Can We Still Trust Docking Results? An Extension of the Applicability of DockBench on PDBbind Database




Abstract
:
1. Introduction
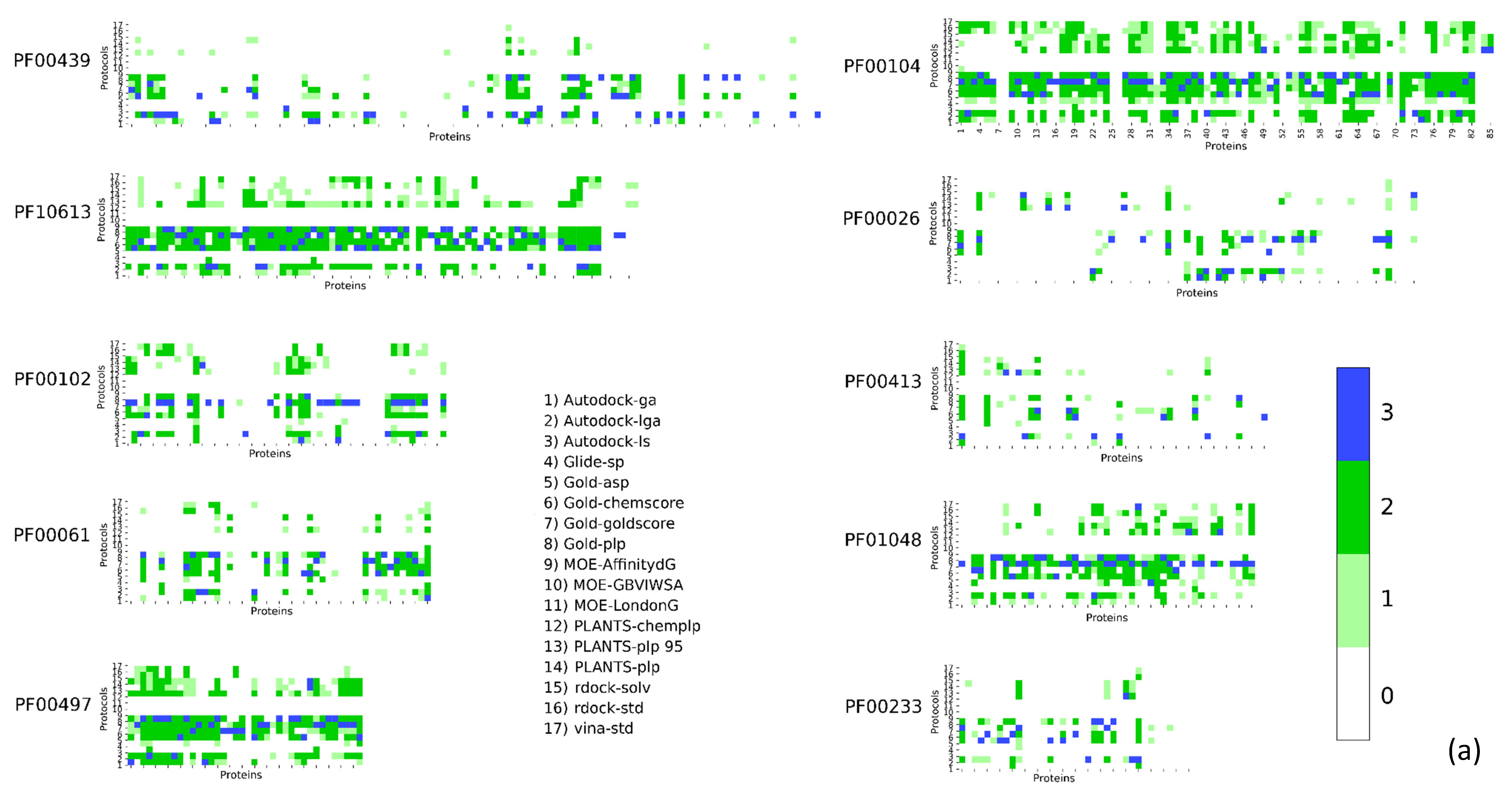
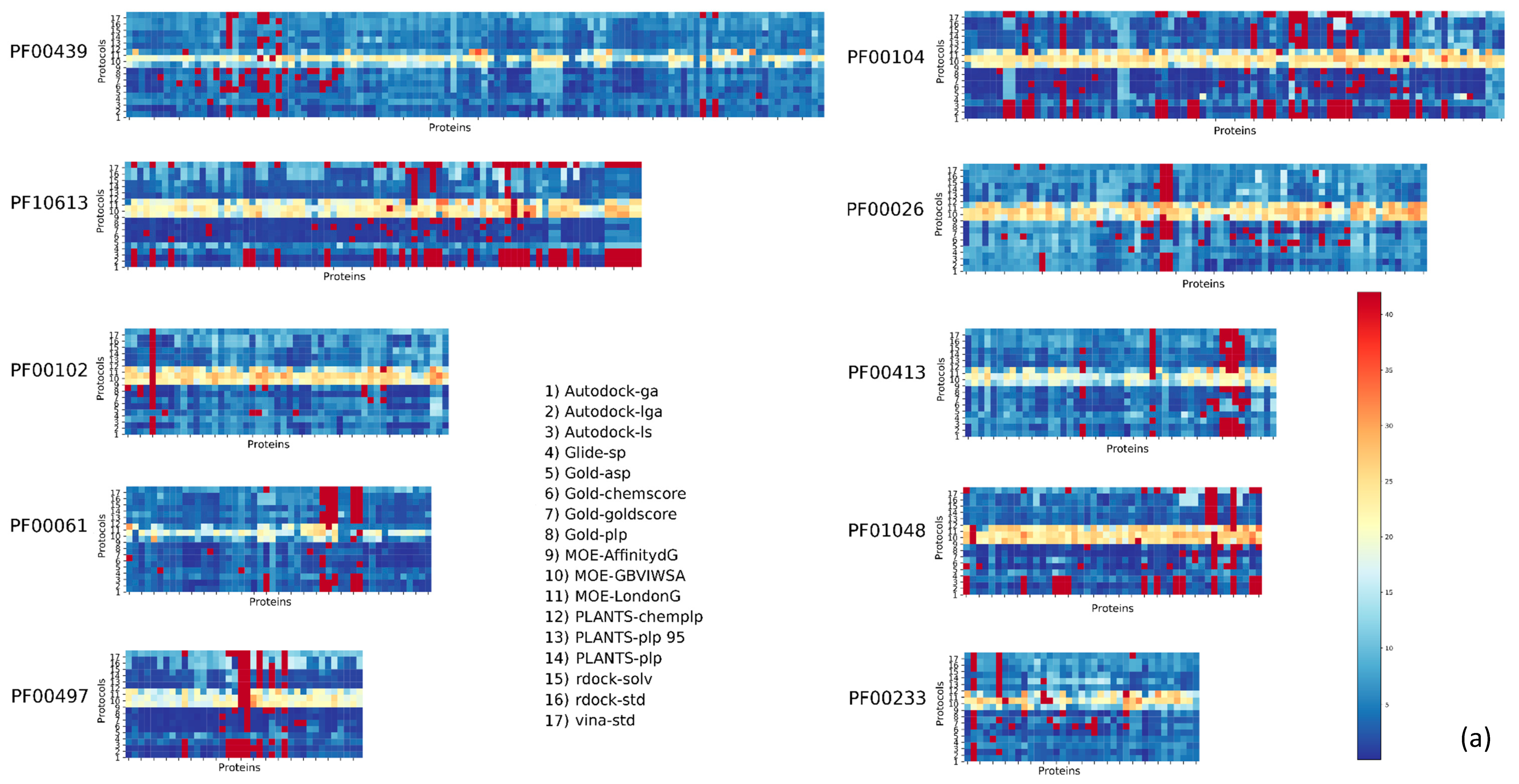
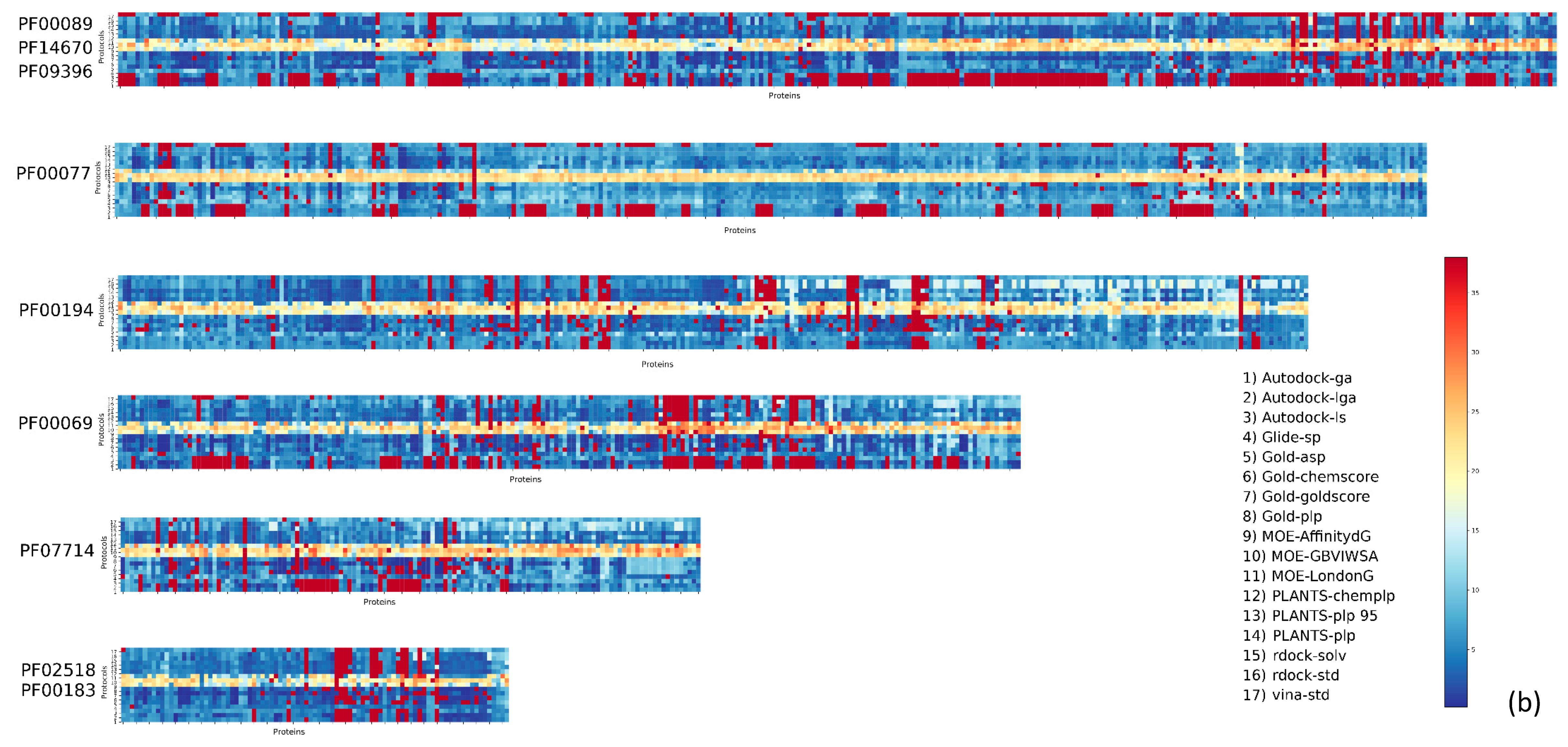
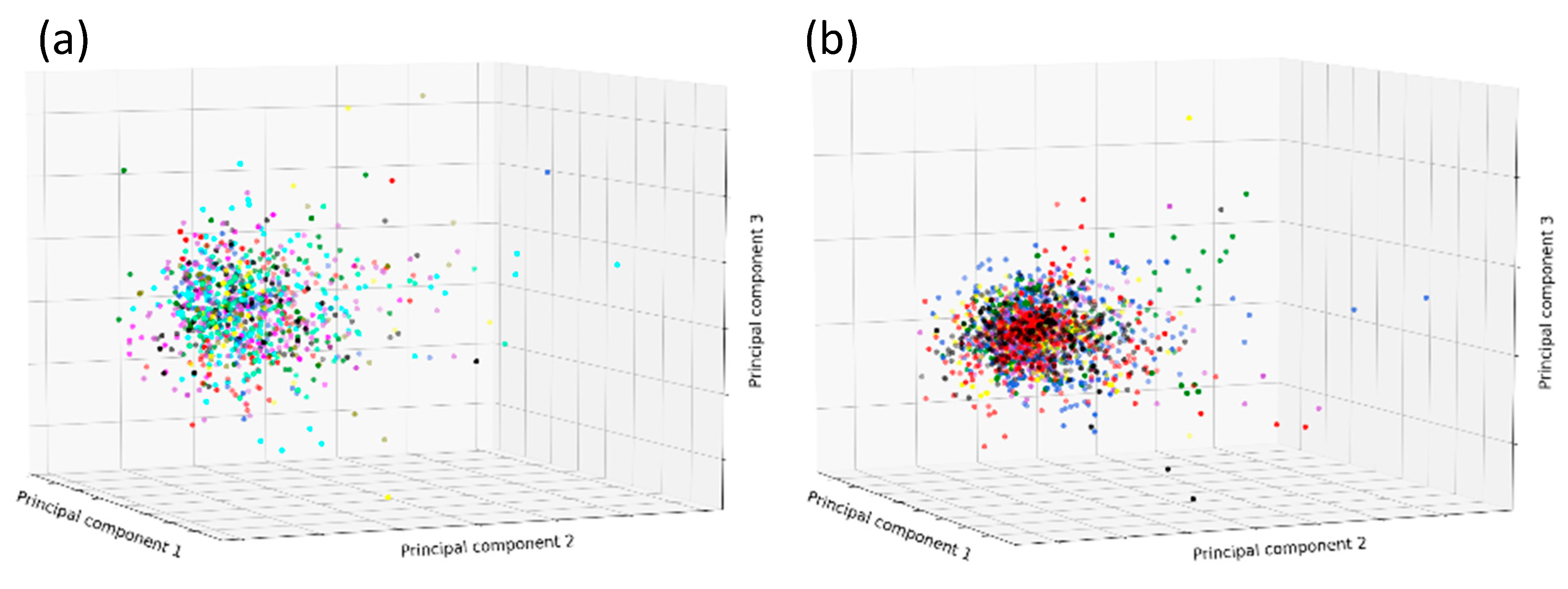
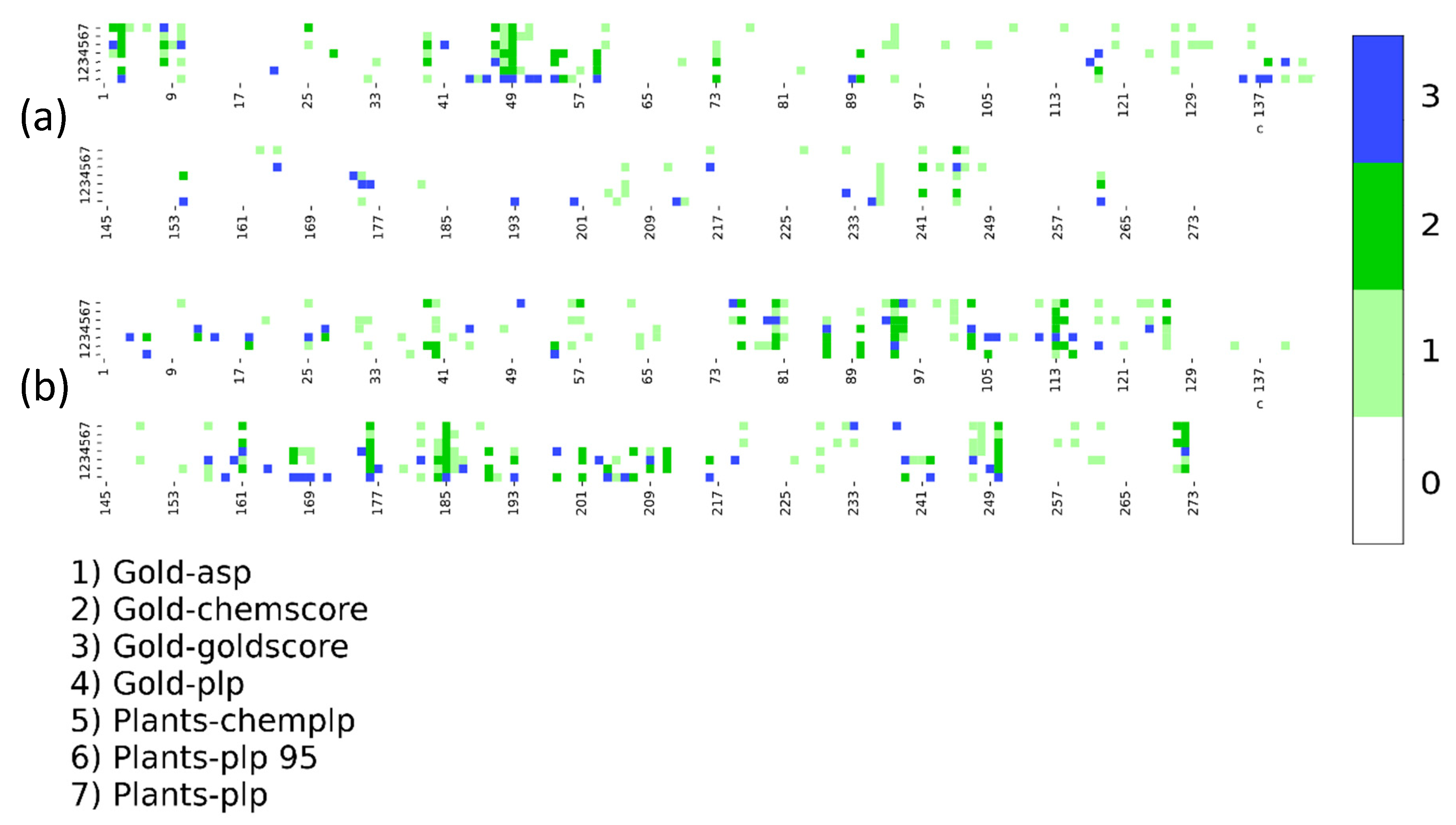
2. Results
3. Discussion
4. Materials and Methods
4.1. Database Preparation
4.2. Benchmark: Software and Hardware
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| SB | Structure-Based |
| VS | Virtual Screening |
| PFAM | Protein Family |
| NMR | Nuclear Magnetic Resona |
| CryoEM | Cryo Electron Microscpy |
| MMFF94 | Merck Molecular Force Field |
| RMSD | Root Main Square Deviation |
References
- Kuntz, I.D.; Blaney, J.M.; Oatley, S.J.; Langridge, R.; Ferrin, T.E. A geometric approach to macromolecule-ligand interactions. J. Mol. Biol. 1982, 161, 269–288. [Google Scholar] [CrossRef]
- Horvath, D. A virtual screening approach applied to the search for trypanothione reductase inhibitors. J. Med. Chem. 1997, 40, 2412–2423. [Google Scholar] [CrossRef] [PubMed]
- Mobley, D.L.; Dill, K.A. Binding of small-molecule ligands to proteins: “What you see” is not always “what you get”. Structure 2009, 17, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Moro, S.; Sturlese, M.; Ciancetta, A.; Floris, M. In silico 3D modeling of binding activities. Methods Mol. Biol. 2016, 1425, 23–35. [Google Scholar] [PubMed]
- Directory of in Silico Drug Design Tools - Docking. Available online: http://www.click2drug.org/directory_Docking.html (accessed on 25 May 2016).
- Houston, D.R.; Walkinshaw, M.D. Consensus docking: improving the reliability of docking in a virtual screening context. J. Chem. Inf. Model. 2013, 53, 384–390. [Google Scholar] [CrossRef] [PubMed]
- Salmaso, V.; Sturlese, M.; Cuzzolin, A.; Moro, S. DockBench as docking selector tool: the lesson learned from D3R Grand Challenge 2015. J. Comput. Aided. Mol. Des. 2016, 30, 773–789. [Google Scholar] [CrossRef]
- Plewczynski, D.; Łaźniewski, M.; Augustyniak, R.; Ginalski, K. Can we trust docking results? Evaluation of seven commonly used programs on PDBbind database. J. Comput. Chem. 2011, 32, 742–755. [Google Scholar] [CrossRef]
- Ciancetta, A.; Cuzzolin, A.; Moro, S. Alternative quality assessment strategy to compare performances of GPCR-ligand docking protocols: the human adenosine A(2A) receptor as a case study. J. Chem. Inf. Model. 2014, 54, 2243–2254. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Fang, X.; Lu, Y.; Yang, C.Y.; Wang, S. The PDBbind database: methodologies and updates. J. Med. Chem. 2005, 48, 4111–4119. [Google Scholar] [CrossRef] [PubMed]
- Chemical Computing Group (CCG) Inc. Molecular Operating Environment (MOE); Chemical Computing Group: Montreal, QC, Canada, 2019. [Google Scholar]
- OpenEye Scientific Software Inc. OEChem; OpenEye Scientific Software Inc.: Santa Fe, NM, USA, 2016. [Google Scholar]
- El-Gebali, S.; Mistry, J.; Bateman, A.; Eddy, S.R.; Luciani, A.; Potter, S.C.; Qureshi, M.; Richardson, L.J.; Salazar, G.A.; Smart, A.; et al. The Pfam protein families database in 2019. Nucleic Acids Res. 2019, 47, D427–D432. [Google Scholar] [CrossRef]
- Shadrick, W.R.; Slavish, P.J.; Chai, S.C.; Waddell, B.; Connelly, M.; Low, J.A.; Tallant, C.; Young, B.M.; Bharatham, N.; Knapp, S.; et al. Exploiting a water network to achieve enthalpy-driven, bromodomain-selective BET inhibitors. Bioorg. Med. Chem. 2018, 26, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comput. Chem. 1996, 17, 490–519. [Google Scholar] [CrossRef]
- Cuzzolin, A.; Sturlese, M.; Malvacio, I.; Ciancetta, A.; Moro, S. DockBench: An Integrated Informatic Platform Bridging the Gap between the Robust Validation of Docking Protocols and Virtual Screening Simulations. Molecules 2015, 20, 9977–9993. [Google Scholar] [CrossRef] [PubMed]
- Salmaso, V.; Sturlese, M.; Cuzzolin, A.; Moro, S. Combining self- and cross-docking as benchmark tools: the performance of DockBench in the D3R Grand Challenge 2. J. Comput. Aided Mol. Des. 2018, 32, 251–264. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Korb, O.; Stützle, T.; Exner, T.E.; Dorigo, M.; Gambardella, L.M.; Birattari, M.; Martinoli, A.; Poli, R.; Hutchison, D.; Kanade, T.; et al. PLANTS: Application of Ant Colony Optimization to Structure-Based Drug Design. In Ant Colony Optimization and Swarm Intelligence; Springer: Berlin/Heidelberg, Germany, 2006; Volume 4150, pp. 247–258. [Google Scholar]
- Ruiz-Carmona, S.; Alvarez-Garcia, D.; Foloppe, N.; Garmendia-Doval, A.B.; Juhos, S.; Schmidtke, P.; Barril, X.; Hubbard, R.E.; Morley, S.D. rDock: A fast, versatile and open source program for docking ligands to proteins and nucleic acids. PLoS Comput. Biol. 2014, 10, e1003571. [Google Scholar] [CrossRef]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef]
- Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.D. Improved protein-ligand docking using GOLD. Proteins 2003, 52, 609–623. [Google Scholar] [CrossRef]
- Cambridge Crystallographic Data Centre. GOLD Suite, version 5.2; Cambridge Crystallographic Data Centre: Cambridge, UK, 2013. [Google Scholar]
- Hunter, J.D. Matplotlib: A 2D Graphics Environment. Comput Sci Eng 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 285–2830. [Google Scholar]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic. Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Program | Search Algorithm/Placing Method | Scoring Function | Protocol Abbreviation |
|---|---|---|---|
| Autodock 4.2 | Local Search | AutoDock SF | AUTODOCK-ls |
| Lamarckian GA | AutoDock SF | AUTODOCK-lga | |
| Genetic Algorithm | AutoDock SF | AUTODOCK-ga | |
| Vina 1.1.2 | Monte Carlo + BFGS local search | Standard Vina SF | VINA-std |
| Glide 6.5 | Glide Algorithm | Standard Precision | GLIDE-sp |
| GOLD 5.4.1 | Genetic Algorithm | Goldscore | GOLD-goldscore |
| Genetic Algorithm | Chemscore | GOLD-chemscore | |
| Genetic Algorithm | ASP | GOLD-asp | |
| Genetic Algorithm | PLP | GOLD-plp | |
| MOE 2019.01 | Triangle Matcher | London-dG | MOE-londondg |
| Triangle Matcher | Affinity-dG | MOE-affinitydg | |
| Triangle Matcher | GBIVIWSA | MOE-gbiviwsa | |
| PLANTS 1.2 | ACO Algorithm | PLP | PLANTS-plp |
| ACO Algorithm | PLP95 | PLANTS-plp95 | |
| ACO Algorithm | ChemPLP | PLANTS-chemplp | |
| rDock 2013.1 | Genetic Algorithm + Monte Carlo + Simplex minimization | Standard rDock master SF | RDOCK-std |
| Genetic Algorithm + Monte Carlo + Simplex minimization | Standard rDock master SF + desolvation potential | RDOCK-solv |
| Pfam Family | Protein Description | Size | Protocol Score Pscore% | ||||
|---|---|---|---|---|---|---|---|
| 0 | 1 | 2 | 3 | >1 | |||
| PF00104 | Ligand-binding domain of nuclear hormone receptor | 85 | 59.34 | 10.24 | 26.57 | 4.84 | 41.66 |
| PF00497 | Bacterial extracellular solute-binding proteins, family 3 | 38 | 59.29 | 9.44 | 25.70 | 5.57 | 40.71 |
| PF10613 | Ligated ion channel l-glutamate- and glycine-binding site | 83 | 67.97 | 6.80 | 20.55 | 4.68 | 32.03 |
| PF01048 | Phosphorylase superfamily | 47 | 70.09 | 8.01 | 16.77 | 5.13 | 29.91 |
| PF00102 | Protein-tyrosine phosphatase | 52 | 79.30 | 5.20 | 11.99 | 3.51 | 20.70 |
| PF00069 | Protein kinase domain | 207 | 80.68 | 5.43 | 10.46 | 3.43 | 19.32 |
| PF00061 | Lipocalin/cytosolic fatty-acid binding protein family | 49 | 82.11 | 4.08 | 10.80 | 3.00 | 17.88 |
| PF02518 PF00183 | Hsp90 protein and GHKL domain | 89 | 82.74 | 5.35 | 8.26 | 3.64 | 17.25 |
| PF07714 | Protein tyrosine kinase | 133 | 83.90 | 5.79 | 6.77 | 3.54 | 16.10 |
| PF00089 PF14670 PF09396 | Trypsin | 330 | 85.54 | 4.65 | 6.84 | 2.96 | 14.45 |
| PF00233 | 3′5′-cyclic nucleotide phosphodiesterase | 37 | 87.92 | 3.82 | 5.41 | 2.86 | 12,08 |
| PF00439 | Bromodomain | 112 | 90.02 | 2.89 | 4.67 | 2.42 | 9.98 |
| PF00026 | Eukaryotic aspartyl protease | 73 | 90.49 | 3.14 | 4.11 | 2.26 | 9.51 |
| PF00413 | Matrixin | 49 | 90.88 | 3.24 | 4.20 | 1.68 | 9.12 |
| PF00077 | Retroviral aspartyl protease | 301 | 95.41 | 2.27 | 1.64 | 0.68 | 4.59 |
| PF00194 | Eukaryotic-type carbonic anhydrase | 273 | 95.63 | 2.28 | 1.17 | 0.91 | 4.37 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bolcato, G.; Cuzzolin, A.; Bissaro, M.; Moro, S.; Sturlese, M. Can We Still Trust Docking Results? An Extension of the Applicability of DockBench on PDBbind Database. Int. J. Mol. Sci. 2019, 20, 3558. https://doi.org/10.3390/ijms20143558
Bolcato G, Cuzzolin A, Bissaro M, Moro S, Sturlese M. Can We Still Trust Docking Results? An Extension of the Applicability of DockBench on PDBbind Database. International Journal of Molecular Sciences. 2019; 20(14):3558. https://doi.org/10.3390/ijms20143558
Chicago/Turabian StyleBolcato, Giovanni, Alberto Cuzzolin, Maicol Bissaro, Stefano Moro, and Mattia Sturlese. 2019. "Can We Still Trust Docking Results? An Extension of the Applicability of DockBench on PDBbind Database" International Journal of Molecular Sciences 20, no. 14: 3558. https://doi.org/10.3390/ijms20143558
APA StyleBolcato, G., Cuzzolin, A., Bissaro, M., Moro, S., & Sturlese, M. (2019). Can We Still Trust Docking Results? An Extension of the Applicability of DockBench on PDBbind Database. International Journal of Molecular Sciences, 20(14), 3558. https://doi.org/10.3390/ijms20143558