Systematic Identification of Characteristic Genes of Ovarian Clear Cell Carcinoma Compared with High-Grade Serous Carcinoma Based on RNA-Sequencing

 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Principal Component Analysis from RNA-Sequencing Data Reveals Candidate Genes Associated with CCC and HGSC
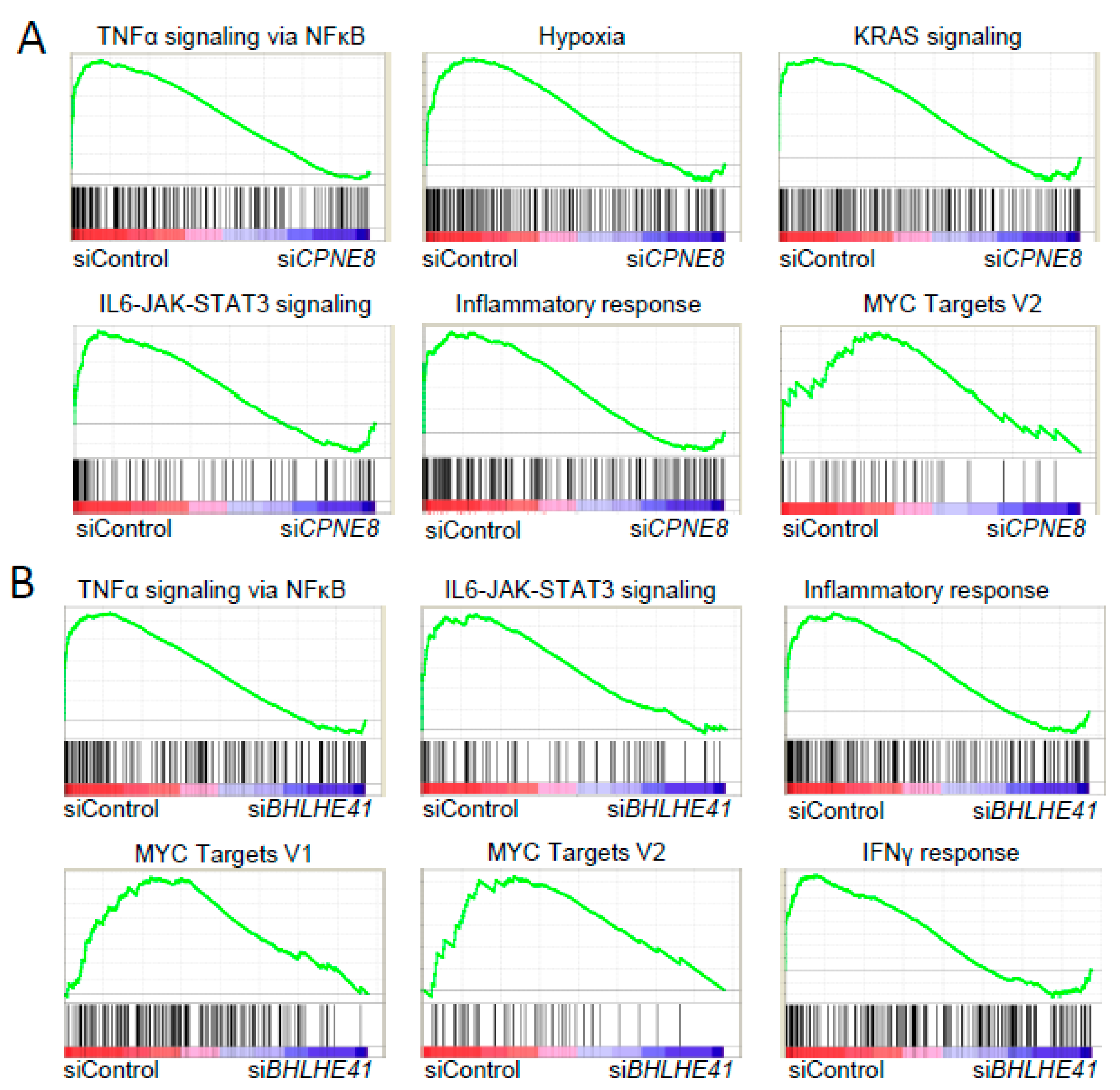
2.2. Functional Analysis of Potential Oncogenes in CCC and HGSC
2.3. Pathway Genes Regulated by CPNE8 and BHLHE41
3. Discussion
4. Materials and Methods
4.1. Clinical Specimens and Cell Lines
4.2. RNA Sequencing
4.3. siRNA and Transfection
4.4. RNA Isolation and Quantitative RT-PCR Analysis
4.5. Cell Proliferation Assay
4.6. Microarray Analysis
4.7. Principal Component Analysis
4.8. Statistical Analysis
4.9. Validation of Gene Expression Levels
4.10. Data Availability Statement
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Center for Cancer Control and Information Services, National Cancer Canter, Japan. Projected Cancer Statistics, 2018. Available online: https://ganjoho.jp/en/public/statistics/short_pred.html (accessed on 15 September 2019).
- Heintz, A.P.; Odicino, F.; Maisonneuve, P.; Quinn, M.A.; Benedet, J.L.; Creasman, W.T.; Ngan, H.Y.; Pecorelli, S.; Beller, U. Carcinoma of the ovary. Int. J. Gynaecol. Obstet. 2006, 95, S161–S192. [Google Scholar] [CrossRef]
- Gurung, A.; Hung, T.; Morin, J.; Gilks, C.B. Molecular abnormalities in ovarian carcinoma: Clinical, morphological and therapeutic correlates. Histopathology 2013, 62, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Reid, B.M.; Permuth, J.B.; Sellers, T.A. Epidemiology of ovarian cancer: A review. Cancer Biol. Med. 2017, 14, 9–32. [Google Scholar] [PubMed]
- Yamagami, W.; Nagase, S.; Takahashi, F.; Ino, K.; Hachisuga, T.; Aoki, D.; Katabuchi, H. Clinical statistics of gynecologic cancers in Japan. J. Gynecol. Oncol. 2017, 28, e32. [Google Scholar] [CrossRef] [PubMed]
- Kotsopoulos, I.C.; Papanikolaou, A.; Lambropoulos, A.F.; Papazisis, K.T.; Tsolakidis, D.; Touplikioti, P.; Tarlatzis, B.C. Serous ovarian cancer signaling pathways. Int. J. Gynecol. Cancer 2014, 24, 410–417. [Google Scholar] [CrossRef]
- Shuang, T.; Wang, M.; Zhou, Y.; Shi, C. Over-expression of nuclear NF-κB1 and c-Rel correlates with chemoresistance and prognosis of serous epithelial ovarian cancer. Exp. Mol. Pathol. 2016, 100, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Park, J.T.; Li, M.; Nakayama, K.; Mao, T.L.; Davidson, B.; Zhang, Z.; Kurman, R.J.; Eberhart, C.G.; Shih, I.e.M.; Wang, T.L. Notch3 gene amplification in ovarian cancer. Cancer Res. 2006, 66, 6312–6318. [Google Scholar] [CrossRef] [PubMed]
- Park, J.T.; Chen, X.; Tropè, C.G.; Davidson, B.; Shih, I.e.M.; Wang, T.L. Notch3 overexpression is related to the recurrence of ovarian cancer and confers resistance to carboplatin. Am. J. Pathol. 2010, 177, 1087–1094. [Google Scholar] [CrossRef] [PubMed]
- Mittempergher, L. Genomic Characterization of High-Grade Serous Ovarian Cancer: Dissecting Its Molecular Heterogeneity as a Road Towards Effective Therapeutic Strategies. Curr. Oncol. Rep. 2016, 18, 44. [Google Scholar] [CrossRef]
- Rhyasen, G.W.; Yao, Y.; Zhang, J.; Dulak, A.; Castriotta, L.; Jacques, K.; Zhao, W.; Gharahdaghi, F.; Hattersley, M.M.; Lyne, P.D.; et al. BRD4 amplification facilitates an oncogenic gene expression program in high-grade serous ovarian cancer and confers sensitivity to BET inhibitors. PLoS ONE 2018, 13, e0200826. [Google Scholar] [CrossRef]
- Brouwer-Visser, J.; Lee, J.; McCullagh, K.; Cossio, M.J.; Wang, Y.; Huang, G.S. Insulin-like growth factor 2 silencing restores taxol sensitivity in drug resistant ovarian cancer. PLoS ONE 2014, 9, e100165. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.C.; Risch, E.; Zhang, M.; Huang, C.; Huang, H.; Lu, L. Association of tRNA methyltransferase NSUN2/IGF-II molecular signature with ovarian cancer survival. Future Oncol. 2017, 13, 1981–1990. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Schwartz, P.; Scarampi, L.; Rutherford, T.; Canuto, E.M.; Yu, H.; Katsaros, D. MicroRNA let-7a: A potential marker for selection of paclitaxel in ovarian cancer management. Gynecol. Oncol. 2011, 122, 366–371. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Katsaros, D.; Wiley, A.; Rigault de la Longrais, I.A.; Puopolo, M.; Yu, H. Expression of MDR1 in epithelial ovarian cancer and its association with disease progression. Oncol. Res. 2007, 16, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Pearce, C.L.; Templeman, C.; Rossing, M.A.; Lee, A.; Near, A.M.; Webb, P.M.; Nagle, C.M.; Doherty, J.A.; Cushing-Haugen, K.L.; Wicklund, K.G.; et al. Association between endometriosis and risk of histological subtypes of ovarian cancer: A pooled analysis of case-control studies. Lancet. Oncol. 2012, 13, 385–394. [Google Scholar] [CrossRef]
- Matsuura, Y.; Robertson, G.; Marsden, D.E.; Kim, S.N.; Gebski, V.; Hacker, N.F. Thromboembolic complications in patients with clear cell carcinoma of the ovary. Gynecol. Oncol. 2007, 104, 406–410. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, A.; Sakamoto, M.; Yasuda, J.; Chuma, M.; Ohta, T.; Ohki, M.; Yasugi, T.; Taketani, Y.; Hirohashi, S. Expression profiling in ovarian clear cell carcinoma: Identification of hepatocyte nuclear factor-1 beta as a molecular marker and a possible molecular target for therapy of ovarian clear cell carcinoma. Am. J. Pathol. 2003, 163, 2503–2512. [Google Scholar] [CrossRef]
- Lee, S.; Garner, E.I.; Welch, W.R.; Berkowitz, R.S.; Mok, S.C. Over-expression of hypoxia-inducible factor 1 alpha in ovarian clear cell carcinoma. Gynecol. Oncol. 2007, 106, 311–317. [Google Scholar] [CrossRef] [Green Version]
- Mosig, R.A.; Lin, L.; Senturk, E.; Shah, H.; Huang, F.; Schlosshauer, P.; Cohen, S.; Fruscio, R.; Marchini, S.; D’Incalci, M.; et al. Application of RNA-Seq transcriptome analysis: CD151 is an Invasion/Migration target in all stages of epithelial ovarian cancer. J. Ovarian Res. 2012, 5, 4. [Google Scholar] [CrossRef]
- Kobayashi, E.; Ueda, Y.; Matsuzaki, S.; Yokoyama, T.; Kimura, T.; Yoshino, K.; Fujita, M.; Kimura, T.; Enomoto, T. Biomarkers for screening, diagnosis, and monitoring of ovarian cancer. Cancer Epidemiol. Biomarkers Prev. 2012, 21, 1902–1912. [Google Scholar] [CrossRef]
- Matulonis, U.A.; Sood, A.K.; Fallowfield, L.; Howitt, B.E.; Sehouli, J.; Karlan, B.Y. Ovarian cancer. Nat. Rev. Dis. Primers 2016, 2, 16061. [Google Scholar] [CrossRef] [PubMed]
- Fridley, B.L.; Dai, J.; Raghavan, R.; Li, Q.; Winham, S.J.; Hou, X.; Weroha, S.J.; Wang, C.; Kalli, K.R.; Cunningham, J.M.; et al. Transcriptomic Characterization of Endometrioid, Clear Cell, and High-Grade Serous Epithelial Ovarian Carcinoma. Cancer Epidemiol. Biomarkers Prev. 2018, 27, 1101–1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zorn, K.K.; Bonome, T.; Gangi, L.; Chandramouli, G.V.; Awtrey, C.S.; Gardner, G.J.; Barrett, J.C.; Boyd, J.; Birrer, M.J. Gene expression profiles of serous, endometrioid, and clear cell subtypes of ovarian and endometrial cancer. Clin. Cancer Res. 2005, 11, 6422–6430. [Google Scholar] [CrossRef] [PubMed]
- Schaner, M.E.; Ross, D.T.; Ciaravino, G.; Sorlie, T.; Troyanskaya, O.; Diehn, M.; Wang, Y.C.; Duran, G.E.; Sikic, T.L.; Caldeira, S.; et al. Gene expression patterns in ovarian carcinomas. Mol. Biol. Cell. 2003, 14, 4376–4786. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, P. Morphologic, Immunophenotypic, and Molecular Features of Epithelial Ovarian Cancer. Oncology (Williston Park) 2016, 30, 166–176. [Google Scholar] [PubMed]
- Pamuła-Piłat, J.; Rubel, T.; Rzepecka, I.K.; Olbryt, M.; Herok, R.; Dansonka-Mieszkowska, A.; Grzybowska, E.; Kupryjańczyk, J. Gene expression profiles in three histologic types, clear-cell, endometrioid and serous ovarian carcinomas. J. Biol. Regul. Homeost Agents 2014, 28, 659–674. [Google Scholar] [PubMed]
- Caudell, E.G.; Caudell, J.J.; Tang, C.H.; Yu, T.K.; Frederick, M.J.; Grimm, E.A. Characterization of human copine III as a phosphoprotein with associated kinase activity. Biochemistry 2000, 39, 13034–13043. [Google Scholar] [CrossRef] [PubMed]
- Tomsig, J.L.; Creutz, C.E. Biochemical characterization of copine: A ubiquitous Ca2+-dependent, phospholipid-binding protein. Biochemistry 2000, 39, 16163–16175. [Google Scholar] [CrossRef] [PubMed]
- Creutz, C.E.; Tomsig, J.L.; Snyder, S.L.; Gautier, M.C.; Skouri, F.; Beisson, J.; Cohen, J. The copines, a novel class of C2 domain-containing, calcium-dependent, phospholipid-binding proteins conserved from Paramecium to humans. J. Biol. Chem. 1998, 273, 1393–1402. [Google Scholar] [CrossRef]
- Bu, D.; Lewis, C.M.; Sarode, V.; Chen, M.; Ma, X.; Lazorwitz, A.M.; Rao, R.; Leitch, M.; Moldrem, A.; Andrews, V.; et al. Identification of breast cancer DNA methylation markers optimized for fine-needle aspiration samples. Cancer Epidemiol. Biomarkers Prev. 2013, 22, 2212–2221. [Google Scholar] [CrossRef]
- Dang, T.T.; Westcott, J.M.; Maine, E.A.; Kanchwala, M.; Xing, C.; Pearson, G.W. ΔNp63α induces the expression of FAT2 and Slug to promote tumor invasion. Oncotarget 2016, 7, 28592–28611. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, P.; Zhang, P.; Zhang, X.; Du, H.; Liu, Q.; Huang, B.; Qian, C.; Zhang, S.; Zhu, W.; et al. An integrative bioinformatics analysis identified miR-375 as a candidate key regulator of malignant breast cancer. J. Appl. Genet. 2019, 1–12, Epub ahead of print. [Google Scholar] [CrossRef]
- Ramsey, C.S.; Yeung, F.; Stoddard, P.B.; Li, D.; Creutz, C.E.; Mayo, M.W. Copine-I represses NF-kappaB transcription by endoproteolysis of p65. Oncogene 2008, 27, 3516–3526. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Tang, H.; Zhu, J.; Ding, H.; Zeng, Y.; Du, W.; Ding, Z.; Song, P.; Zhang, Y.; Liu, Z.; et al. High expression of Copine 1 promotes cell growth and metastasis in human lung adenocarcinoma. Int. J. Oncol. 2018, 53, 2369–2378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinrich, C.; Keller, C.; Boulay, A.; Vecchi, M.; Bianchi, M.; Sack, R.; Lienhard, S.; Duss, S.; Hofsteenge, J.; Hynes, N.E. Copine-III interacts with ErbB2 and promotes tumor cell migration. Oncogene 2010, 29, 1598–1610. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Lin, Y.; Liu, S.; Aziz, F.; Yan, Q. Fucosyltransferase IV (FUT4) as an effective biomarker for the diagnosis of breast cancer. Biomed Pharmacother 2015, 70, 299–304. [Google Scholar] [CrossRef]
- Tian, L.; Shen, D.; Li, X.; Shan, X.; Wang, X.; Yan, Q.; Liu, J. Ginsenoside Rg3 inhibits epithelial-mesenchymal transition (EMT) and invasion of lung cancer by down-regulating FUT4. Oncotarget 2016, 7, 1619–1632. [Google Scholar] [CrossRef]
- Thanasupawat, T.; Glogowska, A.; Burg, M.; Wong, G.W.; Hoang-Vu, C.; Hombach-Klonisch, S.; Klonisch, T. RXFP1 is Targeted by Complement C1q Tumor Necrosis Factor-Related Factor 8 in Brain Cancer. Front. Endocrinol. (Lausanne) 2015, 6, 127. [Google Scholar] [CrossRef] [Green Version]
- Feng, S.; Agoulnik, I.U.; Bogatcheva, N.V.; Kamat, A.A.; Kwabi-Addo, B.; Li, R.; Ayala, G.; Ittmann, M.M.; Agoulnik, A.I. Relaxin promotes prostate cancer progression. Clin. Cancer Res. 2007, 13, 1695–1702. [Google Scholar] [CrossRef]
- Wu, Y.; Sato, F.; Bhawal, U.K.; Kawamoto, T.; Fujimoto, K.; Noshiro, M.; Morohashi, S.; Kato, Y.; Kijima, H. Basic helix-loop-helix transcription factors DEC1 and DEC2 regulate the paclitaxel-induced apoptotic pathway of MCF-7 human breast cancer cells. Int. J. Mol. Med. 2011, 27, 491–495. [Google Scholar]
- Wu, Y.; Sato, H.; Suzuki, T.; Yoshizawa, T.; Morohashi, S.; Seino, H.; Kawamoto, T.; Fujimoto, K.; Kato, Y.; Kijima, H. Involvement of c-Myc in the proliferation of MCF-7 human breast cancer cells induced by bHLH transcription factor DEC2. Int. J. Mol. Med. 2015, 35, 815–820. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Jia, Y.F.; Ma, X.L.; Zheng, Y.; Kong, Y.; Zhang, Y.; Zong, S.; Chen, Z.T.; Wang, Y.S. DEC2 suppresses tumor proliferation and metastasis by regulating ERK/NF-κB pathway in gastric cancer. Am. J. Cancer Res. 2016, 6, 1741–1757. [Google Scholar] [PubMed]
- Liao, Y.; He, X.; Qiu, H.; Che, Q.; Wang, F.; Lu, W.; Chen, Z.; Qiu, M.; Wang, J.; Wang, H.; et al. Suppression of the epithelial-mesenchymal transition by SHARP1 is linked to the NOTCH1 signaling pathway in metastasis of endometrial cancer. BMC Cancer 2014, 14, 487. [Google Scholar] [CrossRef] [PubMed]
- Kurman, R.J.; Shih, I.e.M. The origin and pathogenesis of epithelial ovarian cancer: A proposed unifying theory. Am. J. Surg. Pathol. 2010, 34, 433–443. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Nakayama, K.; Ishikawa, N.; Ishikawa, M.; Sultana, R.; Kiyono, T.; Kyo, S. Reconstitution of high-grade serous ovarian carcinoma from primary fallopian tube secretory epithelial cells. Oncotarget 2017, 9, 12609–12619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guastadisegni, M.C.; Lonoce, A.; Impera, L.; Di Terlizzi, F.; Fugazza, G.; Aliano, S.; Grasso, R.; Cluzeau, T.; Raynaud, S.; Rocchi, M.; et al. CBFA2T2 and C20orf112: Two novel fusion partners of RUNX1 in acute myeloid leukemia. Leukemia 2010, 24, 1516–1519. [Google Scholar] [CrossRef] [PubMed]
- Kawamata, N.; Pennella, M.A.; Woo, J.L.; Berk, A.J.; Koeffler, H.P. Dominant-negative mechanism of leukemogenic PAX5 fusions. Oncogene 2012, 31, 966–977. [Google Scholar] [CrossRef]
- Rustgi, A.K. Familial pancreatic cancer: Genetic advances. Genes Dev. 2014, 28, 1–7. [Google Scholar] [CrossRef]
- Chmielecki, J.; Bailey, M.; He, J.; Elvin, J.; Vergilio, J.A.; Ramkissoon, S.; Suh, J.; Frampton, G.M.; Sun, J.X.; Morley, S.; et al. Genomic Profiling of a Large Set of Diverse Pediatric Cancers Identifies Known and Novel Mutations across Tumor Spectra. Cancer Res. 2017, 77, 509–519. [Google Scholar] [CrossRef] [Green Version]
- Nagasawa, S.; Ikeda, K.; Horie-Inoue, K.; Sato, S.; Itakura, A.; Takeda, S.; Hasegawa, S.; Inoue, S. Systematic Identification of Characteristic Genes of Ovarian Clear Cell Carcinoma Compared with High-Grade Serous Carcinoma Based on RNA-Sequencing. Unpublished work. 2019. [Google Scholar]
- Anglesio, M.S.; Wiegand, K.C.; Melnyk, N.; Chow, C.; Salamanca, C.; Prentice, L.M.; Senz, J.; Yang, W.; Spillman, M.A.; Cochrane, D.R.; et al. Type-specific cell line models for type-specific ovarian cancer research. PLoS ONE 2013, 8, e72162. [Google Scholar] [CrossRef]
- Domcke, S.; Sinha, R.; Levine, D.A.; Sander, C.; Schultz, N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat. Commun. 2013, 4, 2126. [Google Scholar] [CrossRef] [PubMed]
- Beaufort, C.M.; Helmijr, J.C.; Piskorz, A.M.; Hoogstraat, M.; Ruigrok-Ritstier, K.; Besselink, N.; Murtaza, M.; van IJcken, W.F.; Heine, A.A.; Smid, M.; et al. Ovarian cancer cell line panel (OCCP): Clinical importance of in vitro morphological subtypes. PLoS ONE 2014, 9, e103988. [Google Scholar] [CrossRef] [PubMed]
- Shaw, T.J.; Senterman, M.K.; Dawson, K.; Crane, C.A.; Vanderhyden, B.C. Characterization of intraperitoneal, orthotopic, and metastatic xenograft models of human ovarian cancer. Mol. Ther. 2004, 10, 1032–1042. [Google Scholar] [CrossRef] [PubMed]
- Aran, D.; Sirota, M.; Butte, A.J. Systematic pan-cancer analysis of tumour purity. Nat. Commun. 2015, 6, 8971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, J.; Otto, W.R. Fluorimetric DNA assay for cell growth estimation. Anal. Biochem. 1992, 207, 186–192. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nagasawa, S.; Ikeda, K.; Horie-Inoue, K.; Sato, S.; Itakura, A.; Takeda, S.; Hasegawa, K.; Inoue, S. Systematic Identification of Characteristic Genes of Ovarian Clear Cell Carcinoma Compared with High-Grade Serous Carcinoma Based on RNA-Sequencing. Int. J. Mol. Sci. 2019, 20, 4330. https://doi.org/10.3390/ijms20184330
Nagasawa S, Ikeda K, Horie-Inoue K, Sato S, Itakura A, Takeda S, Hasegawa K, Inoue S. Systematic Identification of Characteristic Genes of Ovarian Clear Cell Carcinoma Compared with High-Grade Serous Carcinoma Based on RNA-Sequencing. International Journal of Molecular Sciences. 2019; 20(18):4330. https://doi.org/10.3390/ijms20184330
Chicago/Turabian StyleNagasawa, Saya, Kazuhiro Ikeda, Kuniko Horie-Inoue, Sho Sato, Atsuo Itakura, Satoru Takeda, Kosei Hasegawa, and Satoshi Inoue. 2019. "Systematic Identification of Characteristic Genes of Ovarian Clear Cell Carcinoma Compared with High-Grade Serous Carcinoma Based on RNA-Sequencing" International Journal of Molecular Sciences 20, no. 18: 4330. https://doi.org/10.3390/ijms20184330
APA StyleNagasawa, S., Ikeda, K., Horie-Inoue, K., Sato, S., Itakura, A., Takeda, S., Hasegawa, K., & Inoue, S. (2019). Systematic Identification of Characteristic Genes of Ovarian Clear Cell Carcinoma Compared with High-Grade Serous Carcinoma Based on RNA-Sequencing. International Journal of Molecular Sciences, 20(18), 4330. https://doi.org/10.3390/ijms20184330