Fine Tuning: Effects of Post-Translational Modification on Hsp70 Chaperones

Abstract
:1. Post-Translational Modifications
2. Hsp70 Structure and Allosteric Regulation
3. Hsp70 Co-Chaperones
4. Modulating Chaperone Activity through Post-Translational Modification
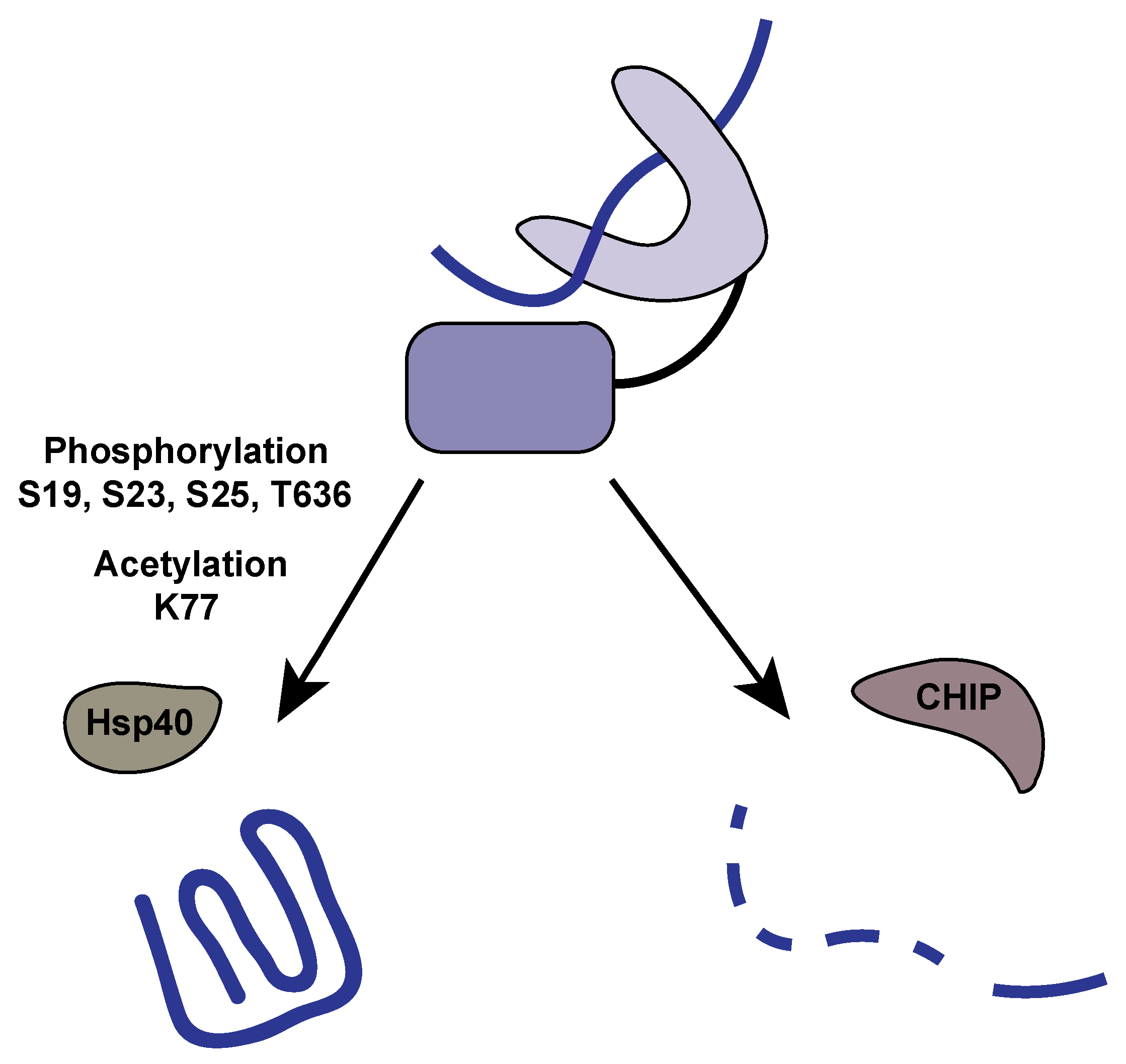
4.1. Phosphorylation
4.2. Acetylation
4.3. Other Identified Post-Translational Modifications
5. Influencing Chaperone Activity Via Pharmaceuticals
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Hsp | Heat Shock Protein |
| PTM | Post-translational Modification |
| CHIP | C-terminal Hsp70 Interacting Protein |
| HOP | Hsp70-Hsp90 Organizing Protein |
References
- Tripodi, F.; Nicastro, R.; Reghellin, V.; Coccetti, P. Post-translational modifications on yeast carbon metabolism: Regulatory mechanisms beyond transcriptional control. Biochim. Biophys. Acta Gen. Subj. 2015, 1850, 620–627. [Google Scholar] [CrossRef] [PubMed]
- Loyet, K.M.; Stults, J.T.; Arnott, D. Mass Spectrometric Contributions to the Practice of Phosphorylation Site Mapping through 2003: A Literature Review. Mol. Cell. Proteom. 2005, 4, 235–245. [Google Scholar] [CrossRef] [PubMed]
- Paik, W.K.; Paik, D.C.; Kim, S. Historical review: The field of protein methylation. Trends Biochem. Sci. 2007, 32, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Kouzarides, T. Acetylation: A regulatory modification to rival phosphorylation? EMBO J. 2000, 19, 1176–1179. [Google Scholar] [CrossRef] [PubMed]
- Monticelli, M.; Ferro, T.; Jaeken, J.; dos Reis Ferreira, V.; Videira, P.A. Immunological aspects of congenital disorders of glycosylation (CDG): A review. J. Inherit. Metab. Dis. 2016, 39, 765–780. [Google Scholar] [CrossRef] [PubMed]
- Ikezawa, H. Glycosylphosphatidylinositol (GPI)-Anchored Proteins. Biol. Pharm. Bull. 2002, 25, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Hershko, A.; Ciechanover, A. THE UBIQUITIN SYSTEM. Annu. Rev. Biochem. 2003, 67, 425–479. [Google Scholar] [CrossRef]
- Ben-Zvi, A.P.; Goloubinoff, P. Review: Mechanisms of disaggregation and refolding of stable protein aggregates by molecular chaperones. J. Struct. Biol. 2001, 135, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Laufen, T.; Mayer, M.P.; Beisel, C.; Klostermeier, D.; Mogk, A.; Reinstein, J.; Bukau, B. Mechanism of regulation of Hsp70 chaperones by DnaJ cochaperones. Proc. Natl. Acad. Sci. USA 1999, 96, 5452–5457. [Google Scholar] [CrossRef] [Green Version]
- Umehara, K.; Hoshikawa, M.; Tochio, N.; Tate, S.-I. Substrate Binding Switches the Conformation at the Lynchpin Site in the Substrate-Binding Domain of Human Hsp70 to Enable Allosteric Interdomain Communication. Molecules 2018, 23, 528. [Google Scholar] [CrossRef]
- Rosenzweig, R.; Nillegoda, N.B.; Mayer, M.P.; Bukau, B. The Hsp70 chaperone network. Nat. Rev. Mol. Cell Biol. 2019, 353, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Kellner, R.; Hofmann, H.; Barducci, A.; Wunderlich, B.; Nettels, D.; Schuler, B. Single-molecule spectroscopy reveals chaperone-mediated expansion of substrate protein. Proc. Natl. Acad. Sci. USA 2014, 111, 13355–13360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taneva, S.G.; Moro, F.; Velázquez-Campoy, A.; Muga, A. Energetics of Nucleotide-Induced DnaK Conformational States. Biochemistry 2010, 49, 1338–1345. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Hendrickson, W.A. Insights into Hsp70 Chaperone Activity from a Crystal Structure of the Yeast Hsp110 Sse1. Cell 2007, 131, 106–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayer, M.P.; Kityk, R. Insights into the molecular mechanism of allostery in Hsp70s. Front. Mol. Biosci. 2015, 2, 4535. [Google Scholar] [CrossRef] [PubMed]
- Sharma, D.; Martineau, C.N.; Le Dall, M.-T.; Reidy, M.; Masison, D.C.; Kabani, M. Function of SSA Subfamily of Hsp70 Within and Across Species Varies Widely in Complementing Saccharomyces cerevisiae Cell Growth and Prion Propagation. PLoS ONE 2009, 4, e6644. [Google Scholar] [CrossRef] [PubMed]
- Gong, W.; Hu, W.; Xu, L.; Wu, H.; Wu, S.; Zhang, H.; Wang, J.; Jones, G.W.; Perrett, S. The C-terminal GGAP motif of Hsp70 mediates substrate recognition and stress response in yeast. J. Biol. Chem. 2018, 293, 17663–17675. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Leu, J.I.-J.; Murphy, M.E.; George, D.L.; Marmorstein, R. Crystal Structure of the Stress-Inducible Human Heat Shock Protein 70 Substrate-Binding Domain in Complex with Peptide Substrate. PLoS ONE 2014, 9, e103518. [Google Scholar] [CrossRef]
- Rüdiger, S.; Germeroth, L.; Schneider-Mergener, J.; Bukau, B. Substrate specificity of the DnaK chaperone determined by screening cellulose-bound peptide libraries. EMBO J. 1997, 16, 1501–1507. [Google Scholar] [CrossRef] [Green Version]
- Ngosuwan, J.; Wang, N.M.; Fung, K.L.; Chirico, W.J. Roles of cytosolic Hsp70 and Hsp40 molecular chaperones in post-translational translocation of presecretory proteins into the endoplasmic reticulum. J. Biol. Chem. 2003, 278, 7034–7042. [Google Scholar] [CrossRef]
- Abrams, J.L.; Verghese, J.; Gibney, P.A.; Morano, K.A. Hierarchical functional specificity of cytosolic heat shock protein 70 (Hsp70) nucleotide exchange factors in yeast. J. Biol. Chem. 2014, 289, 13155–13167. [Google Scholar] [CrossRef] [PubMed]
- Shorter, J.; Lindquist, S. Hsp104, Hsp70 and Hsp40 interplay regulates formation, growth and elimination of Sup35 prions. EMBO J. 2008, 27, 2712–2724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Needham, P.G.; Patel, H.J.; Chiosis, G.; Thibodeau, P.H.; Brodsky, J.L. Mutations in the Yeast Hsp70, Ssa1, at P417 Alter ATP Cycling, Interdomain Coupling, and Specific Chaperone Functions. J. Mol. Biol. 2015, 427, 2948–2965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabani, M.; Martineau, C.N. Multiple Hsp70 Isoforms in the Eukaryotic Cytosol: Mere Redundancy or Functional Specificity? CG 2008, 9, 338–348. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.S.; Golding, G.B. Evolution of HSP70 gene and its implications regarding relationships between archaebacteria, eubacteria, and eukaryotes. J. Mol. Evol. 1993, 37, 573–582. [Google Scholar] [CrossRef]
- Werner-Washburne, M.; Stone, D.E.; Craig, E.A. Complex interactions among members of an essential subfamily of hsp70 genes in Saccharomyces cerevisiae. Mol. Cell. Biol. 1987, 7, 2568–2577. [Google Scholar] [CrossRef]
- Boorstein, W.R.; Ziegelhoffer, T.; Craig, E.A. Molecular evolution of the HSP70 multigene family. J. Mol. Evol. 1994, 38, 1–17. [Google Scholar] [CrossRef]
- Tavaria, M.; Gabriele, T.; Kola, I.; Anderson, R.L. A hitchhiker’s guide to the human Hsp70 family. Cell Stress Chaperones 1994, 1, 23–28. [Google Scholar] [CrossRef]
- Horton, L.E.; James, P.; Craig, E.A.; Hensold, J.O. The yeast hsp70 homologue Ssa is required for translation and interacts with Sis1 and Pab1 on translating ribosomes. J. Biol. Chem. 2001, 276, 14426–14433. [Google Scholar] [CrossRef]
- Boorstein, W.R.; Craig, E.A. Structure and regulation of the SSA4 HSP70 gene of Saccharomyces cerevisiae. J. Biol. Chem. 1990, 265, 18912–18921. [Google Scholar]
- Gautschi, M.; Lilie, H.; Fünfschilling, U.; Mun, A.; Ross, S.; Lithgow, T.; Rücknagel, P.; Rospert, S. RAC, a stable ribosome-associated complex in yeast formed by the DnaK-DnaJ homologs Ssz1p and zuotin. Proc. Natl. Acad. Sci. USA 2001, 98, 3762–3767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, K.D.; Wegrzyn, R.D.; Chernova, T.A.; Müller, S.; Newnam, G.P.; Winslett, P.A.; Wittich, K.B.; Wilkinson, K.D.; Chernoff, Y.O. Hsp70 Chaperones as Modulators of Prion Life Cycle. Genetics 2005, 169, 1227–1242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bush, G.L.; Meyer, D.I. The refolding activity of the yeast heat shock proteins Ssa1 and Ssa2 defines their role in protein translocation. J. Cell Biol. 1996, 135, 1229–1237. [Google Scholar] [CrossRef] [PubMed]
- Nelson, R.J.; Ziegelhoffer, T.; Nicolet, C.; Werner-Washburne, M.; Craig, E.A. The translation machinery and 70 kd heat shock protein cooperate in protein synthesis. Cell 1992, 71, 97–105. [Google Scholar] [CrossRef]
- Lopez, N.; Halladay, J.; Walter, W.; Craig, E.A. SSB, Encoding a Ribosome-Associated Chaperone, Is Coordinately Regulated with Ribosomal Protein Genes. J. Bacteriol. 1999, 181, 3136–3143. [Google Scholar] [PubMed]
- Pfund, C.; Hoyo, N.L.; Ziegelhoffer, T.; Schilke, B.A.; Buesa, P.L.; Walter, W.A.; Wiedmann, M.; Craig, E.A. The molecular chaperone Ssb from Saccharomyces cerevisiae is a component of the ribosome–nascent chain complex. EMBO J. 1998, 17, 3981–3989. [Google Scholar] [CrossRef] [PubMed]
- James, P.; Pfund, C.; Craig, E.A. Functional specificity among Hsp70 molecular chaperones. Science 1997, 275, 387–389. [Google Scholar] [CrossRef] [PubMed]
- Brodsky, J.L.; Hamamoto, S.; Feldheim, D.; Schekman, R. Reconstitution of protein translocation from solubilized yeast membranes reveals topologically distinct roles for BiP and cytosolic Hsc70. J. Cell Biol. 1993, 120, 95–102. [Google Scholar] [CrossRef]
- Brodsky, J.L.; Bäuerle, M.; Horst, M.; McClellan, A.J. Mitochondrial Hsp70 cannot replace BiP in driving protein translocation into the yeast endoplasmic reticulum. FEBS Lett. 1998, 435, 183–186. [Google Scholar] [CrossRef]
- Greene, M.K.; Maskos, K.; Landry, S.J. Role of the J-domain in the cooperation of Hsp40 with Hsp70. Proc. Natl. Acad. Sci. USA 1998, 95, 6108–6113. [Google Scholar] [CrossRef] [Green Version]
- Lu, Z.; Cyr, D.M. Protein folding activity of Hsp70 is modified differentially by the hsp40 co-chaperones Sis1 and Ydj1. J. Biol. Chem. 1998, 273, 27824–27830. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.-Y.; Ren, H.-Y.; Lee, P.; Caplan, A.J.; Cyr, D.M. The type I Hsp40 zinc finger-like region is required for Hsp70 to capture non-native polypeptides from Ydj1. J. Biol. Chem. 2005, 280, 695–702. [Google Scholar] [CrossRef] [PubMed]
- Cyr, D.M.; Lu, X.; Douglas, M.G. Regulation of Hsp70 function by a eukaryotic DnaJ homolog. J. Biol. Chem. 1992, 267, 20927–20931. [Google Scholar] [PubMed]
- Kityk, R.; Kopp, J.; Mayer, M.P. Molecular Mechanism of J-Domain-Triggered ATP Hydrolysis by Hsp70 Chaperones. Mol. Cell 2018, 69, 227–237.e4. [Google Scholar] [CrossRef] [PubMed]
- Cyr, D.M.; Langer, T.; Douglas, M.G. DnaJ-like proteins: Molecular chaperones and specific regulators of Hsp70. Trends Biochem. Sci. 1994, 19, 176–181. [Google Scholar] [CrossRef]
- Lu, Z.; Cyr, D.M. The conserved carboxyl terminus and zinc finger-like domain of the co-chaperone Ydj1 assist Hsp70 in protein folding. J. Biol. Chem. 1998, 273, 5970–5978. [Google Scholar] [CrossRef] [PubMed]
- Cheetham, M.E.; Jackson, A.P.; Anderton, B.H. Regulation of 70-kDa Heat-Shock-Protein ATPase Activity and Substrate Binding by Human DnaJ-Like Proteins, HSJ1a and HSJ1b. Eur. J. Biochem. 1994, 226, 99–107. [Google Scholar] [CrossRef]
- Cyr, D.M.; Douglas, M.G. Differential regulation of Hsp70 subfamilies by the eukaryotic DnaJ homologue YDJ1. J. Biol. Chem. 1994, 269, 9798–9804. [Google Scholar]
- Wall, D.; Zylicz, M.; Georgopoulos, C. The conserved G/F motif of the DnaJ chaperone is necessary for the activation of the substrate binding properties of the DnaK chaperone. J. Biol. Chem. 1995, 270, 2139–2144. [Google Scholar] [CrossRef]
- Hu, J.; Wu, Y.; Li, J.; Qian, X.; Fu, Z.; Sha, B. The crystal structure of the putative peptide-binding fragment from the human Hsp40 protein Hdj1. BMC Struct. Biol. 2008, 8, 3. [Google Scholar] [CrossRef]
- Misselwitz, B.; Staeck, O.; Rapoport, T.A. J Proteins Catalytically Activate Hsp70 Molecules to Trap a Wide Range of Peptide Sequences. Mol. Cell 1998, 2, 593–603. [Google Scholar] [CrossRef]
- Sha, B.; Lee, S.; Cyr, D.M. The crystal structure of the peptide-binding fragment from the yeast Hsp40 protein Sis1. Structure 2000, 8, 799–807. [Google Scholar] [CrossRef] [Green Version]
- Silver, P.A.; Way, J.C. Eukaryotic DnaJ homologs and the specificity of Hsp70 activity. Cell 1993, 74, 5–6. [Google Scholar] [CrossRef]
- Li, J.; Qian, X.; Sha, B. The crystal structure of the yeast Hsp40 Ydj1 complexed with its peptide substrate. Structure 2003, 11, 1475–1483. [Google Scholar] [CrossRef] [PubMed]
- Caplan, A.J.; Tsai, J.; Casey, P.J.; Douglas, M.G. Farnesylation of YDJ1p is required for function at elevated growth temperatures in Saccharomyces cerevisiae. J. Biol. Chem. 1992, 267, 18890–18895. [Google Scholar] [PubMed]
- Fan, C.-Y.; Lee, S.; Ren, H.-Y.; Cyr, D.M. Exchangeable Chaperone Modules Contribute to Specification of Type I and Type II Hsp40 Cellular Function. Mol. Biol. Cell 2004, 15, 761–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, I.; Ghosh, M.K. The E3 Ligase CHIP: Insights into Its Structure and Regulation. BioMed Res. Int. 2014, 2014, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beltrao, P.; Albanèse, V.; Kenner, L.R.; Swaney, D.L.; Burlingame, A.; Villén, J.; Lim, W.A.; Fraser, J.S.; Frydman, J.; Krogan, N.J. Systematic Functional Prioritization of Protein Posttranslational Modifications. Cell 2012, 150, 413–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Truman, A.W.; Kristjansdottir, K.; Wolfgeher, D.; Hasin, N.; Polier, S.; Zhang, H.; Perrett, S.; Prodromou, C.; Jones, G.W.; Kron, S.J. CDK-Dependent Hsp70 Phosphorylation Controls G1 Cyclin Abundance and Cell-Cycle Progression. Cell 2012, 151, 1308–1318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.-J.; Lai, K.-C.; Kuo, H.-H.; Chow, L.-P.; Yih, L.-H.; Lee, T.-C. HSP70 colocalizes with PLK1 at the centrosome and disturbs spindle dynamics in cells arrested in mitosis by arsenic trioxide. Arch. Toxicol. 2014, 88, 1711–1723. [Google Scholar] [CrossRef] [PubMed]
- O’Regan, L.; Sampson, J.; Richards, M.W.; Knebel, A.; Roth, D.; Hood, F.E.; Straube, A.; Royle, S.J.; Bayliss, R.; Fry, A.M. Hsp72 is targeted to the mitotic spindle by Nek6 to promote K-fiber assembly and mitotic progression. J. Cell Biol. 2015, 209, 349–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longshaw, V.M.; Chapple, J.P.; Balda, M.S.; Cheetham, M.E.; Blatch, G.L. Nuclear translocation of the Hsp70/Hsp90 organizing protein mSTI1 is regulated by cell cycle kinases. J. Cell Sci. 2004, 117, 701–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgner, N.; Schmidt, C.; Beilsten-Edmands, V.; Ebong, I.-O.; Patel, N.A.; Clerico, E.M.; Kirschke, E.; Daturpalli, S.; Jackson, S.E.; Agard, D.; et al. Hsp70 Forms Antiparallel Dimers Stabilized by Post-translational Modifications to Position Clients for Transfer to Hsp90. Cell Rep. 2015, 11, 759–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, P.; Ruckova, E.; Halada, P.; Coates, P.J.; Hrstka, R.; Lane, D.P.; Vojtesek, B. C-terminal phosphorylation of Hsp70 and Hsp90 regulates alternate binding to co-chaperones CHIP and HOP to determine cellular protein folding/degradation balances. Oncogene 2013, 32, 3101–3110. [Google Scholar] [CrossRef] [PubMed]
- Assimon, V.A.; Southworth, D.R.; Gestwicki, J.E. Specific Binding of Tetratricopeptide Repeat Proteins to Heat Shock Protein 70 (Hsp70) and Heat Shock Protein 90 (Hsp90) Is Regulated by Affinity and Phosphorylation. Biochemistry 2015, 54, 7120–7131. [Google Scholar] [CrossRef] [PubMed]
- Röhl, A.; Tippel, F.; Bender, E.; Schmid, A.B.; Richter, K.; Madl, T.; Buchner, J. Hop/Sti1 phosphorylation inhibits its co-chaperone function. EMBO Rep. 2015, 16, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Marcu, M.; Yuan, X.; Mimnaugh, E.; Patterson, C.; Neckers, L. Chaperone-dependent E3 ubiquitin ligase CHIP mediates a degradative pathway for c-ErbB2/Neu. Proc. Natl. Acad. Sci. USA 2002, 99, 12847–12852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferns, G.; Shams, S.; Shafi, S. Heat shock protein 27: Its potential role in vascular disease. Int. J. Exp. Pathol. 2006, 87, 253–274. [Google Scholar] [CrossRef] [PubMed]
- Jan, R. Understanding Apoptosis and Apoptotic Pathways Targeted Cancer Therapeutics. Adv. Pharm. Bull. 2019, 9, 205–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elmore, S. Apoptosis: A Review of Programmed Cell Death. Toxicol. Pathol. 2016, 35, 495–516. [Google Scholar] [CrossRef]
- Ravagnan, L.; Gurbuxani, S.; Susin, S.A.; Maisse, C.; Daugas, E.; Zamzami, N.; Mak, T.; Jäättelä, M.; Penninger, J.M.; Garrido, C.; et al. Heat-shock protein 70 antagonizes apoptosis-inducing factor. Nat. Cell Biol. 2001, 3, 839–843. [Google Scholar] [CrossRef] [PubMed]
- Buzzard, K.A.; Giaccia, A.J.; Killender, M.; Anderson, R.L. Heat shock protein 72 modulates pathways of stress-induced apoptosis. J. Biol. Chem. 1998, 273, 17147–17153. [Google Scholar] [CrossRef] [PubMed]
- Saleh, A.; Srinivasula, S.M.; Balkir, L.; Robbins, P.D.; Alnemri, E.S. Negative regulation of the Apaf-1 apoptosome by Hsp70. Nat. Cell Biol. 2000, 2, 476–483. [Google Scholar] [CrossRef] [PubMed]
- Mayer, M.P.; Bukau, B. Hsp70 chaperones: Cellular functions and molecular mechanism. Cell. Mol. Life Sci. 2005, 62, 670–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pezzuto, A.; Citarella, F.; Croghan, I.; Tonini, G. The effects of cigarette smoking extracts on cell cycle and tumor spread: Novel evidence. Future Sci. OA 2019, FSO394. [Google Scholar] [CrossRef] [PubMed]
- Ding, C.-L.; Xu, G.; Tang, H.-L.; Zhu, S.-Y.; Zhao, L.-J.; Ren, H.; Zhao, P.; Qi, Z.-T.; Wang, W. Anchoring of both PKA-RIIα and 14-3-3θ regulates retinoic acid induced 16 mediated phosphorylation of heat shock protein 70. Oncotarget 2015, 6, 15540–15550. [Google Scholar] [CrossRef] [PubMed]
- Bonhoure, A.; Vallentin, A.; Martin, M.; Senff-Ribeiro, A.; Amson, R.; Telerman, A.; Vidal, M. Acetylation of translationally controlled tumor protein promotes its degradation through chaperone-mediated autophagy. Eur. J. Cell Biol. 2017, 96, 83–98. [Google Scholar] [CrossRef]
- Bonam, S.R.; Ruff, M.; Muller, S. HSPA8/HSC70 in Immune Disorders: A Molecular Rheostat that Adjusts Chaperone-Mediated Autophagy Substrates. Cells 2019, 8, 849. [Google Scholar] [CrossRef]
- Kondo, Y.; Kondo, S. Autophagy and cancer therapy. Autophagy 2006, 2, 85–90. [Google Scholar] [CrossRef]
- Singh, R.; Cuervo, A.M. Autophagy in the cellular energetic balance. Cell Metab. 2011, 13, 495–504. [Google Scholar] [CrossRef]
- Gomes, P.; Fleming Outeiro, T.; Cavadas, C. Emerging Role of Sirtuin 2 in the Regulation of Mammalian Metabolism. Trends Pharmacol. Sci. 2015, 36, 756–768. [Google Scholar] [CrossRef] [PubMed]
- Yang, E.; Zha, J.; Jockel, J.; Boise, L.H.; Thompson, C.B.; Korsmeyer, S.J. Bad, a heterodimeric partner for Bcl-xL and Bcl-2, displaces bax and promotes cell death. Cell 1995, 80, 285–291. [Google Scholar] [CrossRef] [Green Version]
- Sun, F.; Jiang, X.; Wang, X.; Bao, Y.; Feng, G.; Liu, H.; Kou, X.; Zhu, Q.; Jiang, L.; Yang, Y. Vincristine ablation of Sirt2 induces cell apoptosis and mitophagy via Hsp70 acetylation in MDA-MB-231 cells. Biochem. Pharmacol. 2019, 162, 142–153. [Google Scholar] [CrossRef] [PubMed]
- Seo, S.U.; Min, K.-J.; Woo, S.M.; Seo, J.H.; Kwon, T.K. HSP70 Acetylation Prevents Combined mTORC1/2 Inhibitor and Curcumin Treatment-Induced Apoptosis. Molecules 2018, 23, 2755. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.H.; Seo, J.H.; Park, J.-H.; Lee, H.S.; Kim, K.-W. Hsp70 acetylation prevents caspase-dependent/independent apoptosis and autophagic cell death in cancer cells. Int. J. Oncol. 2017, 51, 573–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, J.H.; Park, J.-H.; Lee, E.J.; Vo, T.T.L.; Choi, H.; Kim, J.Y.; Jang, J.K.; Wee, H.-J.; Lee, H.S.; Jang, S.H.; et al. ARD1-mediated Hsp70 acetylation balances stress-induced protein refolding and degradation. Nat. Commun. 2016, 7, 12882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polevoda, B.; Sherman, F. Nα-terminal acetylation of eukaryotic proteins. J. Biol. Chem. 2000, 275, 36479–36482. [Google Scholar] [CrossRef]
- Tooley, J.G.; Schaner Tooley, C.E. New roles for old modifications: Emerging roles of N-terminal post-translational modifications in development and disease. Protein Sci. 2014, 23, 1641–1649. [Google Scholar] [CrossRef] [Green Version]
- Holmes, W.M.; Mannakee, B.K.; Gutenkunst, R.N.; Serio, T.R. Loss of amino-terminal acetylation suppresses a prion phenotype by modulating global protein folding. Nat. Commun. 2014, 5, 4383. [Google Scholar] [CrossRef]
- Swaney, D.L.; Beltrao, P.; Starita, L.; Guo, A.; Rush, J.; Fields, S.; Krogan, N.J.; Villén, J. Global analysis of phosphorylation and ubiquitylation cross-talk in protein degradation. Nat. Methods 2013, 10, 676–682. [Google Scholar] [CrossRef]
- Peng, J.; Schwartz, D.; Elias, J.E.; Thoreen, C.C.; Cheng, D.; Marsischky, G.; Roelofs, J.; Finley, D.; Gygi, S.P. A proteomics approach to understanding protein ubiquitination. Nat. Biotechnol. 2003, 21, 921–926. [Google Scholar] [CrossRef] [PubMed]
- Fang, N.N.; Chan, G.T.; Zhu, M.; Comyn, S.A.; Persaud, A.; Deshaies, R.J.; Rotin, D.; Gsponer, J.; Mayor, T. Rsp5/Nedd4 is the main ubiquitin ligase that targets cytosolic misfolded proteins following heat stress. Nat. Cell Biol. 2014, 16, 1227–1237. [Google Scholar] [CrossRef] [PubMed]
- Jakobsson, M.E.; Moen, A.; Bousset, L.; Egge-Jacobsen, W.; Kernstock, S.; Melki, R.; Falnes, P.Ø. Identification and characterization of a novel human methyltransferase modulating Hsp70 protein function through lysine methylation. J. Biol. Chem. 2013, 288, 27752–27763. [Google Scholar] [CrossRef] [PubMed]
- Maltese, W.A. Posttranslational modification of proteins by isoprenoids in mammalian cells. FASEB J. 1990, 4, 3319–3328. [Google Scholar] [CrossRef] [PubMed]
- Caplan, A.J.; Douglas, M.G. Characterization of YDJ1: A yeast homologue of the bacterial dnaJ protein. J. Cell Biol. 1991, 114, 609–621. [Google Scholar] [CrossRef] [PubMed]
- Meacham, G.C.; Lu, Z.; King, S.; Sorscher, E.; Tousson, A.; Cyr, D.M. The Hdj-2/Hsc70 chaperone pair facilitates early steps in CFTR biogenesis. EMBO J. 1999, 18, 1492–1505. [Google Scholar] [CrossRef] [PubMed]
- Summers, D.W.; Douglas, P.M.; Ren, H.-Y.; Cyr, D.M. The type I Hsp40 Ydj1 utilizes a farnesyl moiety and zinc finger-like region to suppress prion toxicity. J. Biol. Chem. 2009, 284, 3628–3639. [Google Scholar] [CrossRef] [PubMed]
- Brownlee, M. ADVANCED PROTEIN GLYCOSYLATION IN DIABETES AND AGING. Annu. Rev. Med. 2003, 46, 223–234. [Google Scholar] [CrossRef] [PubMed]
- Gomes, R.A.; Miranda, H.V.; Silva, M.S.; Graça, G.; Coelho, A.V.; Ferreira, A.E.; Cordeiro, C.; Freire, A.P. Yeast protein glycation in vivo by methylglyoxal. FEBS J. 2006, 273, 5273–5287. [Google Scholar] [CrossRef]
- Vitek, M.P.; Bhattacharya, K.; Glendening, J.M.; Stopa, E.; Vlassara, H.; Bucala, R.; Manogue, K.; Cerami, A. Advanced glycation end products contribute to amyloidosis in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1994, 91, 4766–4770. [Google Scholar] [CrossRef]
- Ahmed, M.U.; Frye, E.B.; Degenhardt, T.P.; Thorpe, S.R.; Baynes, J.W. Nε-(Carboxyethyl)lysine, a product of the chemical modification of proteins by methylglyoxal, increases with age in human lens proteins. Biochem. J. 1997, 324, 565–570. [Google Scholar] [CrossRef] [PubMed]
- Mayer, M.P. Hsp70 chaperone dynamics and molecular mechanism. Trends Biochem. Sci. 2013, 38, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zuiderweg, E.R.P. The 70-kDa heat shock protein chaperone nucleotide-binding domain in solution unveiled as a molecular machine that can reorient its functional subdomains. Proc. Natl. Acad. Sci. USA 2004, 101, 10272–10277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walton-Diaz, A.; Khan, S.; Bourboulia, D.; Trepel, J.B.; Neckers, L.; Mollapour, M. Contributions of co-chaperones and post-translational modifications towards Hsp90 drug sensitivity. Future Med. Chem. 2013, 5, 1059–1071. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.M.; Miyata, Y.; Klinedinst, S.; Peng, H.-M.; Chua, J.P.; Komiyama, T.; Li, X.; Morishima, Y.; Merry, D.E.; Pratt, W.B.; et al. Activation of Hsp70 reduces neurotoxicity by promoting polyglutamine protein degradation. Nat. Chem. Biol. 2013, 9, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Miyata, Y.; Li, X.; Lee, H.-F.; Jinwal, U.K.; Srinivasan, S.R.; Seguin, S.P.; Young, Z.T.; Brodsky, J.L.; Dickey, C.A.; Sun, D.; et al. Synthesis and Initial Evaluation of YM-08, a Blood-Brain Barrier Permeable Derivative of the Heat Shock Protein 70 (Hsp70) Inhibitor MKT-077, Which Reduces Tau Levels. ACS Chem. Neurosci. 2013, 4, 930–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wisén, S.; Bertelsen, E.B.; Thompson, A.D.; Patury, S.; Ung, P.; Chang, L.; Evans, C.G.; Walter, G.M.; Wipf, P.; Carlson, H.A.; et al. Binding of a Small Molecule at a Protein–Protein Interface Regulates the Chaperone Activity of Hsp70–Hsp40. ACS Chem. Biol. 2010, 5, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Yi, F.; Regan, L. A Novel Class of Small Molecule Inhibitors of Hsp90. ACS Chem. Biol. 2008, 3, 645. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
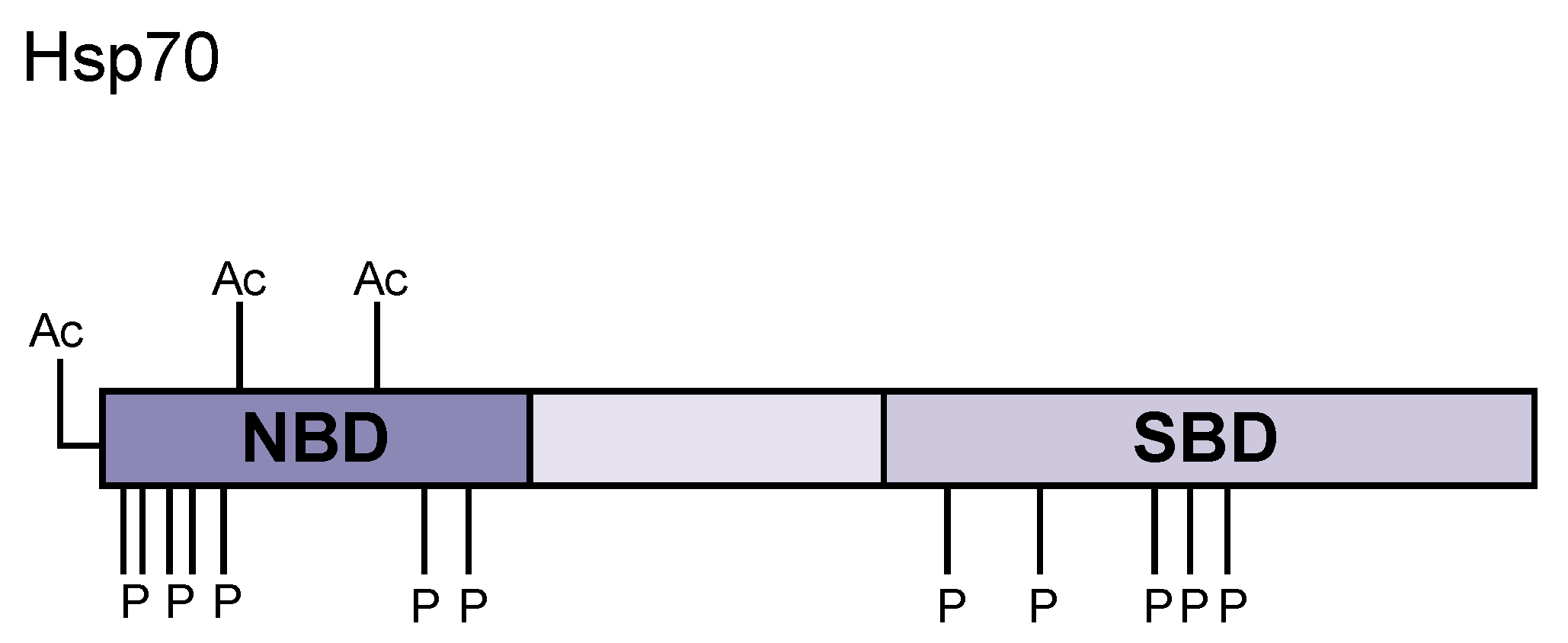
| Chaperone | Position | Modification | Effect |
|---|---|---|---|
| Hsp70 | T13 | Phosphorylation | Centrosome localization |
| S19 | Phosphorylation | Promotes protein refolding and aggregate binding | |
| S23 | Phosphorylation | Promotes protein refolding and aggregate binding | |
| S25 | Phosphorylation | Promotes protein refolding and aggregate binding | |
| T66 | Phosphorylation | Kinetochore microtubule localization | |
| T226 | Phosphorylation | Centrosome localization | |
| S362 | Phosphorylation | Centrosome localization | |
| S486 | Phosphorylation | Stabilize Caspase-3 and inhibit apoptosis | |
| T492 | Phosphorylation | Promotes growth | |
| S495 | Phosphorylation | Promotes growth | |
| T499 | Phosphorylation | Promotes growth | |
| T504 | Phosphorylation | Promotes dimer assembly | |
| S631 | Phosphorylation | Centrosome localization | |
| T633 | Phosphorylation | Centrosome localization | |
| T636 | Phosphorylation | Promotes Hsp40 STi1/HOP binding | |
| N-term | Acetylation | Not specified | |
| K77 | Acetylation | Promotes apoptosis | |
| K126 | Acetylation | Promotes autophagy | |
| Hsp40 | S189 | Sti1 (Hsp40) | Localized to nucleus |
| T198 | STi1 (Hsp40) | Localized to nucleus |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Griffith, A.A.; Holmes, W. Fine Tuning: Effects of Post-Translational Modification on Hsp70 Chaperones. Int. J. Mol. Sci. 2019, 20, 4207. https://doi.org/10.3390/ijms20174207
Griffith AA, Holmes W. Fine Tuning: Effects of Post-Translational Modification on Hsp70 Chaperones. International Journal of Molecular Sciences. 2019; 20(17):4207. https://doi.org/10.3390/ijms20174207
Chicago/Turabian StyleGriffith, Alijah A., and William Holmes. 2019. "Fine Tuning: Effects of Post-Translational Modification on Hsp70 Chaperones" International Journal of Molecular Sciences 20, no. 17: 4207. https://doi.org/10.3390/ijms20174207
APA StyleGriffith, A. A., & Holmes, W. (2019). Fine Tuning: Effects of Post-Translational Modification on Hsp70 Chaperones. International Journal of Molecular Sciences, 20(17), 4207. https://doi.org/10.3390/ijms20174207