Molecular Docking: Shifting Paradigms in Drug Discovery



{kind=link}
{kind=link}
Abstract
:1. Introduction
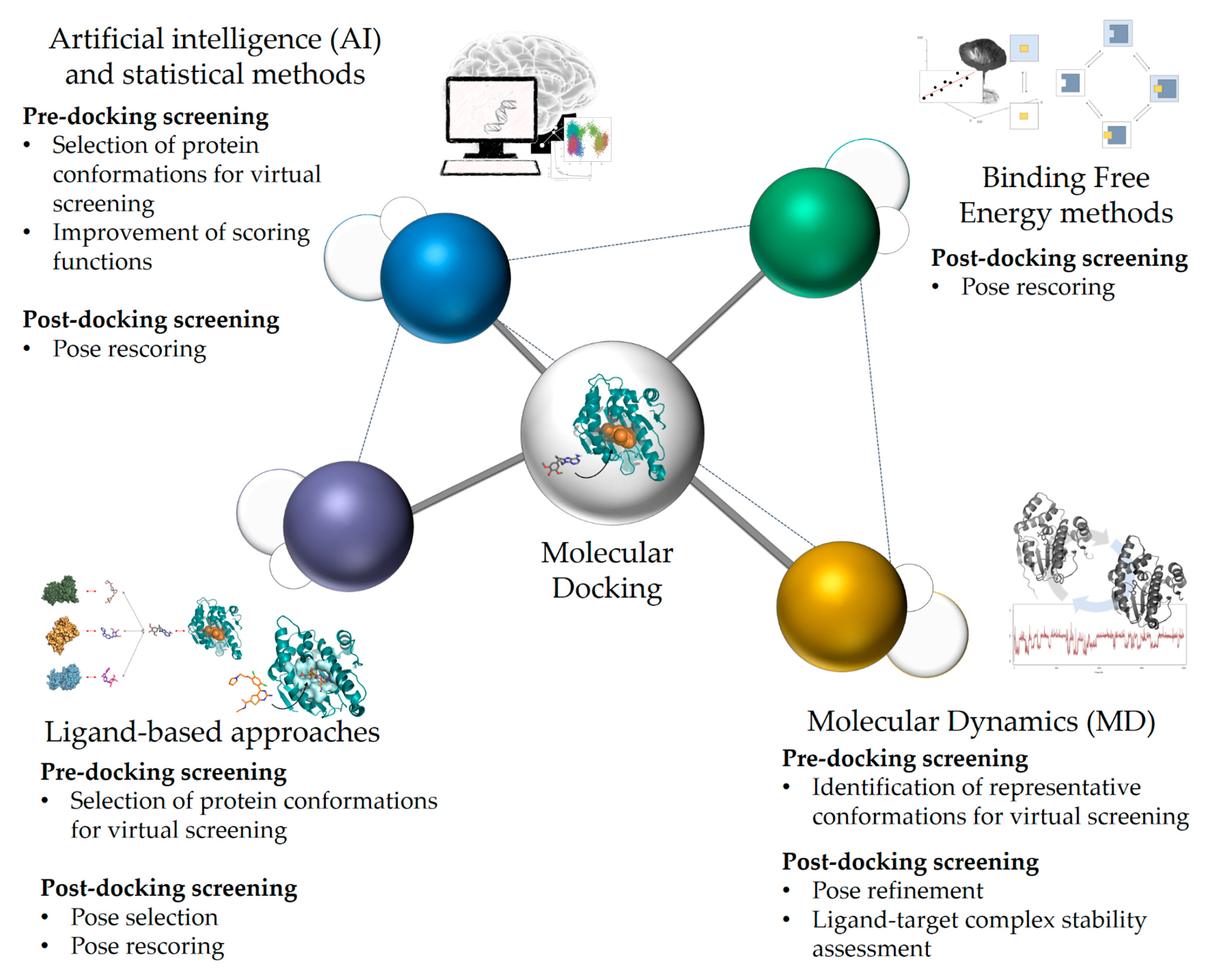
2. Current Rational Design Approaches, Including Docking
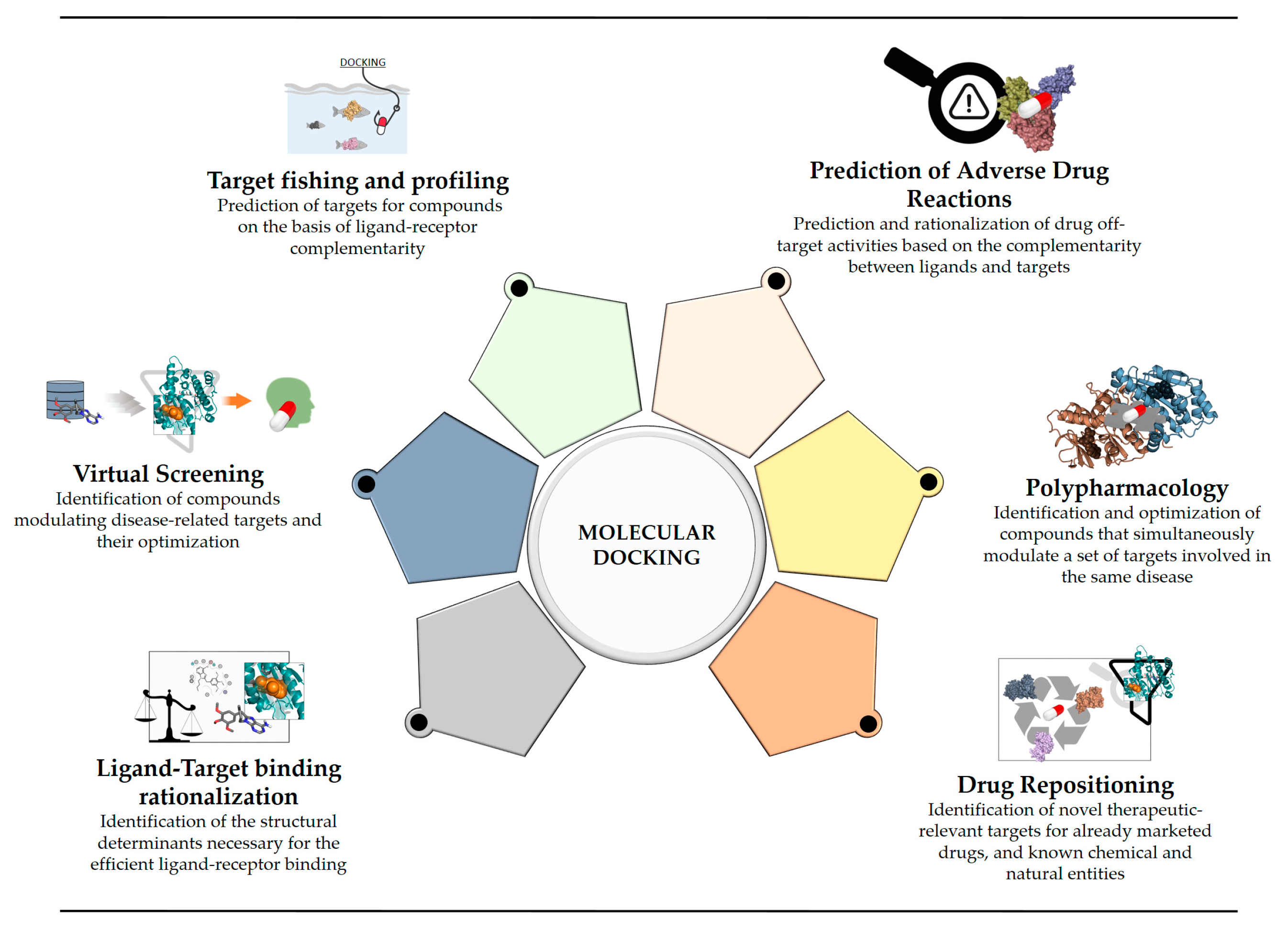
3. Reverse Screening for Target Fishing and Profiling
- (i)
- currently available computational techniques and software in general, which allow to more accurately screen larger databases;
- (ii)
- hardware facilities, which enable a faster screening of ligands to targets, to a larger public, and;
- (iii)
4. Prediction of Adverse Drug Reactions
5. Polypharmacology
6. Drug Repositioning
7. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ADR | Adverse Drug Reaction |
| AI | Artificial Intelligence |
| BEAR | Binding Estimation After Refinement |
| CANDO | Computational Analysis of Novel Drug Opportunities |
| CBC | Cannabichromene |
| CBG | Cannabigerol |
| CNN | Convolutional Neural Networks |
| CPU | Central Processing Unit |
| DL | Deep Learning |
| FDA | Food and Drug Administration |
| FEP | Free Energy Perturbation |
| GPU | Graphics Processing Units |
| GWI | Gulf War Illness |
| HTS | High-Throughput Screening |
| LDA | Linear Discriminant Analysis |
| MD | Molecular Dynamics |
| ML | Machine Learning |
| NN | Neural Networks |
| PADIF | Protein Atom Score Contributions Derived Interaction Fingerprint |
| PLIFs | Protein-Ligand Interaction Fingerprints |
| PNP | Purine Nucleoside Phosphorylase |
| RD | Reverse Docking |
| RF | Random Forest |
| SAR | Structure-Activity Relationships |
| SVM | Support Vector Machines |
| TI | Thermodynamic Integration |
References
- Sliwoski, G.; Kothiwale, S.; Meiler, J.; Lowe, E.W. Computational methods in drug discovery. Pharmacol. Rev. 2014, 66, 334–395. [Google Scholar] [CrossRef] [PubMed]
- Song, C.M.; Lim, S.J.; Tong, J.C. Recent advances in computer-aided drug design. Brief. Bioinform. 2009, 10, 579–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macalino, S.J.Y.; Gosu, V.; Hong, S.; Choi, S. Role of computer-aided drug design in modern drug discovery. Arch. Pharm. Res. 2015, 38, 1686–1701. [Google Scholar] [CrossRef] [PubMed]
- D’Agostino, D.; Clematis, A.; Quarati, A.; Cesini, D.; Chiappori, F.; Milanesi, L.; Merelli, I. Cloud Infrastructures for In Silico Drug Discovery: Economic and Practical Aspects. Biomed Res. Int. 2013, 2013, 138012. [Google Scholar] [PubMed]
- Jorgensen, W.L. The Many Roles of Computation in Drug Discovery. Science 2004, 303, 1813–1818. [Google Scholar] [CrossRef] [PubMed]
- Kapetanovic, I.M. Computer-aided drug discovery and development (CADDD): In silico-chemico-biological approach. Chem. Biol. Interact. 2008, 171, 165–176. [Google Scholar] [CrossRef] [Green Version]
- Kitchen, D.B.; Decornez, H.; Furr, J.R.; Bajorath, J. Docking and scoring in virtual screening for drug discovery: methods and applications. Nat. Rev. Drug Discov. 2004, 3, 935–949. [Google Scholar] [CrossRef]
- DesJarlais, R.L.; Sheridan, R.P.; Dixon, J.S.; Kuntz, I.D.; Venkataraghavan, R. Docking flexible ligands to macromolecular receptors by molecular shape. J. Med. Chem. 1986, 29, 2149–2153. [Google Scholar] [CrossRef] [PubMed]
- Levinthal, C.; Wodak, S.J.; Kahn, P.; Dadivanian, A.K. Hemoglobin interaction in sickle cell fibers. I: Theoretical approaches to the molecular contacts. Proc. Natl. Acad. Sci. USA 1975, 72, 1330–1334. [Google Scholar] [CrossRef]
- Goodsell, D.S.; Olson, A.J. Automated docking of substrates to proteins by simulated annealing. Proteins 1990, 8, 195–202. [Google Scholar] [CrossRef]
- Salemme, F.R. An hypothetical structure for an intermolecular electron transfer complex of cytochromes c and b5. J. Mol. Biol. 1976, 102, 563–568. [Google Scholar] [CrossRef]
- Wodak, S.J.; Janin, J. Computer analysis of protein-protein interaction. J. Mol. Biol. 1978, 124, 323–342. [Google Scholar] [CrossRef]
- Kuntz, I.D.; Blaney, J.M.; Oatley, S.J.; Langridge, R.; Ferrin, T.E. A geometric approach to macromolecule-ligand interactions. J. Mol. Biol. 1982, 161, 269–288. [Google Scholar] [CrossRef]
- Kuhl, F.S.; Crippen, G.M.; Friesen, D.K. A combinatorial algorithm for calculating ligand binding. J. Comput. Chem. 1984, 5, 24–34. [Google Scholar] [CrossRef]
- DesJarlais, R.L.; Sheridan, R.P.; Seibel, G.L.; Dixon, J.S.; Kuntz, I.D.; Venkataraghavan, R. Using shape complementarity as an initial screen in designing ligands for a receptor binding site of known three-dimensional structure. J. Med. Chem. 1988, 31, 722–729. [Google Scholar] [CrossRef] [PubMed]
- Warwicker, J. Investigating protein-protein interaction surfaces using a reduced stereochemical and electrostatic model. J. Mol. Biol. 1989, 206, 381–395. [Google Scholar] [CrossRef]
- Jiang, F.; Kim, S.H. “Soft docking”: Matching of molecular surface cubes. J. Mol. Biol. 1991, 219, 79–102. [Google Scholar] [CrossRef]
- Meng, X.Y.; Zhang, H.X.; Mezei, M.; Cui, M. Molecular docking: A powerful approach for structure-based drug discovery. Curr. Comput. Aided. Drug Des. 2011, 7, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Amaro, R.E.; Baudry, J.; Chodera, J.; Demir, Ö.; McCammon, J.A.; Miao, Y.; Smith, J.C. Ensemble Docking in Drug Discovery. Biophys. J. 2018, 114, 2271–2278. [Google Scholar] [CrossRef]
- Abagyan, R.; Totrov, M. High-throughput docking for lead generation. Curr. Opin. Chem. Biol. 2001, 5, 375–382. [Google Scholar] [CrossRef]
- Carlson, H.A. Protein flexibility and drug design: how to hit a moving target. Curr. Opin. Chem. Biol. 2002, 6, 447–452. [Google Scholar] [CrossRef]
- Greer, J.; Bush, B.L. Macromolecular shape and surface maps by solvent exclusion. Proc. Natl. Acad. Sci. USA 1978, 75, 303–307. [Google Scholar] [CrossRef] [PubMed]
- Rarey, M.; Kramer, B.; Lengauer, T.; Klebe, G. A fast flexible docking method using an incremental construction algorithm. J. Mol. Biol. 1996, 261, 470–489. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, R.; Vajda, S.; DeLisi, C. Flexible docking and design. Annu. Rev. Biophys. Biomol. Struct. 1995, 24, 677–700. [Google Scholar] [CrossRef] [PubMed]
- Sherman, W.; Beard, H.S.; Farid, R. Use of an induced fit receptor structure in virtual screening. Chem. Biol. Drug Des. 2006, 67, 83–84. [Google Scholar] [CrossRef] [PubMed]
- Leach, A.R. Ligand docking to proteins with discrete side-chain flexibility. J. Mol. Biol. 1994, 235, 345–356. [Google Scholar] [CrossRef]
- Ring, C.S.; Sun, E.; McKerrow, J.H.; Lee, G.K.; Rosenthal, P.J.; Kuntz, I.D.; Cohen, F.E. Structure-based inhibitor design by using protein models for the development of antiparasitic agents. Proc. Natl. Acad. Sci. USA 1993, 90, 3583–3587. [Google Scholar] [CrossRef]
- Coupez, B.; Lewis, R.A. Docking and scoring--theoretically easy, practically impossible? Curr. Med. Chem. 2006, 13, 2995–3003. [Google Scholar]
- Ain, Q.U.; Aleksandrova, A.; Roessler, F.D.; Ballester, P.J. Machine-learning scoring functions to improve structure-based binding affinity prediction and virtual screening. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2015, 5, 405–424. [Google Scholar] [CrossRef]
- Guedes, I.A.; Pereira, F.S.S.; Dardenne, L.E. Empirical scoring functions for structure-based virtual screening: Applications, critical aspects, and challenges. Front. Pharmacol. 2018, 9, 1089. [Google Scholar] [CrossRef]
- Elokely, K.M.; Doerksen, R.J. Docking challenge: Protein sampling and molecular docking performance. J. Chem. Inf. Model. 2013, 53, 1934–1945. [Google Scholar] [CrossRef] [PubMed]
- Pantsar, T.; Poso, A. Binding affinity via docking: fact and fiction. Molecules 2018, 23, 1899. [Google Scholar] [CrossRef] [PubMed]
- Salmaso, V.; Moro, S. Bridging molecular docking to molecular dynamics in exploring ligand-protein recognition process: An overview. Front. Pharmacol. 2018, 9, 923. [Google Scholar] [CrossRef] [PubMed]
- De Vivo, M.; Masetti, M.; Bottegoni, G.; Cavalli, A. Role of molecular dynamics and related methods in drug discovery. J. Med. Chem. 2016, 59, 4035–4061. [Google Scholar] [CrossRef] [PubMed]
- Alonso, H.; Bliznyuk, A.A.; Gready, J.E. Combining docking and molecular dynamic simulations in drug design. Med. Res. Rev. 2006, 26, 531–568. [Google Scholar] [CrossRef] [PubMed]
- Bard, J.; Ercolani, K.; Svenson, K.; Olland, A.; Somers, W. Automated systems for protein crystallization. Methods 2004, 34, 329–347. [Google Scholar] [CrossRef] [PubMed]
- Gavira, J.A. Current trends in protein crystallization. Arch. Biochem. Biophys. 2016, 602, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Dauter, Z.; Wlodawer, A. Progress in protein crystallography. Protein Pept. Lett. 2016, 23, 201–210. [Google Scholar] [CrossRef]
- Grimes, J.M.; Hall, D.R.; Ashton, A.W.; Evans, G.; Owen, R.L.; Wagner, A.; McAuley, K.E.; von Delft, F.; Orville, A.M.; Sorensen, T.; et al. Where is crystallography going? Acta Crystallogr. Sect. D 2018, 74, 152–166. [Google Scholar] [CrossRef] [Green Version]
- Durrant, J.D.; McCammon, J.A. Molecular dynamics simulations and drug discovery. BMC Biol. 2011, 9, 71. [Google Scholar] [CrossRef]
- Karplus, M.; McCammon, J.A. Molecular dynamics simulations of biomolecules. Nat. Struct. Biol. 2002, 9, 646–652. [Google Scholar] [CrossRef] [PubMed]
- Rastelli, G.; Degliesposti, G.; Del Rio, A.; Sgobba, M. Binding estimation after refinement, a new automated procedure for the refinement and rescoring of docked ligands in virtual screening. Chem. Biol. Drug Des. 2009, 73, 283–286. [Google Scholar] [CrossRef] [PubMed]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the performance of the molecular mechanics/Poisson Boltzmann surface area and molecular mechanics/generalized Born surface area methods. II. The accuracy of ranking poses generated from docking. J. Comput. Chem. 2011, 32, 866–877. [Google Scholar] [CrossRef] [PubMed]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Pu, C.; Yan, G.; Shi, J.; Li, R. Assessing the performance of docking scoring function, FEP, MM-GBSA, and QM/MM-GBSA approaches on a series of PLK1 inhibitors. Medchemcomm 2017, 8, 1452–1458. [Google Scholar] [CrossRef] [PubMed]
- Rastelli, G.; Pinzi, L. Refinement and rescoring of virtual screening results. Front. Chem. 2019, 7, 498. [Google Scholar] [CrossRef] [PubMed]
- Gschwend, D.A.; Good, A.C.; Kuntz, I.D. Molecular docking towards drug discovery. J. Mol. Recognit. 1996, 9, 175–186. [Google Scholar] [CrossRef]
- De Vivo, M.; Cavalli, A. Recent advances in dynamic docking for drug discovery. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2017, 7, e1320. [Google Scholar] [CrossRef]
- Shoichet, B.K.; McGovern, S.L.; Wei, B.; Irwin, J.J. Lead discovery using molecular docking. Curr. Opin. Chem. Biol. 2002, 6, 439–446. [Google Scholar] [CrossRef]
- Ferreira, L.G.; Dos Santos, R.N.; Oliva, G.; Andricopulo, A.D. Molecular docking and structure-based drug design strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef]
- Kinnings, S.L.; Liu, N.; Buchmeier, N.; Tonge, P.J.; Xie, L.; Bourne, P.E. Drug discovery using chemical systems biology: Repositioning the safe medicine Comtan to treat multi-drug and extensively drug resistant tuberculosis. PLoS Comput. Biol. 2009, 5, e1000423. [Google Scholar] [CrossRef] [PubMed]
- Pinzi, L.; Lherbet, C.; Baltas, M.; Pellati, F.; Rastelli, G. In silico repositioning of cannabigerol as a novel inhibitor of the enoyl acyl carrier protein (ACP) reductase. Molecules 2019, 24, 2567. [Google Scholar] [CrossRef] [PubMed]
- Anighoro, A.; Pinzi, L.; Marverti, G.; Bajorath, J.; Rastelli, G. Heat shock protein 90 and serine/threonine kinase B-Raf inhibitors have overlapping chemical space. RSC Adv. 2017, 7, 31069–31074. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Gao, Z.; Kang, L.; Zhang, H.; Yang, K.; Yu, K.; Luo, X.; Zhu, W.; Chen, K.; Shen, J.; et al. TarFisDock: a web server for identifying drug targets with docking approach. Nucleic Acids Res. 2006, 34, W219–W224. [Google Scholar] [CrossRef] [Green Version]
- Lyu, J.; Wang, S.; Balius, T.E.; Singh, I.; Levit, A.; Moroz, Y.S.; O’Meara, M.J.; Che, T.; Algaa, E.; Tolmachova, K.; et al. Ultra-large library docking for discovering new chemotypes. Nature 2019, 566, 224–229. [Google Scholar] [CrossRef]
- Graziani, D.; Caligari, S.; Callegari, E.; De Toma, C.; Longhi, M.; Frigerio, F.; Dilernia, R.; Menegon, S.; Pinzi, L.; Pirona, L.; et al. Evaluation of amides, carbamates, sulfonamides, and ureas of 4-Prop-2-ynylidenecycloalkylamine as potent, selective, and bioavailable negative allosteric modulators of metabotropic glutamate receptor 5. J. Med. Chem. 2019, 62, 1246–1273. [Google Scholar] [CrossRef]
- Ramsay, R.R.; Popovic-Nikolic, M.R.; Nikolic, K.; Uliassi, E.; Bolognesi, M.L. A perspective on multi-target drug discovery and design for complex diseases. Clin. Transl. Med. 2018, 7, 3. [Google Scholar] [CrossRef]
- Lee, A.; Lee, K.; Kim, D. Using reverse docking for target identification and its applications for drug discovery. Expert Opin. Drug Discov. 2016, 11, 707–715. [Google Scholar] [CrossRef]
- Anighoro, A.; Bajorath, J.; Rastelli, G. Polypharmacology: Challenges and opportunities in drug discovery. J. Med. Chem. 2014, 57, 7874–7887. [Google Scholar] [CrossRef]
- Gloriam, D.E. Bigger is better in virtual drug screens. Nature 2019, 566, 193–194. [Google Scholar] [CrossRef]
- Hazarika, R.R.; Sostaric, N.; Sun, Y.; van Noort, V. Large-scale docking predicts that sORF-encoded peptides may function through protein-peptide interactions in Arabidopsis thaliana. PLoS ONE 2018, 13, e0205179. [Google Scholar] [CrossRef] [PubMed]
- LaBute, M.X.; Zhang, X.; Lenderman, J.; Bennion, B.J.; Wong, S.E.; Lightstone, F.C. Adverse drug reaction prediction using scores produced by large-scale drug-protein target docking on high-performance computing machines. PLoS ONE 2014, 9, e106298. [Google Scholar] [CrossRef] [PubMed]
- de Ruyck, J.; Brysbaert, G.; Blossey, R.; Lensink, M.F. Molecular docking as a popular tool in drug design, an in silico travel. Adv. Appl. Bioinform. Chem. 2016, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Lavecchia, A. Machine-learning approaches in drug discovery: methods and applications. Drug Discov. Today 2015, 20, 318–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- March-Vila, E.; Pinzi, L.; Sturm, N.; Tinivella, A.; Engkvist, O.; Chen, H.; Rastelli, G. On the integration of in silico drug design methods for drug repurposing. Front. Pharmacol. 2017, 8, 298. [Google Scholar] [CrossRef] [PubMed]
- Wilson, G.L.; Lill, M.A. Integrating structure-based and ligand-based approaches for computational drug design. Future Med. Chem. 2011, 3, 735–750. [Google Scholar] [CrossRef]
- Maggiora, G.; Vogt, M.; Stumpfe, D.; Bajorath, J. Molecular similarity in medicinal chemistry. J. Med. Chem. 2014, 57, 3186–3204. [Google Scholar] [CrossRef]
- Sutherland, J.J.; Nandigam, R.K.; Erickson, J.A.; Vieth, M. Lessons in molecular recognition. 2. Assessing and improving cross-docking accuracy. J. Chem. Inf. Model. 2007, 47, 2293–2302. [Google Scholar] [CrossRef]
- Broccatelli, F.; Brown, N. Best of both worlds: on the complementarity of ligand-based and structure-based virtual screening. J. Chem. Inf. Model. 2014, 54, 1634–1641. [Google Scholar] [CrossRef]
- Pinzi, L.; Caporuscio, F.; Rastelli, G. Selection of protein conformations for structure-based polypharmacology studies. Drug Discov. Today 2018, 23, 1889–1896. [Google Scholar] [CrossRef] [Green Version]
- Jain, A.N. Effects of protein conformation in docking: improved pose prediction through protein pocket adaptation. J. Comput. Aided. Mol. Des. 2009, 23, 355–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verdonk, M.L.; Mortenson, P.N.; Hall, R.J.; Hartshorn, M.J.; Murray, C.W. Protein−ligand docking against non-native protein conformers. J. Chem. Inf. Model. 2008, 48, 2214–2225. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Yan, C.; Zou, X. Improving binding mode and binding affinity predictions of docking by ligand-based search of protein conformations: evaluation in D3R grand challenge 2015. J. Comput. Aided. Mol. Des. 2017, 31, 689–699. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Zhang, K.Y. A cross docking pipeline for improving pose prediction and virtual screening performance. J. Comput. Aided. Mol. Des. 2018, 32, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Zhang, K.Y.J. Advances in the development of shape similarity methods and their application in drug discovery. Front. Chem. 2018, 6, 315. [Google Scholar] [CrossRef] [PubMed]
- Perryman, A.L.; Santiago, D.N.; Forli, S.; Martins, D.S.; Olson, A.J. Virtual screening with AutoDock Vina and the common pharmacophore engine of a low diversity library of fragments and hits against the three allosteric sites of HIV integrase: participation in the SAMPL4 protein-ligand binding challenge. J. Comput. Aided. Mol. Des. 2014, 28, 429–441. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Rizzo, R.C. Pharmacophore-based similarity scoring for DOCK. J. Phys. Chem. B 2015, 119, 1083–1102. [Google Scholar] [CrossRef]
- Moustakas, D.T.; Lang, P.T.; Pegg, S.; Pettersen, E.; Kuntz, I.D.; Brooijmans, N.; Rizzo, R.C. Development and validation of a modular, extensible docking program: DOCK 5. J. Comput. Aided. Mol. Des. 2006, 20, 601–619. [Google Scholar] [CrossRef]
- Lang, P.T.; Brozell, S.R.; Mukherjee, S.; Pettersen, E.F.; Meng, E.C.; Thomas, V.; Rizzo, R.C.; Case, D.A.; James, T.L.; Kuntz, I.D. DOCK 6: Combining techniques to model RNA-small molecule complexes. RNA 2009, 15, 1219–1230. [Google Scholar] [CrossRef]
- Anighoro, A.; Bajorath, J. Three-dimensional similarity in molecular docking: prioritizing ligand poses on the basis of experimental binding modes. J. Chem. Inf. Model. 2016, 56, 580–587. [Google Scholar] [CrossRef]
- Kumar, A.; Zhang, K.Y.J. Application of shape similarity in pose selection and virtual screening in CSARdock2014 exercise. J. Chem. Inf. Model. 2016, 56, 965–973. [Google Scholar] [CrossRef] [PubMed]
- Jasper, J.B.; Humbeck, L.; Brinkjost, T.; Koch, O. A novel interaction fingerprint derived from per atom score contributions: exhaustive evaluation of interaction fingerprint performance in docking based virtual screening. J. Cheminform. 2018, 10, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da, C.; Kireev, D. Structural protein–ligand interaction fingerprints (SPLIF) for structure-based virtual screening: method and benchmark study. J. Chem. Inf. Model. 2014, 54, 2555–2561. [Google Scholar] [CrossRef]
- Liu, J.; Su, M.; Liu, Z.; Li, J.; Li, Y.; Wang, R. Enhance the performance of current scoring functions with the aid of 3D protein-ligand interaction fingerprints. BMC Bioinform. 2017, 18, 343. [Google Scholar] [CrossRef] [PubMed]
- Caporuscio, F.; Rastelli, G. Exploiting computationally derived out-of-the-box protein conformations for drug design. Future Med. Chem. 2016, 8, 1887–1897. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Y.; Li, L.; Chen, T.; Chen, W.; Xu, Y. Microsecond molecular dynamics simulation of Aβ42 and identification of a novel dual inhibitor of Aβ42 aggregation and BACE1 activity. Acta Pharmacol. Sin. 2013, 34, 1243–1250. [Google Scholar] [CrossRef] [PubMed]
- Spyrakis, F.; Benedetti, P.; Decherchi, S.; Rocchia, W.; Cavalli, A.; Alcaro, S.; Ortuso, F.; Baroni, M.; Cruciani, G. A pipeline to enhance ligand virtual screening: Integrating molecular dynamics and fingerprints for ligand and proteins. J. Chem. Inf. Model. 2015, 55, 2256–2274. [Google Scholar] [CrossRef]
- Park, H.S.; Jun, C.H. A simple and fast algorithm for K-medoids clustering. Expert Syst. Appl. 2009, 36, 3336–3341. [Google Scholar] [CrossRef]
- Baroni, M.; Cruciani, G.; Sciabola, S.; Perruccio, F.; Mason, J.S. A common reference framework for analyzing/comparing proteins and ligands. fingerprints for ligands and proteins (FLAP): Theory and application. J. Chem. Inf. Model. 2007, 47, 279–294. [Google Scholar] [CrossRef]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of useful decoys, enhanced (DUD-E): Better ligands and decoys for better benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef]
- Torrie, G.M.; Valleau, J.P. Nonphysical sampling distributions in Monte Carlo free-energy estimation: Umbrella sampling. J. Comput. Phys. 1977, 23, 187–199. [Google Scholar] [CrossRef]
- Laio, A.; Parrinello, M. Escaping free-energy minima. Proc. Natl. Acad. Sci. 2002, 99, 12562–12566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugita, Y.; Okamoto, Y. Replica-exchange molecular dynamics method for protein folding. Chem. Phys. Lett. 1999, 314, 141–151. [Google Scholar] [CrossRef]
- Oleinikovas, V.; Saladino, G.; Cossins, B.P.; Gervasio, F.L. Understanding cryptic pocket formation in protein targets by enhanced sampling simulations. J. Am. Chem. Soc. 2016, 138, 14257–14263. [Google Scholar] [CrossRef] [PubMed]
- Leontiadou, H.; Galdadas, I.; Athanasiou, C.; Cournia, Z. Insights into the mechanism of the PIK3CA E545K activating mutation using MD simulations. Sci. Rep. 2018, 8, 15544. [Google Scholar] [CrossRef]
- Yang, L.Q.; Sang, P.; Tao, Y.; Fu, Y.X.; Zhang, K.Q.; Xie, Y.H.; Liu, S.Q. Protein dynamics and motions in relation to their functions: several case studies and the underlying mechanisms. J. Biomol. Struct. Dyn. 2014, 32, 372–393. [Google Scholar] [CrossRef]
- Herbert, C.; Schieborr, U.; Saxena, K.; Juraszek, J.; De Smet, F.; Alcouffe, C.; Bianciotto, M.; Saladino, G.; Sibrac, D.; Kudlinzki, D.; et al. Molecular mechanism of SSR128129E, an extracellularly acting, small-molecule, allosteric inhibitor of FGF receptor signaling. Cancer Cell 2013, 23, 489–501. [Google Scholar] [CrossRef]
- D’Abramo, M.; Besker, N.; Chillemi, G.; Grottesi, A. Modeling conformational transitions in kinases by molecular dynamics simulations: achievements, difficulties, and open challenges. Front. Genet. 2014, 5, 128. [Google Scholar] [CrossRef] [Green Version]
- Meng, Y.; Lin, Y.; Roux, B. Computational study of the “DFG-flip” conformational transition in c-Abl and c-Src tyrosine kinases. J. Phys. Chem. B. 2015, 119, 1443–1456. [Google Scholar] [CrossRef]
- Berteotti, A.; Cavalli, A.; Branduardi, D.; Gervasio, F.L.; Recanatini, M.; Parrinello, M. Protein conformational transitions: The closure mechanism of a kinase explored by atomistic simulations. J. Am. Chem. Soc. 2009, 131, 244–250. [Google Scholar] [CrossRef]
- Morando, M.A.; Saladino, G.; D’Amelio, N.; Pucheta-Martinez, E.; Lovera, S.; Lelli, M.; López-Méndez, B.; Marenchino, M.; Campos-Olivas, R.; Gervasio, F.L. Conformational selection and induced fit mechanisms in the binding of an anticancer drug to the c-src kinase. Sci. Rep. 2016, 6, 24439. [Google Scholar] [CrossRef] [PubMed]
- Comitani, F.; Gervasio, F.L. Exploring cryptic pockets formation in targets of pharmaceutical interest with SWISH. J. Chem. Theory Comput. 2018, 14, 3321–3331. [Google Scholar] [CrossRef] [PubMed]
- Pisani, P.; Caporuscio, F.; Carlino, L.; Rastelli, G. Molecular dynamics simulations and classical multidimensional scaling unveil new metastable states in the conformational landscape of CDK2. PLoS ONE 2016, 11, e0154066. [Google Scholar] [CrossRef] [PubMed]
- Gioia, D.; Bertazzo, M.; Recanatini, M.; Masetti, M.; Cavalli, A. Dynamic docking: A paradigm shift in computational drug discovery. Molecules 2017, 22, 2029. [Google Scholar] [CrossRef] [PubMed]
- Degliesposti, G.; Portioli, C.; Parenti, M.D.; Rastelli, G. BEAR, a novel virtual screening methodology for drug discovery. J. Biomol. Screen. 2011, 16, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Parenti, M.D.; Rastelli, G. Advances and applications of binding affinity prediction methods in drug discovery. Biotechnol. Adv. 2012, 30, 244–250. [Google Scholar] [CrossRef]
- Rastelli, G. Emerging topics in structure-based virtual screening. Pharm. Res. 2013, 30, 1458–1463. [Google Scholar] [CrossRef]
- Gilson, M.K.; Zhou, H.X. Calculation of protein-ligand binding affinities. Annu. Rev. Biophys. Biomol. Struct. 2007, 36, 21–42. [Google Scholar] [CrossRef]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W.; et al. Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Thomas, L.L. Perspective on free-energy perturbation calculations for chemical equilibria. J. Chem. Theory Comput. 2008, 4, 869–876. [Google Scholar] [CrossRef]
- Limongelli, V.; Bonomi, M.; Parrinello, M. Funnel metadynamics as accurate binding free-energy method. Proc. Natl. Acad. Sci. USA. 2013, 110, 6358–6363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, A.J.; Tiwary, P.; Borrelli, K.; Feng, S.; Miller, E.B.; Abel, R.; Friesner, R.A.; Berne, B.J. Prediction of protein–ligand binding poses via a combination of induced fit docking and metadynamics simulations. J. Chem. Theory Comput. 2016, 12, 2990–2998. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.S.; Jo, S.; Lim, H.S.; Im, W. Application of binding free energy calculations to prediction of binding modes and affinities of MDM2 and MDMX inhibitors. J. Chem. Inf. Model. 2012, 52, 1821–1832. [Google Scholar] [CrossRef] [PubMed]
- Bhati, A.P.; Wan, S.; Wright, D.W.; Coveney, P.V. Rapid, accurate, precise, and reliable relative free energy prediction using ensemble based thermodynamic integration. J. Chem. Theory Comput. 2017, 13, 210–222. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Greene, D.; Xiao, L.; Qi, R.; Luo, R. Recent developments and applications of the MMPBSA method. Front. Mol. Biosci. 2018, 4, 87. [Google Scholar] [CrossRef] [PubMed]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Cortes, C.; Vapnik, V. Support-vector networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef]
- Wojcikowski, M.; Ballester, P.J.; Siedlecki, P.; Wójcikowski, M.; Ballester, P.J.; Siedlecki, P. Performance of machine-learning scoring functions in structure-based virtual screening. Sci. Rep. 2017, 7, 46710. [Google Scholar] [CrossRef]
- Xie, Q.Q.; Zhong, L.; Pan, Y.L.; Wang, X.Y.; Zhou, J.P.; Di-wu, L.; Huang, Q.; Wang, Y.L.; Yang, L.L.; Xie, H.Z.; et al. Combined SVM-based and docking-based virtual screening for retrieving novel inhibitors of c-Met. Eur. J. Med. Chem. 2011, 46, 3675–3680. [Google Scholar] [CrossRef]
- Leong, M.K.; Syu, R.G.; Ding, Y.L.; Weng, C.F. Prediction of N-methyl-D-aspartate receptor GluN1-ligand binding affinity by a novel SVM-Pose/SVM-score combinatorial ensemble docking scheme. Sci. Rep. 2017, 7, 40053. [Google Scholar] [CrossRef]
- Ballester, P.J.; Mitchell, J.B. A machine learning approach to predicting protein–ligand binding affinity with applications to molecular docking. Bioinformatics 2010, 26, 1169–1175. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Fang, X.; Lu, Y.; Wang, S. The PDBbind database: collection of binding affinities for protein−ligand complexes with known three-dimensional structures. J. Med. Chem. 2004, 47, 2977–2980. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhang, Y. Improving scoring-docking-screening powers of protein-ligand scoring functions using random forest. J. Comput. Chem. 2017, 38, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Pereira, J.C.; Caffarena, E.R.; dos Santos, C.N. Boosting docking-based virtual screening with deep learning. J. Chem. Inf. Model. 2016, 56, 2495–2506. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Wang, L.; Xie, X.Q. ProSelection: A novel algorithm to select proper protein structure subsets for in silico target identification and drug discovery research. J. Chem. Inf. Model. 2017, 57, 2686–2698. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Huang, M.; Zou, X. Docking-based inverse virtual screening: methods, applications, and challenges. Biophys. Rep. 2018, 4, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Z.; Li, H.; Zhang, H.; Liu, X.; Kang, L.; Luo, X.; Zhu, W.; Chen, K.; Wang, X.; Jiang, H. PDTD: a web-accessible protein database for drug target identification. BMC Bioinform. 2008, 9, 104. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Kellenberger, E.; Muller, P.; Schalon, C.; Bret, G.; Foata, N.; Rognan, D. sc-PDB: An annotated database of druggable binding sites from the Protein Data Bank. J. Chem. Inf. Model. 2006, 46, 717–727. [Google Scholar] [CrossRef]
- Kufareva, I.; Ilatovskiy, A.V.; Abagyan, R. Pocketome: An encyclopedia of small-molecule binding sites in 4D. Nucleic Acids Res. 2012, 40, D535–D540. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Ji, Z.L.; Chen, Y.Z. TTD: Therapeutic Target Database. Nucleic Acids Res. 2002, 30, 412–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef] [PubMed]
- Park, K.; Cho, A.E. Using reverse docking to identify potential targets for ginsenosides. J. Ginseng Res. 2017, 41, 534–539. [Google Scholar] [CrossRef] [PubMed]
- Nag, S.A.; Qin, J.J.; Wang, W.; Wang, M.H.; Wang, H.; Zhang, R. Ginsenosides as anticancer agents: in vitro and in vivo activities, structure-activity relationships, and molecular mechanisms of action. Front. Pharmacol. 2012, 3, 25. [Google Scholar] [CrossRef] [PubMed]
- Luo, Q.; Zhao, L.; Hu, J.; Jin, H.; Liu, Z.; Zhang, L. The scoring bias in reverse docking and the score normalization strategy to improve success rate of target fishing. PLoS ONE 2017, 12, e0171433. [Google Scholar] [CrossRef]
- Luo, H.; Fokoue-Nkoutche, A.; Singh, N.; Yang, L.; Hu, J.; Zhang, P. Molecular docking for prediction and interpretation of adverse drug reactions. Comb. Chem. High. Throughput Screen. 2018, 21, 314–322. [Google Scholar] [CrossRef]
- Nogueira, M.S.; Koch, O. The development of target-specific machine learning models as scoring functions for docking-based target prediction. J. Chem. Inf. Model. 2019, 59, 1238–1252. [Google Scholar] [CrossRef]
- Gaulton, A.; Bellis, L.J.; Bento, A.P.; Chambers, J.; Davies, M.; Hersey, A.; Light, Y.; McGlinchey, S.; Michalovich, D.; Al-Lazikani, B.; et al. ChEMBL: A large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2011, 40, D1100–D1107. [Google Scholar] [CrossRef]
- Perez-Sanchez, H.; Wenzel, W. Optimization methods for virtual screening on novel computational architectures. Curr. Comput. Aided. Drug Des. 2011, 7, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Dong, D.; Xu, Z.; Zhong, W.; Peng, S. Parallelization of molecular docking: A review. Curr. Top. Med. Chem. 2018, 18, 1015–1028. [Google Scholar] [CrossRef] [PubMed]
- Lapillo, M.; Tuccinardi, T.; Martinelli, A.; Macchia, M.; Giordano, A.; Poli, G. Extensive reliability evaluation of docking-based target-fishing strategies. Int. J. Mol. Sci. 2019, 20, 1023. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.Z.; Zhi, D.G. Ligand–protein inverse docking and its potential use in the computer search of protein targets of a small molecule. Proteins 2001, 43, 217–226. [Google Scholar] [CrossRef]
- Xie, T.; Zhang, L.; Zhang, S.; Ouyang, L.; Cai, H.; Liu, B. ACTP: A webserver for predicting potential targets and relevant pathways of autophagy-modulating compounds. Oncotarget 2016, 7, 10015–10022. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.C.; Chu, P.Y.; Chen, C.M.; Lin, J.H. idTarget: A web server for identifying protein targets of small chemical molecules with robust scoring functions and a divide-and-conquer docking approach. Nucleic Acids Res. 2012, 40, W393–W399. [Google Scholar] [CrossRef]
- Wang, Z.Y.; Kang, H.; Ji, L.L.; Yang, Y.Q.; Liu, T.; Cao, Z.W.; Morahan, G.; Wang, Z.T. Proteomic characterization of the possible molecular targets of pyrrolizidine alkaloid isoline-induced hepatotoxicity. Environ. Toxicol. Pharmacol. 2012, 34, 608–617. [Google Scholar] [CrossRef]
- Ji, Z.L.; Wang, Y.; Yu, L.; Han, L.Y.; Zheng, C.J.; Chen, Y.Z. In silico search of putative adverse drug reaction related proteins as a potential tool for facilitating drug adverse effect prediction. Toxicol. Lett. 2006, 164, 104–112. [Google Scholar] [CrossRef]
- Eric, S.; Ke, S.; Barata, T.; Solmajer, T.; Antic Stankovic, J.; Juranic, Z.; Savic, V.; Zloh, M. Target fishing and docking studies of the novel derivatives of aryl-aminopyridines with potential anticancer activity. Bioorg. Med. Chem. 2012, 20, 5220–5228. [Google Scholar] [CrossRef]
- Zhang, L.; Fu, L.; Zhang, S.; Zhang, J.; Zhao, Y.; Zheng, Y.; He, G.; Yang, S.; Ouyang, L.; Liu, B. Discovery of a small molecule targeting ULK1-modulated cell death of triple negative breast cancer in vitro and in vivo. Chem. Sci. 2017, 8, 2687–2701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassan, A.H.E.; Yoo, S.Y.; Lee, K.W.; Yoon, Y.M.; Ryu, H.W.; Jeong, Y.; Shin, J.S.; Kang, S.Y.; Kim, S.Y.; Lee, H.H.; et al. Repurposing mosloflavone/5,6,7-trimethoxyflavone-resveratrol hybrids: Discovery of novel p38-α MAPK inhibitors as potent interceptors of macrophage-dependent production of proinflammatory mediators. Eur. J. Med. Chem. 2019, 180, 253–267. [Google Scholar] [CrossRef] [PubMed]
- Chang, D.T.H.; Oyang, Y.J.; Lin, J.H. MEDock: A web server for efficient prediction of ligand binding sites based on a novel optimization algorithm. Nucleic Acids Res. 2005, 33, W233–W238. [Google Scholar] [CrossRef] [PubMed]
- Diller, D.J.; Merz, K.M.J. High throughput docking for library design and library prioritization. Proteins 2001, 43, 113–124. [Google Scholar] [CrossRef]
- Muhammed, M.T.; Aki-Yalcin, E. Homology modeling in drug discovery: Overview, current applications, and future perspectives. Chem. Biol. Drug Des. 2019, 93, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Daga, P.R.; Patel, R.Y.; Doerksen, R.J. Template-based protein modeling: recent methodological advances. Curr. Top. Med. Chem. 2010, 10, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Klein, E.; Bourdette, D. Postmarketing adverse drug reactions: A duty to report? Neurol. Clin. Pract. 2013, 3, 288–294. [Google Scholar] [CrossRef] [Green Version]
- Yoo, S.; Noh, K.; Shin, M.; Park, J.; Lee, K.H.; Nam, H.; Lee, D. In silico profiling of systemic effects of drugs to predict unexpected interactions. Sci. Rep. 2018, 8, 1612. [Google Scholar] [CrossRef] [Green Version]
- Dimitri, G.M.; Lió, P. DrugClust: A machine learning approach for drugs side effects prediction. Comput. Biol. Chem. 2017, 68, 204–210. [Google Scholar] [CrossRef]
- Li, Z.; Han, P.; You, Z.H.; Li, X.; Zhang, Y.; Yu, H.; Nie, R.; Chen, X. In silico prediction of drug-target interaction networks based on drug chemical structure and protein sequences. Sci. Rep. 2017, 7, 11174. [Google Scholar] [CrossRef]
- Ekins, S. Predicting undesirable drug interactions with promiscuous proteins in silico. Drug Discov. Today 2004, 9, 276–285. [Google Scholar] [CrossRef]
- Chen, Y.Z.; Ung, C.Y. Prediction of potential toxicity and side effect protein targets of a small molecule by a ligand-protein inverse docking approach. J. Mol. Graph. Model. 2001, 20, 199–218. [Google Scholar] [CrossRef]
- Yang, L.; Chen, J.; He, L. Harvesting candidate genes responsible for serious adverse drug reactions from a chemical-protein interactome. PLoS Comput. Biol. 2009, 5, e1000441. [Google Scholar] [CrossRef] [PubMed]
- Li, C.Y.; Yu, Q.; Ye, Z.Q.; Sun, Y.; He, Q.; Li, X.M.; Zhang, W.; Luo, J.; Gu, X.; Zheng, X.; et al. A nonsynonymous SNP in human cytosolic sialidase in a small Asian population results in reduced enzyme activity: Potential link with severe adverse reactions to oseltamivir. Cell Res. 2007, 17, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, M.; Letunic, I.; Jensen, L.J.; Bork, P. The SIDER database of drugs and side effects. Nucleic Acids Res. 2016, 44, D1075–D1079. [Google Scholar] [CrossRef] [PubMed]
- Jaundoo, R.; Bohmann, J.; Gutierrez, G.E.; Klimas, N.; Broderick, G.; Craddock, T.J.A. Using a consensus docking approach to predict adverse drug reactions in combination drug therapies for gulf war illness. Int. J. Mol. Sci. 2018, 19, 3355. [Google Scholar] [CrossRef]
- Kola, I.; Landis, J. Can the pharmaceutical industry reduce attrition rates? Nat. Rev. Drug Discov. 2004, 3, 711–716. [Google Scholar] [CrossRef]
- Zhang, W.; Bai, Y.; Wang, Y.; Xiao, W. Polypharmacology in drug discovery: A review from systems pharmacology perspective. Curr. Pharm. Des. 2016, 22, 3171–3181. [Google Scholar] [CrossRef]
- Lepailleur, A.; Freret, T.; Lemaître, S.; Boulouard, M.; Dauphin, F.; Hinschberger, A.; Dulin, F.; Lesnard, A.; Bureau, R.; Rault, S. Dual histamine H3R/serotonin 5-HT4R ligands with antiamnesic properties: pharmacophore-based virtual screening and polypharmacology. J. Chem. Inf. Model. 2014, 54, 1773–1784. [Google Scholar] [CrossRef] [PubMed]
- Wei, D.; Jiang, X.; Zhou, L.; Chen, J.; Chen, Z.; He, C.; Yang, K.; Liu, Y.; Pei, J.; Lai, L. Discovery of multitarget inhibitors by combining molecular docking with common pharmacophore matching. J. Med. Chem. 2008, 51, 7882–7888. [Google Scholar] [CrossRef] [PubMed]
- Rastelli, G.; Pinzi, L. Computational polypharmacology comes of age. Front. Pharmacol. 2015, 6, 157. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Pei, J.; Lai, L. Computational multitarget drug design. J. Chem. Inf. Model. 2017, 57, 403–412. [Google Scholar] [CrossRef] [PubMed]
- Selvam, B.; Porter, S.L.; Tikhonova, I.G. Addressing selective polypharmacology of antipsychotic drugs targeting the bioaminergic receptors through receptor dynamic conformational ensembles. J. Chem. Inf. Model. 2013, 53, 1761–1774. [Google Scholar] [CrossRef] [PubMed]
- Minie, M.; Chopra, G.; Sethi, G.; Horst, J.; White, G.; Roy, A.; Hatti, K.; Samudrala, R. CANDO and the infinite drug discovery frontier. Drug Discov. Today 2014, 19, 1353–1363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chopra, G.; Samudrala, R. Exploring polypharmacology in drug discovery and repurposing using the CANDO platform. Curr. Pharm. Des. 2016, 22, 3109–3123. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Chen, J.; Shi, L.; Mikailov, M.; Zhu, H.; Wang, K.; He, L.; Yang, L. DRAR-CPI: A server for identifying drug repositioning potential and adverse drug reactions via the chemical-protein interactome. Nucleic Acids Res. 2011, 39, W492–W498. [Google Scholar] [CrossRef]
- Luo, H.; Zhang, P.; Cao, X.H.; Du, D.; Ye, H.; Huang, H.; Li, C.; Qin, S.; Wan, C.; Shi, L.; et al. DPDR-CPI, a server that predicts drug positioning and drug repositioning via chemical-protein interactome. Sci. Rep. 2016, 6, 35996. [Google Scholar] [CrossRef]
- Hurle, M.R.; Yang, L.; Xie, Q.; Rajpal, D.K.; Sanseau, P.; Agarwal, P. Computational drug repositioning: From data to therapeutics. Clin. Pharmacol. Ther. 2013, 93, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Keiser, M.J.; Setola, V.; Irwin, J.J.; Laggner, C.; Abbas, A.I.; Hufeisen, S.J.; Jensen, N.H.; Kuijer, M.B.; Matos, R.C.; Tran, T.B.; et al. Predicting new molecular targets for known drugs. Nature 2009, 462, 175–181. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Zhu, F.; Ma, X.H.; Shi, Z.; Yang, S.Y.; Wei, Y.Q.; Chen, Y.Z. Predicting targeted polypharmacology for drug repositioning and multi- target drug discovery. Curr. Med. Chem. 2013, 20, 1646–1661. [Google Scholar] [CrossRef]
- Nugent, T.; Plachouras, V.; Leidner, J.L. Computational drug repositioning based on side-effects mined from social media. PeerJ Comput. Sci. 2016, 2, e46. [Google Scholar] [CrossRef]
- Defranchi, E.; Schalon, C.; Messa, M.; Onofri, F.; Benfenati, F.; Rognan, D. Binding of protein kinase inhibitors to synapsin I inferred from pair-wise binding site similarity measurements. PLoS ONE 2010, 5, e12214. [Google Scholar] [CrossRef]
- Barneh, F.; Jafari, M.; Mirzaie, M. Updates on drug-target network; facilitating polypharmacology and data integration by growth of DrugBank database. Brief. Bioinform. 2016, 17, 1070–1080. [Google Scholar] [CrossRef] [PubMed]
- Kharkar, P.S.; Warrier, S.; Gaud, R.S. Reverse docking: A powerful tool for drug repositioning and drug rescue. Future Med. Chem. 2014, 6, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Wenning, G.K.; O’Connell, M.T.; Patsalos, P.N.; Quinn, N.P. A clinical and pharmacokinetic case study of an interaction of levodopa and antituberculous therapy in Parkinson’s disease. Mov. Disord. 1995, 10, 664–667. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.H.; Chou, C.H.; Liu, F.C.; Lin, T.Y.; Huang, W.Y.; Wang, Y.C.; Kao, C.H. Association Between Tuberculosis and Parkinson Disease: A Nationwide, Population-Based Cohort Study. Medicine 2016, 95, e2883. [Google Scholar] [CrossRef] [PubMed]
- Dakshanamurthy, S.; Issa, N.T.; Assefnia, S.; Seshasayee, A.; Peters, O.J.; Madhavan, S.; Uren, A.; Brown, M.L.; Byers, S.W. Predicting new indications for approved drugs using a proteochemometric method. J. Med. Chem. 2012, 55, 6832–6848. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Shrivastava, S.; Hassanali, M.; Stothard, P.; Chang, Z.; Woolsey, J. DrugBank: A comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006, 34, D668–D672. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Lin, Y.; Wen, X.; Jorissen, R.N.; Gilson, M.K. BindingDB: A web-accessible database of experimentally determined protein–ligand binding affinities. Nucleic Acids Res. 2007, 35, D198–D201. [Google Scholar] [CrossRef] [PubMed]
- Koehn, F.E.; Carter, G.T. The evolving role of natural products in drug discovery. Nat. Rev. Drug Discov. 2005, 4, 206–220. [Google Scholar] [CrossRef]
- Harvey, A.L. Natural products in drug discovery. Drug Discov. Today 2008, 13, 894–901. [Google Scholar] [CrossRef] [PubMed]
- Thomford, N.E.; Senthebane, D.A.; Rowe, A.; Munro, D.; Seele, P.; Maroyi, A.; Dzobo, K. Natural products for drug discovery in the 21st century: innovations for novel drug discovery. Int. J. Mol. Sci. 2018, 19, 1578. [Google Scholar] [CrossRef] [PubMed]
- Kinnings, S.L.; Liu, N.; Tonge, P.J.; Jackson, R.M.; Xie, L.; Bourne, P.E. A machine learning-based method to improve docking scoring functions and its application to drug repurposing. J. Chem. Inf. Model. 2011, 51, 408–419. [Google Scholar] [CrossRef] [PubMed]
- Labbé, C.M.; Rey, J.; Lagorce, D.; Vavruša, M.; Becot, J.; Sperandio, O.; Villoutreix, B.O.; Tufféry, P.; Miteva, M.A. MTiOpenScreen: A web server for structure-based virtual screening. Nucleic Acids Res. 2015, 43, W448–W454. [Google Scholar] [CrossRef] [PubMed]
- Lagarde, N.; Rey, J.; Gyulkhandanyan, A.; Tufféry, P.; Miteva, M.A.; Villoutreix, B.O. Online structure-based screening of purchasable approved drugs and natural compounds: Retrospective examples of drug repositioning on cancer targets. Oncotarget 2018, 9, 32346–32361. [Google Scholar] [CrossRef] [PubMed]
- Grosdidier, A.; Zoete, V.; Michielin, O. SwissDock, a protein-small molecule docking web service based on EADock DSS. Nucleic Acids Res. 2011, 39, W270–W277. [Google Scholar] [CrossRef] [Green Version]
- Irwin, J.J.; Shoichet, B.K.; Mysinger, M.M.; Huang, N.; Colizzi, F.; Wassam, P.; Cao, Y. Automated docking screens: A feasibility study. J. Med. Chem. 2009, 52, 5712–5720. [Google Scholar] [CrossRef]
- Gawehn, E.; Hiss, J.A.; Brown, J.B.; Schneider, G. Advancing drug discovery via GPU-based deep learning. Expert Opin. Drug Discov. 2018, 13, 579–582. [Google Scholar] [CrossRef] [Green Version]
- Loukatou, S.; Papageorgiou, L.; Fakourelis, P.; Filntisi, A.; Polychronidou, E.; Bassis, I.; Megalooikonomou, V.; Makałowski, W.; Vlachakis, D.; Kossida, S. Molecular dynamics simulations through GPU video games technologies. J. Mol. Biochem. 2014, 3, 64–71. [Google Scholar]
- Chen, X.W.; Lin, X. Big data deep learning: challenges and perspectives. IEEE Access 2014, 2, 514–525. [Google Scholar] [CrossRef]
- Stone, J.E.; Hardy, D.J.; Ufimtsev, I.S.; Schulten, K. GPU-accelerated molecular modeling coming of age. J. Mol. Graph. Model. 2010, 29, 116–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dowden, H.; Munro, J. Trends in clinical success rates and therapeutic focus. Nat. Rev. Drug Discov. 2019, 18, 495–496. [Google Scholar] [CrossRef] [PubMed]
- Kinch, M.S.; Haynesworth, A.; Kinch, S.L.; Hoyer, D. An overview of FDA-approved new molecular entities: 1827–2013. Drug Discov. Today 2014, 19, 1033–1039. [Google Scholar] [CrossRef] [PubMed]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pinzi, L.; Rastelli, G. Molecular Docking: Shifting Paradigms in Drug Discovery. Int. J. Mol. Sci. 2019, 20, 4331. https://doi.org/10.3390/ijms20184331
Pinzi L, Rastelli G. Molecular Docking: Shifting Paradigms in Drug Discovery. International Journal of Molecular Sciences. 2019; 20(18):4331. https://doi.org/10.3390/ijms20184331
Chicago/Turabian StylePinzi, Luca, and Giulio Rastelli. 2019. "Molecular Docking: Shifting Paradigms in Drug Discovery" International Journal of Molecular Sciences 20, no. 18: 4331. https://doi.org/10.3390/ijms20184331
APA StylePinzi, L., & Rastelli, G. (2019). Molecular Docking: Shifting Paradigms in Drug Discovery. International Journal of Molecular Sciences, 20(18), 4331. https://doi.org/10.3390/ijms20184331