.; Viera, L.; Russell, D.W.; et al. Fibroblast growth factor 23 and Klotho contribute to airway inflammation. Eur. Respir. J. 2018, 52, 1800236. [Google Scholar] [CrossRef] [Green Version]
- Krick, S.; Baumlin, N.; Aller, S.P.; Aguiar, C.; Grabner, A.; Sailland, J.; Mendes, E.; Schmid, A.; Qi, L.; David, N.V.; et al. Klotho Inhibits Interleukin-8 Secretion from Cystic Fibrosis Airway Epithelia. Sci. Rep. 2017, 7, 14388. [Google Scholar] [CrossRef] [Green Version]
- Courtney, J.M.; Ennis, M.; Elborn, J.S. Cytokines and inflammatory mediators in cystic fibrosis. J. Cyst. Fibros. 2004, 3, 223–231. [Google Scholar] [CrossRef] [Green Version]
- Han, X.; Li, L.; Yang, J.; King, G.; Xiao, Z.; Quarles, L.D. Counter-regulatory paracrine actions of FGF-23 and 1,25(OH)2D in macrophages. FEBS Lett. 2016, 590, 53–67. [Google Scholar] [CrossRef]
- Rao, A.; Luo, C.; Hogan, P. Transcription Factors of the NFAT Family: Regulation and Function. Annu. Rev. Immunol. 1997, 15, 707–747. [Google Scholar] [CrossRef]
- Rossaint, J.; Unruh, M.; Zarbock, A. Fibroblast growth factor 23 actions in inflammation: A key factor in CKD outcomes. Nephrol. Dial. Transpl. 2017, 32, 1448–1453. [Google Scholar] [CrossRef]
- Yamada, S.; Tokumoto, M.; Tatsumoto, N.; Taniguchi, M.; Noguchi, H.; Nakano, T.; Masutani, K.; Ooboshi, H.; Tsuruya, K.; Kitazono, T. Phosphate overload directly induces systemic inflammation and malnutrition as well as vascular calcification in uremia. Am. J. Physiol. Ren. Physiol. 2014, 306, F1418–F1428. [Google Scholar] [CrossRef]
- Sugihara, K.; Masuda, M.; Nakao, M.; Taketani, Y. Dietary phosphate exacerbates intestinal inflammation in experimental colitis. J. Clin. Biochem. 2017, 61, 91–99. [Google Scholar] [CrossRef] [Green Version]
- Iglehart, J. Bundled Payment for ESRD—Including ESAs in Medicare’s Dialysis Package. N. Engl. J. Med. 2011, 94, 38–39. [Google Scholar] [CrossRef]
- Coresh, J.; Selvin, E.; Stevens, L.; Levey, A. Prevalence of Chronic Kidney Disease in the United States. JAMA. 2007, 298, 2038–2047. [Google Scholar] [CrossRef] [Green Version]
- United States Renal Data System. USRDS 2010 Annual report: Atlas of Chronic Kidney Disease and End Stage-Renal Disease in the United States; National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Disease: Bethesda, MD, USA, 2010; pp. 1–2. [Google Scholar]
- Keith, D.; Nichols, G.; Smith, D. Longitudinal Follow-up and Outcomes Among a Population with Chronic Kidney Disease in a Large Managed Care Organization. JAMA 2004, 164, 659–663. [Google Scholar] [CrossRef]
- Webster, A.C.; Nagler, E.V.; Morton, R.L.; Masson, P. Chronic Kidney Disease. Lancet 2017, 389, 1238–1252. [Google Scholar] [CrossRef]
- Razmaria, A. Chronic Kidney Disease. JAMA 2016, 315, 2248. [Google Scholar] [CrossRef]
- MD, J.A.V.; MD, R.C.; MSED, B.J.T.M.; MHS, R.C.G.M.; MD, M.C.; MPH, T.D.S.M. Practical Approach to Detection and Management of Chronic Kidney Disease for the Primary Care Clinician. Am. J. Med. 2016, 129, 153–162. [Google Scholar]
- Thomas, R.; Kanso, A.; Sedor, J. Chronic Kidney Disease and Its Complications. Prim. Care 2008, 35, 329–344. [Google Scholar] [CrossRef] [Green Version]
- Helal, I.; Fick-Brosnahan, G.M.; Reed-Gitomer, B.; Schrier, R.W. Glomerular hyperfiltration: Definitions, mechanisms and clinical implications. Nat. Rev. Nephrol. 2012, 8, 293–300. [Google Scholar] [CrossRef]
- Ruggenenti, P.; Porrini, E.; Gaspari, F. Glomerular Hyperfiltration and Renal Disease Progression in Type 2 Diabetes. Diabetes Care 2012, 35, 2061–2068. [Google Scholar] [CrossRef] [Green Version]
- Palatini, P. Glomerular hyperfiltration: A marker of early renal damage in pre-diabetes and pre-hypertension. Nephrol. Dial. Transplant. 2012, 27, 1708–1714. [Google Scholar] [CrossRef]
- Din, U.A.A.S.E.; Salem, M.M.; Abdulazim, D.O. Stop chronic kidney disease progression: Time is approaching. World J. Nephrol. 2016, 5, 258–273. [Google Scholar] [CrossRef] [Green Version]
- Stenvinkel, P. Inflammation in end-stage renal disease: The hidden enemy (Review Article). Nephrology 2006, 11, 36–41. [Google Scholar] [CrossRef]
- Kimmel, P.L.; Phillips, T.M.; Simmens, S.J.; Peterson, R.A.; Weihs, K.L.; Alleyne, S.; Cruz, I.; Yanovski, J.A.; Veis, J.H. Immunologic function and survival in hemodialysis patients. Kidney Int. 1998, 54, 236–244. [Google Scholar] [CrossRef] [Green Version]
- Akchurin, O.M.; Kaskel, F. Update on inflammation in chronic kidney disease. Blood Purif. 2015, 39, 84–92. [Google Scholar] [CrossRef]
- Yao, Q.; Axelsson, J.; Stenvinkel, P.; Lindholm, B. Chronic Systemic Inflammation in Dialysis Patients: An Update on Causes and Consequences. ASAIO J. 2004, 50, Iii–Ivii. [Google Scholar] [CrossRef]
- Eming, S.A.; Wynn, T.A.; Martin, P. Inflammation and metabolism in tissue repair and regeneration. Science 2017, 356, 1026–1030. [Google Scholar] [CrossRef] [Green Version]
- Cavaillon, J.M.; Singer, M. Inflammation—From Molecular and Cellular Mechanisms to the Clinic; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2017; pp. 1–3. [Google Scholar]
- Koh, T.J.; DiPietro, L.A. Inflammation and wound healing: The role of the macrophage. Expert Rev. Mol. Med. 2011, 13, e23. [Google Scholar] [CrossRef]
- Salles, G.F.; Fiszman, R.; Cardoso, C.R.L.; Muxfeldt, E.S. Relation of Left Ventricular Hypertrophy with Systemic Inflammation and Endothelial Damage in Resistant Hypertension. Hypertension 2007, 50, 723–728. [Google Scholar] [CrossRef]
- Raj, D.S.; Pecoits-Filho, R.; Kimmel, P.L. Inflammation in Chronic Kidney Disease; Elsevier Inc.: Amsterdam, Netherlands, 2014; pp. 199–212. [Google Scholar]
- Vaziri, N.D. Roles of oxidative stress and antioxidant therapy in chronic kidney disease and hypertension. Curr. Opin. Nephrol. Hypertens. 2004, 13, 93–99. [Google Scholar] [CrossRef]
- Sung, C.-C.; Hsu, Y.-C.; Chen, C.-C.; Lin, Y.-F.; Wu, C.-C. Oxidative Stress and Nucleic Acid Oxidation in Patients with Chronic Kidney Disease. Oxidative Med. Cell. Longev. 2013, 2013, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Nuhu, F.; Bhandari, S. Oxidative Stress and Cardiovascular Complications in Chronic Kidney Disease, the Impact of Anemia. Pharmaceuticals 2018, 11, 103. [Google Scholar] [CrossRef]
- Panth, N.; Paudel, K.R.; Parajuli, K. Review Article Reactive Oxygen Species: A Key Hallmark of Cardiovascular Disease. Adv. Med. 2016, 2016, 9152732. [Google Scholar] [CrossRef]
- Zargari, M.; Sedighi, O. Influence of Hemodialysis on Lipid Peroxidation, Enzymatic and Non-Enzymatic Antioxidant Capacity in Chronic Renal Failure Patients. Nephro. Urol. Mon. 2015, 7, e28526. [Google Scholar] [CrossRef] [Green Version]
- Kao, M.P.C.; Ang, D.S.C.; Pall, A.; Struthers, A.D. Oxidative stress in renal dysfunction: Mechanisms, clinical sequelae and therapeutic options. J. Hum. Hypertens. 2009, 24, 1–8. [Google Scholar] [CrossRef]
- Taetzsch, T.; Levesque, S. Redox regulation of NF-kB p50 and M1 polarization in microglia. Glia 2019, 1–3. [Google Scholar]
- Meijers, B.K.I.; Evenepoel, P. The gut-kidney axis: Indoxyl sulfate, p-cresyl sulfate and CKD progression. Nephrol. Dial. Transplant. 2011, 26, 759–761. [Google Scholar] [CrossRef]
- Evenepoel, P.; Meijers, B.K.I.; Bammens, B.R.M.; Verbeke, K. Uremic toxins originating from colonic microbial metabolism. Kidney Int. 2009, 76, S12–S19. [Google Scholar] [CrossRef] [Green Version]
- Andersen, K.; Kesper, M.S.; Marschner, J.A.; Konrad, L.; Ryu, M.; Kumar VR, S.; Kulkarni, O.P.; Mulay, S.R.; Romoli, S.; Demleitner, J.; et al. Intestinal Dysbiosis, Barrier Dysfunction, and Bacterial Translocation Account for CKD-Related Systemic Inflammation. J. Am. Soc. Nephrol. 2017, 28, 76–83. [Google Scholar] [CrossRef]
- Turner, M.D.; Nedjai, B.; Hurst, T.; Pennington, D.J. Cytokines and chemokines: At the crossroads of cell signaling and inflammatory disease. BBA - Mol. Cell Res. 2014, 1843, 2563–2582. [Google Scholar] [CrossRef]
- Sokol, C.L.; Luster, A.D. The chemokine system in innate immunity. Cold Spring Harb. Perspect. Biol. 2015, 7, a016303. [Google Scholar] [CrossRef]
- Duque, G.; Descoteaux, A. Macrophage cytokines: Involvement in immunity and infectious diseases. Front. Immunol. 2014, 5, 491. [Google Scholar]
- Surmi, B.K.; Hasty, A.H. The role of chemokines in recruitment of immune cells to the artery wall and adipose tissue. Vasc. Pharmacol. 2010, 52, 27–36. [Google Scholar] [CrossRef] [Green Version]
- Bode, J.G.; Albrecht, U.; Häussinger, D.; Heinrich, P.C.; Schaper, F. Hepatic acute phase proteins—Regulation by IL-6- and IL-1-type cytokines involving STAT3 and its crosstalk with NF-κB-dependent signaling. Eur. J. Cell Biol. 2011, 91, 496–505. [Google Scholar] [CrossRef]
- Panwar, B.; Gutiérrez, O.M. Disorders of Iron Metabolism and Anemia in Chronic Kidney Disease. Semin. Nephrol. 2016, 36, 252–261. [Google Scholar] [CrossRef]
- Gruys, E.; Toussaint, M.J.M.; Niewold, T.A.; Koopmans, S.J. Acute phase reaction and acute phase proteins. J. Zhejiang Univ. Sci. 2005, 6B, 1045–1056. [Google Scholar] [CrossRef]
- Nemeth, E.; Valore, E.; Ganz, T. Hepcidin, a putative mediator of anemia of inflammation, is a type II acute-phase protein. Blood 2003, 101, 2461–2463. [Google Scholar] [CrossRef]
- Malaponte, G.; Bevelacqua, V.; Mazzarino, M. IL-1b, TNF-a and IL-6 release from monocytes in haemodialysis patients in relation to dialytic age. Nephrol. Dial. Transplant. 2002, 17, 1964–1970. [Google Scholar] [CrossRef]
- Schütze, S.; Machleidt, T.; Krönke, M. Mechanisms of tumor necrosis factor action. Semin. Oncol. 1992, 19, 16–24. [Google Scholar]
- Reid, M.; Li, Y.-P. Tumor necrosis factor-α and muscle wasting: A cellular perspective. Respir. Res. 2001, 2, 269–272. [Google Scholar] [CrossRef]
- Bhatnagar, S.; Panguluri, S.K.; Gupta, S.K.; Dahiya, S.; Lundy, R.F.; Kumar, A. Tumor Necrosis Factor-α Regulates Distinct Molecular Pathways and Gene Networks in Cultured Skeletal Muscle Cells. PLoS ONE 2010, 5, e13262. [Google Scholar] [CrossRef]
- Li, Y.-P.; Reid, M. NF-kB mediates the protein loss induced by TNF-α in differentiated skeletal muscle myotubes. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2000, 279, R1165–R1170. [Google Scholar] [CrossRef]
- Aghagolzadeh, P.; Bachtler, M.; Bijarnia, R.; Jackson, C.; Smith, E.R.; Odermatt, A.; Radpour, R.; Pasch, A. Calcification of vascular smooth muscle cells is induced by secondary calciprotein particles and enhanced by tumor necrosis factor-α. Atherosclerosis 2016, 251, 404–414. [Google Scholar] [CrossRef]
- Tintut, Y.; Patel, J.; Parhami, F.; Demer, L.L. Tumor necrosis factor-alpha promotes in vitro calcification of vascular cells via the cAMP pathway. Circulation 2000, 102, 2636–2642. [Google Scholar] [CrossRef]
- Al-Aly, Z. Arterial calcification: A tumor necrosis factor-alpha mediated vascular Wnt-opathy. Transl. Res. 2008, 151, 233–239. [Google Scholar] [CrossRef]
- Kishimoto, T. Interleukin-6 and its Receptor in Autoimmunity. J. Immun. 1992, 5, 123–132. [Google Scholar] [CrossRef]
- Scheller, J.; Chalaris, A.; Schmidt-Arras, D.; Rose-John, S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. BBA - Mol. Cell Res. 2011, 1813, 878–888. [Google Scholar] [CrossRef] [Green Version]
- Mauer, J.; Denson, J.L.; Brüning, J.C. Versatile functions for IL-6 in metabolism and cancer. Trends Immunol. 2015, 36, 92–101. [Google Scholar] [CrossRef]
- Hénaut, L.; Massy, Z.A. New insights into the key role of interleukin 6 in vascular calcification of chronic kidney disease. Nephrol. Dial. Transplant. 2018, 33, 543–548. [Google Scholar] [CrossRef] [Green Version]
- Su, H.; Lei, C.-T.; Zhang, C. Interleukin-6 Signaling Pathway and Its Role in Kidney Disease: An Update. Front. Immunol. 2017, 8, 405. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Cheng, G.; Jin, R.; Dawn, B. Deletion of Interleukin-6 Attenuates Pressure Overload- Induced Left Ventricular Hypertrophy and Dysfunction. Circ. Res. 2016, 118, 1918–1929. [Google Scholar] [CrossRef]
- Erten, Y.; Tulmac, M.; Derici, U.; Pasaoglu, H.; Reis, K.A.; Bali, M.; Arinsoy, T.; Cengel, A.; Sindel, S. An Association Between Inflammatory State and Left Ventricular Hypertrophy in Hemodialysis Patients. Renal Fail. 2009, 27, 581–589. [Google Scholar] [CrossRef]
- Kozak, W.; Kluger, M.J.; Soszynski, D.; Conn, C.A.; Rudolph, K.; Leon, L.R.; Zheng, H. IL-6 and IL-1 beta in Fever: Studies Using Cytokine-Deficient (Knockout) Mice. Ann. N. Y. Acad. Sci. 1998, 856, 33–47. [Google Scholar] [CrossRef]
- Ren, K.; Torres, R. Role of interleukin-1β during pain and inflammation. Brain Res. Rev. 2009, 60, 57–64. [Google Scholar] [CrossRef]
- Wang, J.; Pantopoulos, K. Regulation of cellular iron metabolism. Biochem. J. 2011, 434, 365–381. [Google Scholar] [CrossRef] [Green Version]
- Hayes, W. Measurement of iron status in chronic kidney disease. Pediatric Nephrology 2019, 34, 605–613. [Google Scholar] [CrossRef]
- Andrews, N.C. Disorders of Iron Metabolism. N. Engl. J. Med. 2000, 342, 364. [Google Scholar]
- Ganz, T. Systemic iron homeostasis. Physiol. Rev. 2013, 93, 1721–1741. [Google Scholar] [CrossRef]
- Hentze, M.W.; Muckenthaler, M.U.; Galy, B.; Camaschella, C. Two to Tango: Regulation of Mammalian Iron Metabolism. Cell 2010, 142, 24–38. [Google Scholar] [CrossRef] [Green Version]
- Hentze, M.W.; Muckenthaler, M.; Andrews, N.C. Balancing Acts: Molecular Control of Review Mammalian Iron Metabolism. Cell 2004, 117, 1–13. [Google Scholar] [CrossRef]
- Ganz, T. Molecular control of iron transport. Journal of the American Society of Nephrology 2007, 18, 394–400. [Google Scholar] [CrossRef]
- Boas, F.; Forman, L.; Beutler, E. Phosphatidylserine exposure and red cell viability in red cell aging and in hemolytic anemia. Proc. Natl. Acad. Sci. USA 1998, 95, 3077–3081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Bruggen, R. Of macrophages and red blood cells; a complex love story. Front. Physiol. 2014, 5, 9. [Google Scholar]
- Schroit, A.J.; Tanaka, Y.; Madsen, J.; Fidler, I.J. The recognition of red blood cells by macrophages: Role of phosphatidylserine and possible implications of membrane phospholipid asymmetry. Biol. Cell 2012, 51, 227–238. [Google Scholar] [CrossRef]
- Schmidt, R. The Current Status of Anemia Management: KDIGO Guidelines. ASN Kidney News 2019, 11, 5. [Google Scholar]
- Astor, B.; Muntner, P.; Levin, A.; Eustace, J.; Coresh, J. Association of Kidney Function with Anemia: The Third National Health and Nutrition Examination Survey (1988–1994). JAMA. Intern. Med. 2002, 162, 1401–1408. [Google Scholar] [CrossRef] [PubMed]
- Stauffer, M.; Fan, T. Prevalence of Anemia in Chronic Kidney Disease in the United States. PLoS ONE 2014, 9, e84943. [Google Scholar] [CrossRef] [PubMed]
- KDOQI National Kidney Foundation: KDOQI Clinical Practice Guidelines and Clinical Practice Recommendations for Anemia in Chronic Kidney Disease. Am. J. Kidney Dis. 2006, 47, S11–S15.
- Babitt, J.L.; Lin, H.Y. Mechanisms of Anemia in CKD. J. Am. Soc. Nephrol. 2012, 23, 1631–1634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dziegala, M.; Josiak, K.; Kasztura, M.; Kobak, K.; Haehling, von, S.; Banasiak, W.; Anker, S.D.; Ponikowski, P.; Jankowska, E. Iron deficiency as energetic insult to skeletal muscle in chronic diseases. J. Cachexia Sarcopenia Muscle 2018, 9, 802–815. [Google Scholar] [CrossRef]
- Wish, J.B. Assessing iron status: Beyond serum ferritin and transferrin saturation. Clin. J. Am. Soc. Nephrol. 2006, 1, S4–S8. [Google Scholar] [CrossRef]
- Eschbach, J.W. The anemia of chronic renal failure: Pathophysiology and the effects of recombinant erythropoietin. Kidney Int. 1989, 35, 134–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caro, J.; Brown, S.; Miller, O.; Murray, T.; Erslev, A.J. Erythropoietin levels in uremic nephric and anephric patients. Journal of Lab. Clin. Med. 1979, 93, 449–458. [Google Scholar]
- Radtke, H.; Claussner, A.; Erbes, P.; Koch, K. Serum Erythropoietin Concentration in Chronic Renal Failure: Relationship to Degree of Anemia and Excretory Renal Function. Blood 1979, 54, 877–884. [Google Scholar] [PubMed]
- Erslev, A.J.; Besarab, A. Erythropoietin in the pathogenesis and treatment of the anemia of chronic renal failure. Kidney Int. 1997, 51, 622–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGonigle, R.J.S.; Husserl, F.; Wallin, J.D.; Fisher, J.W. Hemodialysis and continuous ambulatory peritoneal dialysis effects on erythropoiesis in renal failure. Kidney Int. 1984, 25, 430–436. [Google Scholar] [CrossRef] [Green Version]
- Wallner, S.; Kurnick, J.; Ward, H.; Alfrey, A. The Anemia of Chronic Renal Failure and Chronic Diseases: In Vitro Studies of Erythropoiesis. Blood 1976, 47, 561–569. [Google Scholar]
- Rege, A.B.; Ohno, Y.; Barona, J.; Fisher, J.W. Inhibitors of erythroid colony forming cells in sera of azotemic patients with anemia of renal disease. Clin. Dial. Transplant. Forum 1978, 8, 189–193. [Google Scholar]
- Wallner, S.F.; Kurnick, J.E.; Vautrin, R.; Ward, H.P. The effect of serum from uremic patients on erythropoietin. Am. J. Hematol. 1977, 3, 45–55. [Google Scholar] [CrossRef]
- Macdougall, I.C. Role of uremic toxins in exacerbating anemia in renal failure. Kidney Int. 2001, 59, S67–S72. [Google Scholar] [CrossRef] [Green Version]
- Loge, J.P.; Lange, R.D.; Moore, C.V. Characterization of the anemia associated with chronic renal insufficiency. Am. J. Med. 1958, 24, 4–18. [Google Scholar] [CrossRef]
- Koury, M.J.; Haase, V.H. Anemia in kidney disease: Harnessing hypoxia responses for therapy. Nat. Rev. Nephrol. 2015, 11, 394–410. [Google Scholar] [CrossRef]
- Lacombe, C.; Da Silva, J.L.; Bruneval, P.; Fournier, J.G.; Wendling, F.; Casadevall, N.; Camilleri, J.P.; Bariety, J.; Varet, B.; Tambourin, P. Peritubular cells are the site of erythropoietin synthesis in the murine hypoxic kidney. J. Clin. Invest. 1988, 81, 620–623. [Google Scholar] [CrossRef]
- Koury, S.; Bondurant, M.; Koury, M. Localization of Erythropoietin Synthesizing Cells in Murine Kidneys by In Situ Hybridization. Blood 1987, 71, 524–527. [Google Scholar]
- Schrier, R. Anemia in renal disease: Diseases of the Kidney and Urinary Tract; Philadelphia, Lippincott Williams and Wilkins: Philadelphia, PA, USA, 2007; pp. 2406–2430. [Google Scholar]
- Przybylowski, P.; Wasilewski, G.; Golabek, K.; Bachorzewska-Gajewska, H.; Dobrzycki, S.; Koc-Zorawska, E.; Malyszko, J. Absolute and Functional Iron Deficiency Is a Common Finding in Patients with Heart Failure and After Heart Transplantation. Transplant. Proc. 2016, 48, 173–176. [Google Scholar] [CrossRef]
- Ganz, T.; Nemeth, E. Iron Balance and the Role of Hepcidin in Chronic Kidney Disease. Semin. Nephrol. 2016, 36, 87–93. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, P.J. Regulation of Iron Metabolism by Hepcidin under Conditions of Inflammation. J. Biol. Chem. 2015, 290, 18975–18983. [Google Scholar] [CrossRef] [Green Version]
- Park, C.H.; Valore, E.V.; Waring, A.J.; Ganz, T. Hepcidin, a Urinary Antimicrobial Peptide Synthesized in the Liver. J. Biol. Chem. 2001, 276, 7806–7810. [Google Scholar] [CrossRef] [Green Version]
- Nemeth, E. Hepcidin Regulates Cellular Iron Efflux by Binding to Ferroportin and Inducing Its Internalization. Science 2004, 306, 2090–2093. [Google Scholar] [CrossRef] [Green Version]
- Petzer, V.; Theurl, I.; Weiss, G. Established and Emerging Concepts to Treat Imbalances of Iron Homeostasis in Inflammatory Diseases. Pharmaceuticals 2018, 11, 135. [Google Scholar] [CrossRef]
- Sebastiani, G.; Wilkinson, N.; Pantopoulos, K. Pharmacological Targeting of the Hepcidin/Ferroportin Axis. Front. Pharmacol. 2016, 7, 160. [Google Scholar] [CrossRef]
- Imel, E.A.; Peacock, M.; Gray, A.K.; Padgett, L.R.; Hui, S.L.; Econs, M.J. Iron Modifies Plasma FGF23 Differently in Autosomal Dominant Hypophosphatemic Rickets and Healthy Humans. J. Clin. Endocrinol. Metab. 2011, 96, 3541–3549. [Google Scholar] [CrossRef]
- Durham, B.; Joseph, F.; Bailey, L.; Fraser, W. The association of circulating ferritin with serum concentrations of fibroblast growth factor-23 measured by three commercial assays. Assoc. Clin. Biochem. 2007, 44, 463–466. [Google Scholar] [CrossRef] [Green Version]
- Tsai, M.-H.; Leu, J.-G.; Fang, Y.-W.; Liou, H.-H. High Fibroblast Growth Factor 23 Levels Associated with Low Hemoglobin Levels in Patients with Chronic Kidney Disease Stages 3 and 4. Medicine 2016, 95, e3049. [Google Scholar] [CrossRef]
- Eser, B.; Yayar, O.; Buyukbakkal, M.; Erdogan, B.; Ercan, Z.; Merhametsiz, O.; haspulat, A.; Oğuz, E.G.; Dogan, İ.; Canbakan, B.; et al. Fibroblast growth factor is associated to left ventricular mass index, anemia and low values of transferrin saturation. Nefrologia 2015, 35, 465–472. [Google Scholar] [CrossRef] [Green Version]
- Lewerin, C.; Ljunggren, Ö.; Nilsson-Ehle, H.; Karlsson, M.K.; Herlitz, H.; Lorentzon, M.; Ohlsson, C.; Mellström, D. Low serum iron is associated with high serum intact FGF23 in elderly men: The Swedish MrOS study. Bone 2017, 98, 1–8. [Google Scholar] [CrossRef]
- Coe, L.M.; Madathil, S.V.; Casu, C.; Lanske, B.; Rivella, S.; Sitara, D. FGF-23 is a negative regulator of prenatal and postnatal erythropoiesis. J. Biol. Chem. 2014, 289, 9795–9810. [Google Scholar] [CrossRef]
- Ilizarov, G.A. Osteogenesis and Hematopoiesis. In Transosseous Osteosynthesis; Springer Berlin Heidelberg: Berlin, Germany, 1992; pp. 1–2. [Google Scholar]
- Zhang, J.; Niu, C.; Ye, L.; Huang, H.; He, X.; Tong, W.G.; Ross, J.; Haug, J.; Johnson, T.; Feng, J.Q.; et al. Identification of the hematopoietic stem cell niche and control of the niche size. Nature 2003, 425, 836–841. [Google Scholar] [CrossRef]
- Zhu, J.; Garrett, R.; Jung, Y.; Emerson, S. Osteoblasts support B-lymphocyte commitment and differentiation from hematopoietic stem cells. Blood 2007, 109, 3706–3712. [Google Scholar] [CrossRef] [Green Version]
- Adams, G.B.; Chabner, K.T.; Alley, I.R.; Olson, D.P.; Szczepiorkowski, Z.M.; Poznansky, M.C.; Kos, C.H.; Pollak, M.R.; Brown, E.M.; Scadden, D.T. Stem cell engraftment at the endosteal niche is specified by the calcium-sensing receptor. Nature 2005, 439, 599–603. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Czaya, B.; Faul, C. The Role of Fibroblast Growth Factor 23 in Inflammation and Anemia. Int. J. Mol. Sci. 2019, 20, 4195. https://doi.org/10.3390/ijms20174195
Czaya B, Faul C. The Role of Fibroblast Growth Factor 23 in Inflammation and Anemia. International Journal of Molecular Sciences. 2019; 20(17):4195. https://doi.org/10.3390/ijms20174195
Chicago/Turabian StyleCzaya, Brian, and Christian Faul. 2019. "The Role of Fibroblast Growth Factor 23 in Inflammation and Anemia" International Journal of Molecular Sciences 20, no. 17: 4195. https://doi.org/10.3390/ijms20174195
APA StyleCzaya, B., & Faul, C. (2019). The Role of Fibroblast Growth Factor 23 in Inflammation and Anemia. International Journal of Molecular Sciences, 20(17), 4195. https://doi.org/10.3390/ijms20174195