Supplementation with Nicotinamide Riboside Reduces Brain Inflammation and Improves Cognitive Function in Diabetic Mice


Abstract
:1. Introduction
2. Results
2.1. Effects of NR on Body Weight, Brain Weight, Food Intake, and 2 h Oral Glucose Tolerance Test Area under the Curve (OGTT AUC)
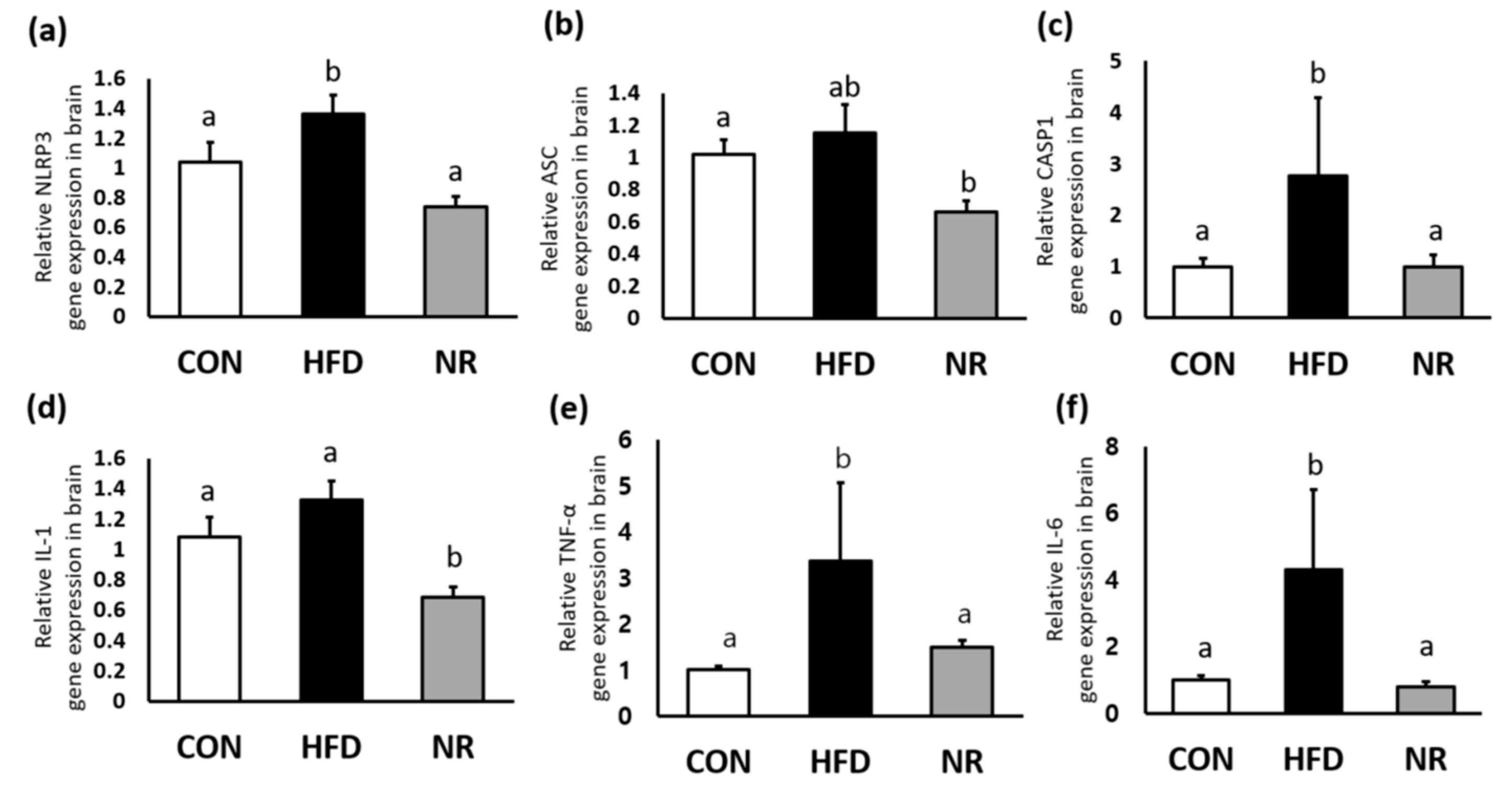
2.2. NR Reduces Levels of Inflammatory Markers in Whole Brains of Mice
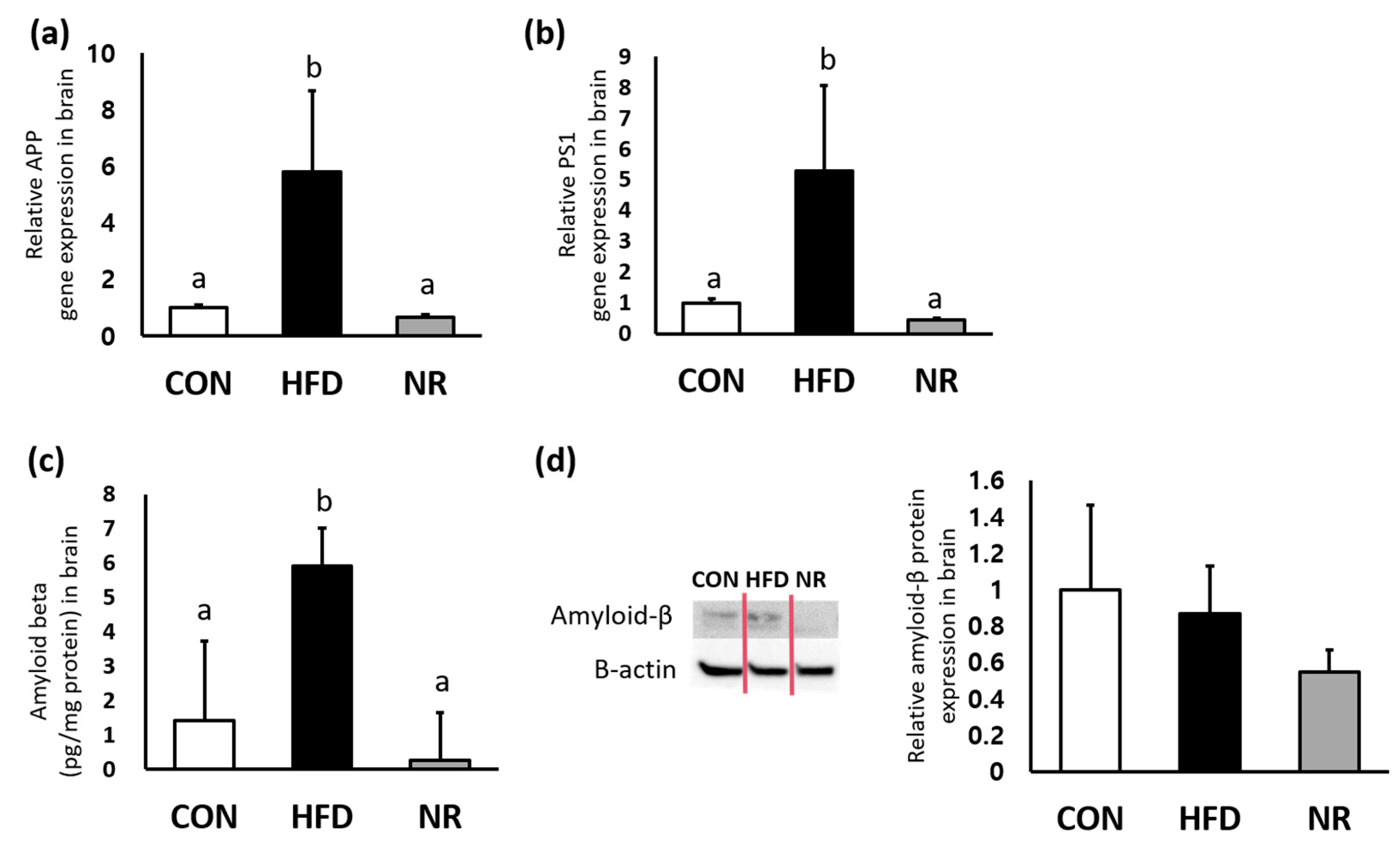
2.3. NR Supplementation Reduces Amyloid-β Concentrations in Whole Brains of Mice
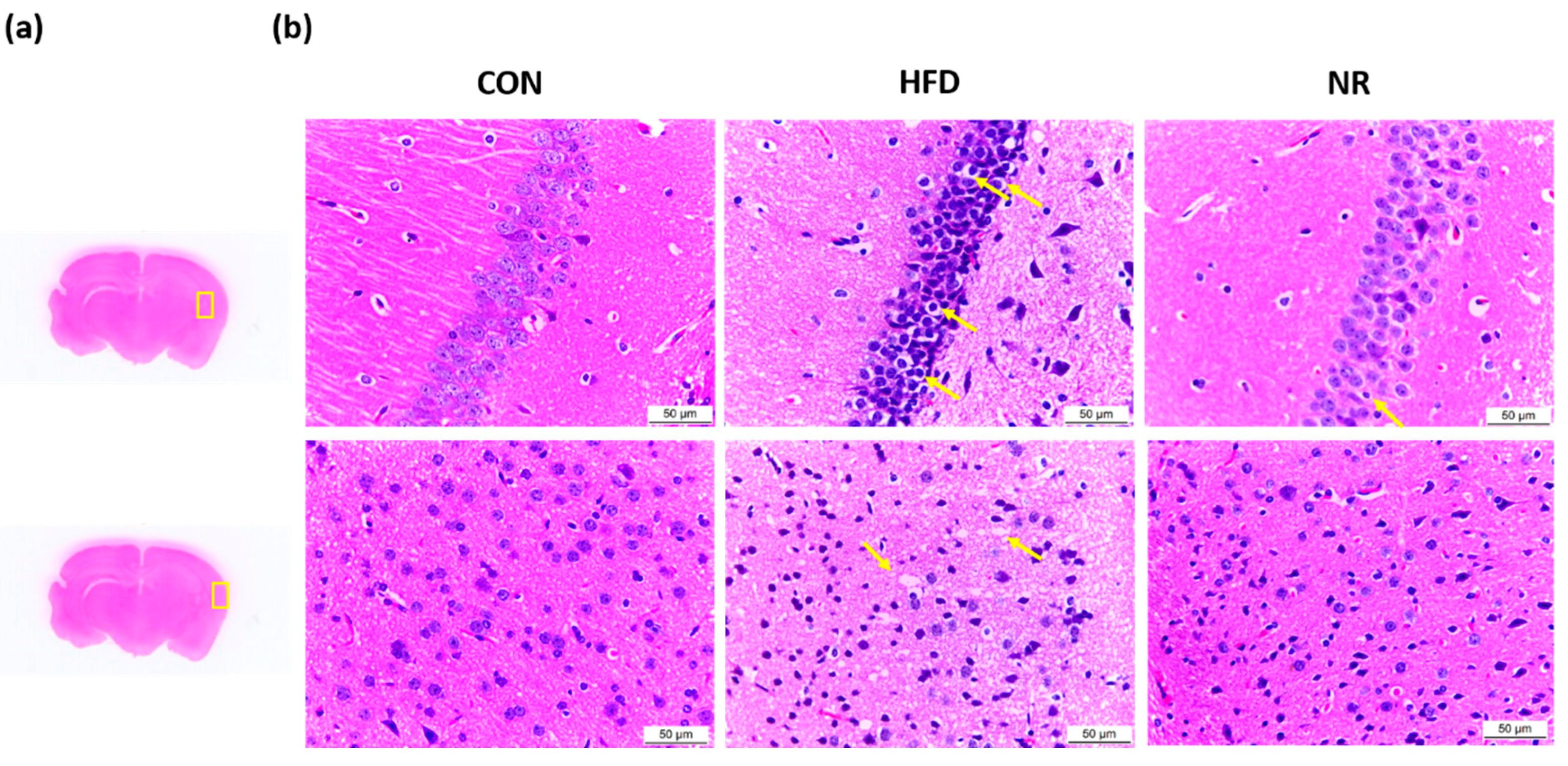
2.4. NR-Mediated Morphological Changes in Brain Tissues of HF Diet-Fed Mice
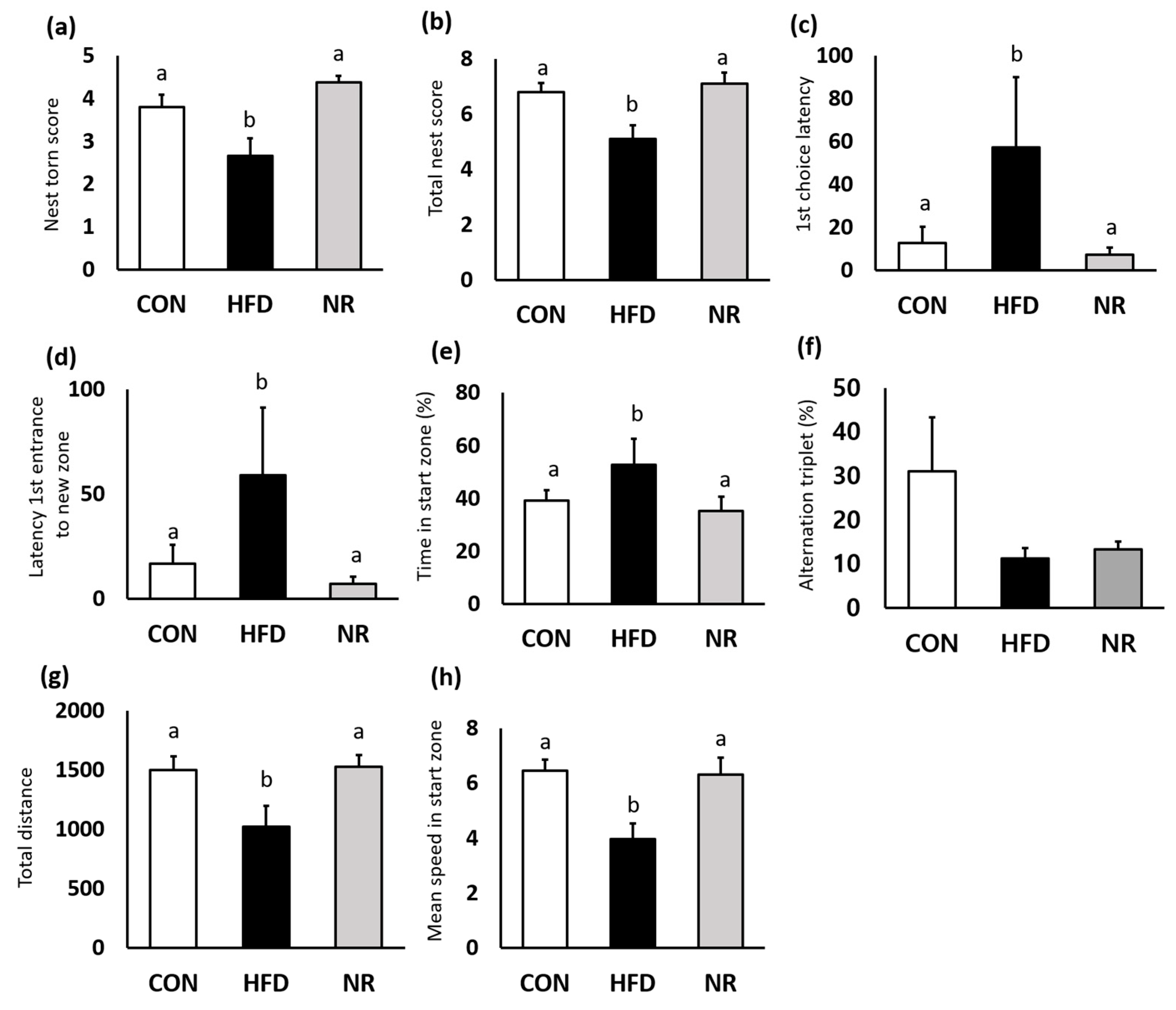
2.5. Nest Construction Test and Y-maze Test
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. OGTT
4.3. Nest Construction Test
4.4. Y-maze Test
4.5. Histological Study
4.6. RNA Extraction and Gene Expression Determination
4.7. Western Blot Analysis
4.8. ELISA
4.9. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| APP | amyloid-beta precursor protein |
| ASC | apoptosis-associated speck-like protein containing a caspase recruitment domain |
| NLRP3 | nucleotide binding and oligomerization domains-like receptor family: pyrin domain containing 3 |
| NR | nicotinamide riboside |
| PS1 | presenilin1 |
References
- Eid, A.; Mhatre, I.; Richardson, J.R. Gene-environment interactions in Alzheimer’s disease: A potential path to precision medicine. Pharmacol. Ther. 2019, 199, 173–187. [Google Scholar] [CrossRef] [PubMed]
- Naseri, N.N.; Wang, H.; Guo, J.; Sharma, M.; Luo, W. The complexity of tau in Alzheimer’s disease. Neurosci. Lett. 2019, 705, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Bostanciklioglu, M. An update on the interactions between Alzheimer’s disease, autophagy and inflammation. Gene 2019, 705, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Shou, Y.; Pan, J.; Du, Y.; Liu, C.; Wang, H. The relationship between cholesterol level and Alzheimer’s disease-associated APP proteolysis/Abeta metabolism. Nutr. Neurosci. 2019, 22, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Zoltowska, K.M.; Berezovska, O. Dynamic Nature of presenilin1/gamma-Secretase: Implication for Alzheimer’s Disease Pathogenesis. Mol. Neurobiol. 2018, 55, 2275–2284. [Google Scholar] [CrossRef]
- Bustos, V.; Pulina, M.V.; Kelahmetoglu, Y.; Sinha, S.C.; Gorelick, F.S.; Flajolet, M.; Greengard, P. Bidirectional regulation of Abeta levels by Presenilin 1. Proc. Natl. Acad. Sci. USA. 2017, 114, 7142–7147. [Google Scholar] [CrossRef] [PubMed]
- Andreeva, T.V.; Lukiw, W.J.; Rogaev, E.I. Biological Basis for Amyloidogenesis in Alzheimer’s Disease. Biochemistry 2017, 82, 122–139. [Google Scholar] [CrossRef]
- Alonso, A.; Zaidi, T.; Novak, M.; Grundke-Iqbal, I.; Iqbal, K. Hyperphosphorylation induces self-assembly of tau into tangles of paired helical filaments/straight filaments. Proc. Natl. Acad. Sci. USA 2001, 98, 6923–6928. [Google Scholar] [CrossRef]
- Kubis-Kubiak, A.M.; Rorbach-Dolata, A.; Piwowar, A. Crucial players in Alzheimer’s disease and diabetes mellitus: Friends or foes? Mech. Ageing Dev. 2019, 181, 7–21. [Google Scholar] [CrossRef]
- Chen, Y.; Yu, Q.; Gong, C.X. Molecular Connection Between Diabetes and Dementia. Adv. Exp. Med. Biol. 2019, 1128, 103–131. [Google Scholar]
- Ninomiya, T. Epidemiological Evidence of the Relationship Between Diabetes and Dementia. Adv. Exp. Med. Biol. 2019, 1128, 13–25. [Google Scholar] [PubMed]
- Hanyu, H. Diabetes-Related Dementia. Adv. Exp. Med. Biol. 2019, 1128, 147–160. [Google Scholar] [PubMed]
- Hatanaka, H.; Hanyu, H.; Fukasawa, R.; Sato, T.; Shimizu, S.; Sakurai, H. Peripheral oxidative stress markers in diabetes-related dementia. Geriatr. Gerontol. Int. 2016, 16, 1312–1318. [Google Scholar] [CrossRef] [PubMed]
- Tsugawa, A.; Ogawa, Y.; Takenoshita, N.; Kaneko, Y.; Hatanaka, H.; Jaime, E.; Fukasawa, R.; Hanyu, H. Decreased Muscle Strength and Quality in Diabetes-Related Dementia. Dement. Geriatr. Cogn. Dis. Extra 2017, 7, 454–462. [Google Scholar] [CrossRef] [PubMed]
- Crane, P.K.; Walker, R.; Hubbard, R.A.; Li, G.; Nathan, D.M.; Zheng, H.; Haneuse, S.; Craft, S.; Montine, T.J.; Kahn, S.E.; et al. Glucose levels and risk of dementia. N. Engl. J. Med. 2013, 369, 540–548. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Kim, C.H.; Jung, H.; Kim, E.; Song, H.T.; Lee, J.E. Agmatine ameliorates type 2 diabetes induced-Alzheimer’s disease-like alterations in high-fat diet-fed mice via reactivation of blunted insulin signalling. Neuropharmacology 2017, 113, 467–479. [Google Scholar] [CrossRef] [PubMed]
- Kullmann, S.; Heni, M.; Hallschmid, M.; Fritsche, A.; Preissl, H.; Häring, H.U. Brain Insulin Resistance at the Crossroads of Metabolic and Cognitive Disorders in Humans. Physiol. Rev. 2016, 96, 1169–1209. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.J.; Seo, H.I.; Cha, H.Y.; Yang, Y.J.; Kwon, S.H.; Yang, S.J. Diabetes and Alzheimer’s Disease: Mechanisms and Nutritional Aspects. Clin. Nutr. Res. 2018, 7, 229–240. [Google Scholar] [CrossRef]
- Su, M.; Naderi, K.; Samson, N.; Youssef, I.; Fulop, L.; Bozso, Z.; Laroche, S.; Delatour, B.; Davis, S. Mechanisms Associated with Type 2 Diabetes as a Risk Factor for Alzheimer-Related Pathology. Mol. Neurobiol. 2019, 56, 5815–5834. [Google Scholar] [CrossRef]
- Ragy, M.M.; Kamal, N.N. Linking senile dementia to type 2 diabetes: Role of oxidative stress markers, C-reactive protein and tumor necrosis factor-alpha. Neurol. Res. 2017, 39, 587–595. [Google Scholar] [CrossRef]
- Ahmad, W.; Ijaz, B.; Shabbiri, K.; Ahmed, F.; Rehman, S. Oxidative toxicity in diabetes and Alzheimer’s disease: Mechanisms behind ROS/ RNS generation. J. Biomed. Sci. 2017, 24, 76. [Google Scholar] [CrossRef] [PubMed]
- Nichols, M.R.; St-Pierre, M.K.; Wendeln, A.C.; Makoni, N.J.; Gouwens, L.K.; Garrad, E.C.; Sohrabi, M.; Neher, J.J.; Tremblay, M.E.; Combs, C.K. Inflammatory mechanisms in neurodegeneration. J. Neurochem. 2019, 149, 562–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Combs, C.K.; Karlo, J.C.; Kao, S.C.; Landreth, G.E. beta-Amyloid stimulation of microglia and monocytes results in TNFalpha-dependent expression of inducible nitric oxide synthase and neuronal apoptosis. J. Neurosci. 2001, 21, 1179–1188. [Google Scholar] [CrossRef] [PubMed]
- Yates, S.L.; Burgess, L.H.; Kocsis-Angle, J.; Antal, J.M.; Dority, M.D.; Embury, P.B.; Piotrkowski, A.M.; Brunden, K.R. Amyloid beta and amylin fibrils induce increases in proinflammatory cytokine and chemokine production by THP-1 cells and murine microglia. J. Neurochem. 2000, 74, 1017–1025. [Google Scholar] [CrossRef] [PubMed]
- Brunden, K.R.; Kocsis-Angle, J.; Embury, P.; Yates, S.L. Abeta-Induced Proinflammatory Cytokine Release from Differentiated Human THP-1 Monocytes. Methods Mol. Med. 2000, 32, 101–112. [Google Scholar] [PubMed]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef]
- Halle, A.; Hornung, V.; Petzold, G.C.; Stewart, C.R.; Monks, B.G.; Reinheckel, T.; Fitzgerald, K.A.; Latz, E.; Moore, K.J.; Golenbock, D.T. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat. Immunol. 2008, 9, 857–865. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Baglietto-Vargas, D.; Shi, J.; Yaeger, D.M.; Ager, R.; LaFerla, F.M. Diabetes and Alzheimer’s disease crosstalk. Neurosci. Biobehav. Rev. 2016, 64, 272–287. [Google Scholar] [CrossRef]
- Trammell, S.A.; Yu, L.; Redpath, P.; Migaud, M.E.; Brenner, C. Nicotinamide Riboside Is a Major NAD+ Precursor Vitamin in Cow Milk. J. Nutr. 2016, 146, 957–963. [Google Scholar] [CrossRef] [Green Version]
- Vaur, P.; Brugg, B.; Mericskay, M.; Li, Z.; Schmidt, M.S.; Vivien, D.; Orset, C.; Jacotot, E.; Brenner, C.; Duplus, E. Nicotinamide riboside, a form of vitamin B3, protects against excitotoxicity-induced axonal degeneration. FASEB J. 2017, 31, 5440–5452. [Google Scholar] [CrossRef]
- Gong, B.; Pan, Y.; Vempati, P.; Zhao, W.; Knable, L.; Ho, L.; Wang, J.; Sastre, M.; Ono, K.; Sauve, A.A.; et al. Nicotinamide riboside restores cognition through an upregulation of proliferator-activated receptor-gamma coactivator 1alpha regulated beta-secretase 1 degradation and mitochondrial gene expression in Alzheimer’s mouse models. Neurobiol. Aging 2013, 34, 1581–1588. [Google Scholar] [CrossRef]
- Trammell, S.A.; Weidemann, B.J.; Chadda, A.; Yorek, M.S.; Holmes, A.; Coppey, L.J.; Obrosov, A.; Kardon, R.H.; Yorek, M.A.; Brenner, C. Nicotinamide Riboside Opposes Type 2 Diabetes and Neuropathy in Mice. Sci. Rep. 2016, 6, 26933. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Li, H.; Zhang, L.; Li, J.; Wang, R.; Wang, M. Effects of troxerutin on cognitive deficits and glutamate cysteine ligase subunits in the hippocampus of streptozotocin-induced type 1 diabetes mellitus rats. Brain Res. 2017, 1657, 355–360. [Google Scholar] [CrossRef]
- Lin, W.T.; Chen, R.C.; Lu, W.W.; Liu, S.H.; Yang, F.Y. Protective effects of low-intensity pulsed ultrasound on aluminum-induced cerebral damage in Alzheimer’s disease rat model. Sci. Rep. 2015, 5, 9671. [Google Scholar] [CrossRef]
- Jiang, L.Y.; Tang, S.S.; Wang, X.Y.; Liu, L.P.; Long, Y.; Hu, M.; Liao, M.X.; Ding, Q.L.; Hu, W.; Li, J.C.; et al. PPARgamma agonist pioglitazone reverses memory impairment and biochemical changes in a mouse model of type 2 diabetes mellitus. CNS Neurosci. Ther. 2012, 18, 659–666. [Google Scholar] [CrossRef]
- Hong, H.; Liu, L.P.; Liao, J.M.; Wang, T.S.; Ye, F.Y.; Wu, J.; Wang, Y.Y.; Wang, Y.; Li, Y.Q.; Long, Y.; et al. Downregulation of LRP1 [correction of LPR1] at the blood-brain barrier in streptozotocin-induced diabetic mice. Neuropharmacology 2009, 56, 1054–1059. [Google Scholar] [CrossRef]
- Sims-Robinson, C.; Kim, B.; Rosko, A.; Feldman, E.L. How does diabetes accelerate Alzheimer disease pathology? Nat. Rev. Neurol. 2010, 6, 551–559. [Google Scholar] [CrossRef] [Green Version]
- Roriz-Filho, J.S.; Sá-Roriz, T.M.; Rosset, I.; Camozzato, A.L.; Santos, A.C.; Chaves, M.L.; Moriguti, J.C.; Roriz-Cruz, M. (Pre)diabetes, brain aging, and cognition. Biochim. Biophys. Acta 2009, 1792, 432–443. [Google Scholar] [CrossRef] [Green Version]
- Querfurth, H.W.; LaFerla, F.M. Alzheimer’s disease. N. Engl. J. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef]
- Dollerup, O.L.; Christensen, B.; Svart, M.; Schmidt, M.S.; Sulek, K.; Ringgaard, S.; Stødkilde-Jørgensen, H.; Møller, N.; Brenner, C.; Treebak, J.T.; et al. A randomized placebo-controlled clinical trial of nicotinamide riboside in obese men: Safety, insulin-sensitivity, and lipid-mobilizing effects. Am. J. Clin. Nutr. 2018, 108, 343–353. [Google Scholar] [CrossRef]
- Akiyama, H.; Barger, S.; Barnum, S.; Bradt, B.; Bauer, J.; Cole, G.M.; Cooper, N.R.; Eikelenboom, P.; Emmerling, M.; Fiebich, B.L.; et al. Inflammation and Alzheimer’s disease. Neurobiol. Aging 2000, 21, 383–421. [Google Scholar] [CrossRef]
- Marioni, R.E.; Strachan, M.W.; Reynolds, R.M.; Lowe, G.D.; Mitchell, R.J.; Fowkes, F.G.; Frier, B.M.; Lee, A.J.; Butcher, I.; Rumley, A.; et al. Association between raised inflammatory markers and cognitive decline in elderly people with type 2 diabetes: The Edinburgh Type 2 Diabetes Study. Diabetes 2010, 59, 710–713. [Google Scholar] [CrossRef]
- Miao, Y.; He, T.; Zhu, Y.; Li, W.; Wang, B.; Zhong, Y. Activation of Hippocampal CREB by Rolipram Partially Recovers Balance Between TNF-alpha and IL-10 Levels and Improves Cognitive Deficits in Diabetic Rats. Cell Mol. Neurobiol. 2015, 35, 1157–1164. [Google Scholar] [CrossRef]
- Dutheil, S.; Ota, K.T.; Wohleb, E.S.; Rasmussen, K.; Duman, R.S. High-Fat Diet Induced Anxiety and Anhedonia: Impact on Brain Homeostasis and Inflammation. Neuropsychopharmacology 2016, 41, 1874–1887. [Google Scholar] [CrossRef]
- Kothari, V.; Luo, Y.; Tornabene, T.; O’Neill, A.M.; Greene, M.W.; Geetha, T.; Babu, J.R. High fat diet induces brain insulin resistance and cognitive impairment in mice. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 499–508. [Google Scholar] [CrossRef]
- Glass, C.K.; Saijo, K.; Winner, B.; Marchetto, M.C.; Gage, F.H. Mechanisms underlying inflammation in neurodegeneration. Cell 2010, 140, 918–934. [Google Scholar] [CrossRef]
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef]
- Vandanmagsar, B.; Youm, Y.H.; Ravussin, A.; Galgani, J.E.; Stadler, K.; Mynatt, R.L.; Ravussin, E.; Stephens, J.M.; Dixit, V.D. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med. 2011, 17, 179–188. [Google Scholar] [CrossRef]
- Morbelli, S.; Piccardo, A.; Villavecchia, G.; Dessi, B.; Brugnolo, A.; Piccini, A.; Caroli, A.; Frisoni, G.; Rodriguez, G.; Nobili, F. Mapping brain morphological and functional conversion patterns in amnestic MCI: A voxel-based MRI and FDG-PET study. Eur. J. Nucl. Med. Mol. Imaging 2010, 37, 36–45. [Google Scholar] [CrossRef]
- Feng, Y.; Chu, A.; Luo, Q.; Wu, M.; Shi, X.; Chen, Y. The Protective Effect of Astaxanthin on Cognitive Function via Inhibition of Oxidative Stress and Inflammation in the Brains of Chronic T2DM Rats. Front. Pharmacol. 2018, 9, 748. [Google Scholar] [CrossRef]
- Ghasemi, A.; Khalifi, S.; Jedi, S. Streptozotocin-nicotinamide-induced rat model of type 2 diabetes. Acta Physiol. Hung. 2014, 101, 408–420. [Google Scholar] [CrossRef]
- Deacon, R.M. Assessing nest building in mice. Nat. Protoc. 2006, 1, 1117–1119. [Google Scholar] [CrossRef]
- Kraeuter, A.K.; Guest, P.C.; Sarnyai, Z. The Nest Building Test in Mice for Assessment of General Well-Being. Methods Mol. Biol. 2019, 1916, 87–91. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CON | HFD | NR | |
|---|---|---|---|
| BW (g) | 44.69 ± 1.31 | 45.90 ± 2.84 | 44.05 ± 2.61 |
| Brain weight (g) | 0.55 ± 0.01 ab | 0.52 ± 0.01 a | 0.59 ± 0.01 b |
| Brain weight (% BW) | 1.27 ± 0.05 ab | 1.14 ± 0.05 a | 1.36 ± 0.10 b |
| Food intake (g/day) | 3.82 ± 0.15 a | 3.16 ± 0.11 ab | 2.28 ± 0.40 b |
| Fasting blood glucose (mg/dL) | 123 ± 6 a | 389 ± 41 b | 383 ± 50 b |
| 2 h OGTT AUC | 35099 ± 2538 a | 65091 ± 2619 b | 68648 ± 861 b |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, H.J.; Yang, S.J. Supplementation with Nicotinamide Riboside Reduces Brain Inflammation and Improves Cognitive Function in Diabetic Mice. Int. J. Mol. Sci. 2019, 20, 4196. https://doi.org/10.3390/ijms20174196
Lee HJ, Yang SJ. Supplementation with Nicotinamide Riboside Reduces Brain Inflammation and Improves Cognitive Function in Diabetic Mice. International Journal of Molecular Sciences. 2019; 20(17):4196. https://doi.org/10.3390/ijms20174196
Chicago/Turabian StyleLee, Hee Jae, and Soo Jin Yang. 2019. "Supplementation with Nicotinamide Riboside Reduces Brain Inflammation and Improves Cognitive Function in Diabetic Mice" International Journal of Molecular Sciences 20, no. 17: 4196. https://doi.org/10.3390/ijms20174196
APA StyleLee, H. J., & Yang, S. J. (2019). Supplementation with Nicotinamide Riboside Reduces Brain Inflammation and Improves Cognitive Function in Diabetic Mice. International Journal of Molecular Sciences, 20(17), 4196. https://doi.org/10.3390/ijms20174196