Autophagy: New Insights into Mechanisms of Action and Resistance of Treatment in Acute Promyelocytic leukemia


Abstract
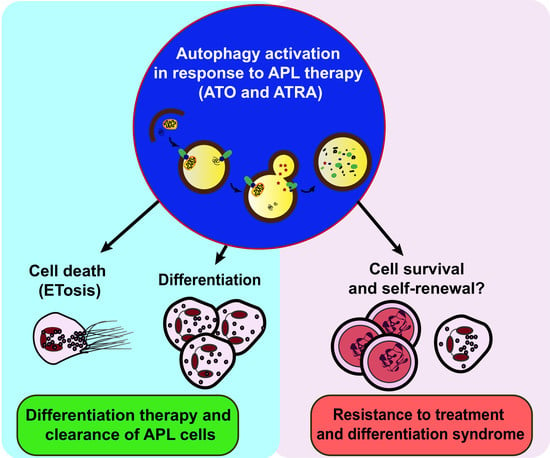
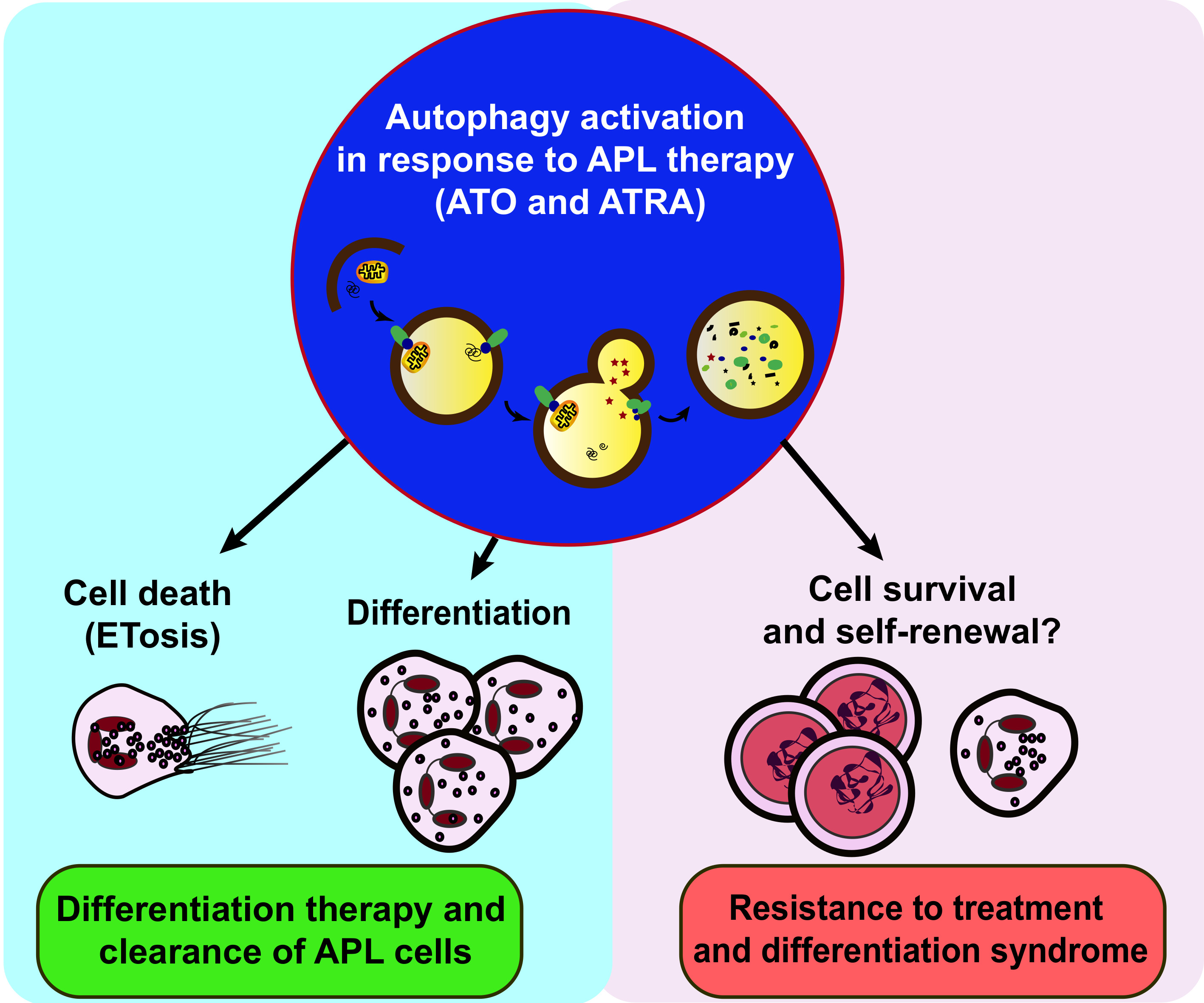
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
1.1. Acute Promyelocytic Leukemia
1.2. The Modes of Action of APL Therapy
1.3. All-Trans Retinoic Acid (ATRA)
1.4. Arsenic Trioxide
1.5. Differentiation Syndrome
2. The Autophagy Machinery and its Role in Cancer
3. Regulation of Autophagy in APL Cells at Basal Level and in Response to Treatment
4. Regulation of Autophagy during Differentiation of APL Cells by ATRA
5. Regulation of Autophagy in APL Cells Following Arsenic Trioxide Treatment
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AML | Acute myeloid leukemia |
| AMPK | AMP-activated protein kinase |
| APL | Acute promyelocytic leukemia |
| ARID1B | AT rich interactive domain 1B |
| ATG | Autophagy-related gene |
| ATO | Arsenic trioxide |
| ATRA | All-trans retinoid acid |
| BCL-2 | B-cell lymphoma 2 |
| BNIP3 | BCL2-interacting protein 3 |
| DRAM1 | DNA damage regulated autophagy modulator 1 |
| ERK | Extracellular signal-regulated kinase |
| FAM134B | Family with sequence similarity 134 member B |
| FIP200 | Focal adhesion kinase family interacting protein of 200 kD |
| FLT3 | Fms-like tyrosine kinase 3 |
| GABARAP | Gamma-aminobutyric acid receptor-associated protein |
| HMGB1 | High–mobility group box 1 |
| LncRNA | Long-noncoding RNA |
| MAM | Mitochondrial-associated membrane |
| MAP1S | Microtubule-associated protein 1S |
| MEK | Mitogen-activated protein kinase kinase |
| MTOR | Mechanistic target of rapamycin kinase |
| NBR1 | Next to BRCA1 gene 1 protein |
| NCoR1 | Nuclear receptor corepressor |
| NDP52 | Nuclear dot protein 52 kDa |
| NEDD8 E1 | NEDD8 activating E1 enzyme |
| NET (ET) | Neutrophil extracellular trap |
| NF-kappaB | Nuclear factor kappa B |
| PI3K | Phosphoinositide 3-kinase |
| PLZF-RARα | Leukemia zinc finger-retinoic acid receptor α |
| PML | Promyelocytic leukemia protein |
| RAR | Retinoid acid receptor |
| RARE | Retinoid acid response element |
| RXR | Retinoid X receptor |
| SMRT | Silencing mediator of retinoic acid and thyroid hormone receptor |
| SQSMT1 | Sequestosome 1 |
| TNF | Tumor necrosis alpha |
| ULK1 | Unc-51 like autophagy activating kinase 1 |
| WIPI | WD-repeat protein interacting with phosphoinositides |
| WT1 | Wilms’ tumor 1 |
References
- De Kouchkovsky, I.; Abdul-Hay, M. Acute myeloid leukemia: A comprehensive review and 2016 update. Blood Cancer J. 2016, 6, e441. [Google Scholar] [CrossRef] [PubMed]
- Döhner, H.; Weisdorf, D.J.; Bloomfield, C.D. Acute Myeloid Leukemia. N. Engl. J. Med. 2015, 373, 1136–1152. [Google Scholar] [CrossRef] [PubMed]
- De Thé, H.; Le Bras, M.; Lallemand-Breitenbach, V. The cell biology of disease: Acute promyelocytic leukemia, arsenic, and PML bodies. J. Cell Biol. 2012, 198, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Thomas, X. Acute Promyelocytic Leukemia: A History over 60 Years—From the Most Malignant to the most Curable Form of Acute Leukemia. Oncol. Ther. 2019, 7, 33–65. [Google Scholar] [CrossRef]
- De Thé, H.; Pandolfi, P.P.; Chen, Z. Acute Promyelocytic Leukemia: A Paradigm for Oncoprotein-Targeted Cure. Cancer Cell 2017, 32, 552–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ronchini, C.; Brozzi, A.; Riva, L.; Luzi, L.; Gruszka, A.M.; Melloni, G.E.M.; Scanziani, E.; Dharmalingam, G.; Mutarelli, M.; Belcastro, V.; et al. PML-RARA-associated cooperating mutations belong to a transcriptional network that is deregulated in myeloid leukemias. Leukemia 2017, 31, 1975–1986. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, H.; Chen, L.; Wang, M.; Xi, J.; Liu, X.; Xie, M.; Li, D.; Gulati, E.S.; Gong, S.; et al. Arsenic trioxide and all-trans retinoic acid (ATRA) treatment for acute promyelocytic leukemia in all risk groups: Study protocol for a randomized controlled trial. Trials 2018, 19, 476. [Google Scholar] [CrossRef] [PubMed]
- Tomita, A.; Kiyoi, H.; Naoe, T. Mechanisms of action and resistance to all-trans retinoic acid (ATRA) and arsenic trioxide As2O3 in acute promyelocytic leukemia. Int. J. Hematol. 2013, 97, 717–725. [Google Scholar] [CrossRef]
- Zhang, S.P.; Niu, Y.N.; Yuan, N.; Zhang, A.H.; Chao, D.; Xu, Q.P.; Wang, L.J.; Zhang, X.G.; Zhao, W.L.; Zhao, Y.; et al. Role of autophagy in acute myeloid leukemia therapy. Chin. J. Cancer 2013, 32, 130–135. [Google Scholar] [CrossRef] [Green Version]
- di Masi, A.; Leboffe, L.; De Marinis, E.; Pagano, F.; Cicconi, L.; Rochette-Egly, C.; Lo-Coco, F.; Ascenzi, P.; Nervi, C. Retinoic acid receptors: From molecular mechanisms to cancer therapy. Mol. Asp. Med. 2015, 41, 1–115. [Google Scholar] [CrossRef]
- Germain, P.; Iyer, J.; Zechel, C.; Gronemeyer, H. Co-regulator recruitment and the mechanism of retinoic acid receptor synergy. Nature 2002, 415, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Orfali, N.; O’Donovan, T.R.; Nyhan, M.J.; Britschgi, A.; Tschan, M.P.; Cahill, M.R.; Mongan, N.P.; Gudas, L.J.; McKenna, S.L. Induction of autophagy is a key component of all-trans-retinoic acid-induced differentiation in leukemia cells and a potential target for pharmacologic modulation. Exp. Hematol. 2015, 43, 781–793. [Google Scholar] [CrossRef] [PubMed]
- Perissi, V.; Staszewski, L.M.; McInerney, E.M.; Kurokawa, R.; Krones, A.; Rose, D.W.; Lambert, M.H.; Milburn, M.V.; Glass, C.K.; Rosenfeld, M.G. Molecular determinants of nuclear receptor-corepressor interaction. Genes Dev. 1999, 13, 3198–3208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isakson, P.; Bjørås, M.; Bøe, S.O.; Simonsen, A. Autophagy contributes to therapy-induced degradation of the PML/RARA oncoprotein. Blood 2010, 116, 2324–2331. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, R.E.; Moser, B.K.; Racevskis, J.; Poiré, X.; Bloomfield, C.D.; Carroll, A.J.; Ketterling, R.P.; Roulston, D.; Schachter-Tokarz, E.; Zhou, D.; et al. Treatment-influenced associations of PML-RARα mutations, FLT3 mutations, and additional chromosome abnormalities in relapsed acute promyelocytic leukemia. Blood 2012, 120, 2098–2108. [Google Scholar] [CrossRef] [PubMed]
- Bally, C.; Lehmann-Che, J.; Cassinat, B.; Ades, L.; Letouze, E.; Hirsch, P.; Mozziconacci, M.J.; Raynaud, S.; Delabesse, E.; Uzunov, M.; et al. Whole Exome Analysis of Relapsing Patients with Acute Promyelocytic Leukemia. Blood 2016, 128, 2892–2892. [Google Scholar]
- Madan, V.; Shyamsunder, P.; Han, L.; Mayakonda, A.; Nagata, Y.; Sundaresan, J.; Kanojia, D.; Yoshida, K.; Ganesan, S.; Hattori, N.; et al. Comprehensive mutational analysis of primary and relapse acute promyelocytic leukemia. Leukemia 2016, 30, 1672–1681. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Brand, N.J.; Chen, A.; Chen, S.J.; Tong, J.H.; Wang, Z.Y.; Waxman, S.; Zelent, A. Fusion between a novel Krüppel-like zinc finger gene and the retinoic acid receptor-alpha locus due to a variant t (11; 17) translocation associated with acute promyelocytic leukaemia. EMBO J. 1993, 12, 1161–1167. [Google Scholar] [CrossRef]
- Ruthardt, M.; Testa, U.; Nervi, C.; Ferrucci, P.F.; Grignani, F.; Puccetti, E.; Grignani, F.; Peschle, C.; Pelicci, P.G. Opposite effects of the acute promyelocytic leukemia PML-retinoic acid receptor alpha (RAR alpha) and PLZF-RAR alpha fusion proteins on retinoic acid signalling. Mol. Cell. Biol. 1997, 17, 4859–4869. [Google Scholar] [CrossRef] [Green Version]
- Voisset, E.; Moravcsik, E.; Stratford, E.W.; Jaye, A.; Palgrave, C.J.; Hills, R.K.; Salomoni, P.; Kogan, S.C.; Solomon, E.; Grimwade, D. Pml nuclear body disruption cooperates in APL pathogenesis and impairs DNA damage repair pathways in mice. Blood 2018, 131, 636–648. [Google Scholar] [CrossRef]
- Zhu, J.; Chen, Z.; Lallemand-Breitenbach, V.; de Thé, H. How acute promyelocytic leukaemia revived arsenic. Nat. Rev. Cancer 2002, 2, 705. [Google Scholar] [CrossRef] [PubMed]
- Mumford, J.L.; Wu, K.; Xia, Y.; Kwok, R.; Yang, Z.; Foster, J.; Sanders, W.E., Jr. Chronic arsenic exposure and cardiac repolarization abnormalities with QT interval prolongation in a population-based study. Environ. Health Perspect. 2007, 115, 690–694. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.B.; Zhao, W.L.; Wang, Z.Y.; Chen, S.J.; Chen, Z. Retinoic acid and arsenic for treating acute promyelocytic leukemia. PLoS Med. 2005, 2, e12. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.Q.; Shi, X.G.; Tang, W.; Xiong, S.M.; Zhu, J.; Cai, X.; Han, Z.G.; Ni, J.H.; Shi, G.Y.; Jia, P.M. Use of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia (APL): I. As2O3 exerts dose-dependent dual effects on APL cells. Blood 1997, 89, 3345–3353. [Google Scholar] [PubMed]
- Zheng, X.; Seshire, A.; Rüster, B.; Bug, G.; Beissert, T.; Puccetti, E.; Hoelzer, D.; Henschler, R.; Ruthardt, M. Arsenic but not all-trans retinoic acid overcomes the aberrant stem cell capacity of PML/RARα-positive leukemic stem cells. Haematologica 2007, 92, 323–331. [Google Scholar] [CrossRef]
- Li, T.; Ma, R.; Zhang, Y.; Mo, H.; Yang, X.; Hu, S.; Wang, L.; Novakovic, V.A.; Chen, H.; Kou, J.; et al. Arsenic trioxide promoting ETosis in acute promyelocytic leukemia through mTOR-regulated autophagy. Cell Death Dis. 2018, 9, 75. [Google Scholar] [CrossRef]
- Alex, A.A.; Chendamarai, E.; Ganesan, S.; Balasundaram, N.; Palani, H.K.; David, S.; Mathews, V. Arsenic Trioxide Resistance: More to It Than Mutations in PML-RARα. Blood 2014, 124, 3605. [Google Scholar]
- Hattori, H.; Ishikawa, Y.; Kawashima, N.; Akashi, A.; Yamaguchi, Y.; Harada, Y.; Hirano, D.; Adachi, Y.; Miyao, K.; Ushijima, Y. Identification of the novel deletion-type PML-RARA mutation associated with the retinoic acid resistance in acute promyelocytic leukemia. PLoS ONE 2018, 13, e0204850. [Google Scholar] [CrossRef]
- Balasundaram, N.; Ganesan, S.; Palani, H.K.; Alex, A.A.; David, S.; Korula, A.; George, B.; Chomienne, C.; Balasubramanian, P.; Mathews, V. Metabolic Rewiring Drives Resistance to Arsenic Trioxide in Acute Promyelocytic Leukemia. Blood 2016, 128, 3956. [Google Scholar]
- Chendamarai, E.; Ganesan, S.; Alex, A.A.; Kamath, V.; Nair, S.C.; Nellickal, A.J.; Janet, N.B.; Srivastava, V.; Lakshmi, K.M.; Viswabandya, A. Comparison of newly diagnosed and relapsed patients with acute promyelocytic leukemia treated with arsenic trioxide: Insight into mechanisms of resistance. PLoS ONE 2015, 10, e0121912. [Google Scholar] [CrossRef]
- Chen, S.-J.; Zhou, G.-B.; Zhang, X.-W.; Mao, J.-H.; Chen, Z. From an old remedy to a magic bullet: Molecular mechanisms underlying the therapeutic effects of arsenic in fighting leukemia. Blood 2011, 117, 6425–6437. [Google Scholar] [CrossRef] [PubMed]
- Sanz, M.A.; Montesinos, P. How we prevent and treat differentiation syndrome in patients with acute promyelocytic leukemia. Blood 2014, 123, 2777–2782. [Google Scholar] [CrossRef] [PubMed]
- Montesinos, P.; Sanz, M.A. The differentiation syndrome in patients with acute promyelocytic leukemia: Experience of the pethema group and review of the literature. Mediterr. J. Hematol. Infect. Dis. 2011, 3, e2011059. [Google Scholar] [CrossRef] [PubMed]
- Jin, B.; Hou, K.; Liu, Y.; Yu, P. Leukocytosis and retinoic acid syndrome in patients with acute promyelocytic leukemia treated with arsenic trioxide. Chin. Med. Sci. J. Chung-Kuo Hsueh Ko Hsueh Tsa Chih 2006, 21, 171–174. [Google Scholar]
- Rogers, J.E.; Yang, D. Differentiation syndrome in patients with acute promyelocytic leukemia. J. Oncol. Pharm. Pract. 2012, 18, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Cardinale, L.; Asteggiano, F.; Moretti, F.; Torre, F.; Ulisciani, S.; Fava, C.; Rege-Cambrin, G. Pathophysiology, clinical features and radiological findings of differentiation syndrome/all-trans-retinoic acid syndrome. World J. Radiol. 2014, 6, 583. [Google Scholar] [CrossRef] [PubMed]
- Larson, R.S.; Tallman, M.S. Retinoic acid syndrome: Manifestations, pathogenesis, and treatment. Best Pract. Res. Clin. Haematol. 2003, 16, 453–461. [Google Scholar] [CrossRef]
- Frankel, S.R.; Eardley, A.; Lauwers, G.; Weiss, M.; Warrell, R.P. The retinoic acid syndrome in acute promyelocytic leukemia. Ann. Intern. Med. 1992, 117, 292–296. [Google Scholar] [CrossRef] [PubMed]
- Tallman, M. Retinoic acid syndrome: A problem of the past? Leukemia 2002, 16, 160. [Google Scholar] [CrossRef]
- Luesink, M.; Pennings, J.L.; Wissink, W.M.; Linssen, P.C.; Muus, P.; Pfundt, R.; de Witte, T.J.; van der Reijden, B.A.; Jansen, J.H. Chemokine induction by all-trans retinoic acid and arsenic trioxide in acute promyelocytic leukemia: Triggering the differentiation syndrome. Blood 2009, 114, 5512–5521. [Google Scholar] [CrossRef]
- Dubois, C.; Schlageter, M.H.; de Gentile, A.; Guidez, F.; Balitrand, N.; Toubert, M.E.; Krawice, I.; Fenaux, P.; Castaigne, S.; Najean, Y. Hematopoietic growth factor expression and ATRA sensitivity in acute promyelocytic blast cells. Blood 1994, 83, 3264–3270. [Google Scholar] [PubMed]
- Seale, J.; Delva, L.; Renesto, P.; Balitrand, N.; Dombret, H.; Scrobohaci, M.; Degos, L.; Paul, P.; Chomienne, C. All-trans retinoic acid rapidly decreases cathepsin G synthesis and mRNA expression in acute promyelocytic leukemia. Leukemia 1996, 10, 95–101. [Google Scholar] [PubMed]
- Rego, E.M.; De Santis, G.C. Differentiation syndrome in promyelocytic leukemia: Clinical presentation, pathogenesis and treatment. Mediterr. J. Hematol. Infect. Dis. 2011, 3, e2011048. [Google Scholar] [CrossRef] [PubMed]
- Wiley, J.; Firkin, F. Reduction of pulmonary toxicity by prednisolone prophylaxis during all-trans retinoic acid treatment of acute promyelocytic leukemia. Australian Leukaemia Study Group. Leukemia 1995, 9, 774–778. [Google Scholar] [PubMed]
- Tallman, M.S.; Andersen, J.W.; Schiffer, C.A.; Appelbaum, F.R.; Feusner, J.H.; Ogden, A.; Shepherd, L.; Rowe, J.M.; François, C.; Larson, R.S.; et al. Clinical description of 44 patients with acute promyelocytic leukemia who developed the retinoic acid syndrome. Blood 2000, 95, 90–95. [Google Scholar] [PubMed]
- Sanz, M.A.; Montesinos, P.; Vellenga, E.; Rayón, C.; de la Serna, J.; Parody, R.; Bergua, J.M.; León, A.; Negri, S.; González, M.; et al. Risk-adapted treatment of acute promyelocytic leukemia with all-trans retinoic acid and anthracycline monochemotherapy: Long-term outcome of the LPA 99 multicenter study by the PETHEMA Group. Blood 2008, 112, 3130–3134. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; He, D.; Yao, Z.; Klionsky, D.J. The machinery of macroautophagy. Cell Res. 2014, 24, 24–41. [Google Scholar] [CrossRef]
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular definitions of autophagy and related processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef]
- Boya, P.; Reggiori, F.; Codogno, P. Emerging regulation and functions of autophagy. Nat. Cell Biol. 2013, 15, 713–720. [Google Scholar] [CrossRef]
- Kroemer, G.; Mariño, G.; Levine, B. Autophagy and the Integrated Stress Response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef] [Green Version]
- Klionsky, D.J.; Codogno, P. The mechanism and physiological function of macroautophagy. J. Innate Immun. 2013, 5, 427–433. [Google Scholar] [CrossRef]
- Levine, B.; Kroemer, G. Biological Functions of Autophagy Genes: A Disease Perspective. Cell 2019, 176, 11–42. [Google Scholar] [CrossRef] [Green Version]
- Gatica, D.; Lahiri, V.; Klionsky, D.J. Cargo recognition and degradation by selective autophagy. Nat. Cell Biol. 2018, 20, 233–242. [Google Scholar] [CrossRef]
- Johansen, T.; Lamark, T. Selective autophagy mediated by autophagic adapter proteins. Autophagy 2011, 7, 279–296. [Google Scholar] [CrossRef]
- Khaminets, A.; Behl, C.; Dikic, I. Ubiquitin-Dependent and Independent Signals In Selective Autophagy. Trends Cell Biol. 2016, 26, 6–16. [Google Scholar] [CrossRef]
- Clarke, A.J.; Simon, A.K. Autophagy in the renewal, differentiation and homeostasis of immune cells. Nat. Rev. Immunol. 2019, 19, 170. [Google Scholar] [CrossRef]
- Djavaheri-Mergny, M.; Giuriato, S.; Tschan, M.P.; Humbert, M. Therapeutic Modulation of Autophagy in Leukaemia and Lymphoma. Cells 2019, 8, 103. [Google Scholar] [CrossRef]
- Orsini, M.; Morceau, F.; Dicato, M.; Diederich, M. Autophagy as a pharmacological target in hematopoiesis and hematological disorders. Biochem. Pharmacol. 2018, 152, 347–361. [Google Scholar] [CrossRef]
- White, E. The role for autophagy in cancer. J. Clin. Investig. 2015, 125, 42–46. [Google Scholar] [CrossRef] [Green Version]
- Levine, B.; Packer, M.; Codogno, P. Development of autophagy inducers in clinical medicine. J. Clin. Investig. 2015, 125, 14–24. [Google Scholar] [CrossRef] [Green Version]
- Thorburn, A.; Thamm, D.H.; Gustafson, D.L. Autophagy and Cancer Therapy. Mol. Pharmacol. 2014, 85, 830–838. [Google Scholar] [CrossRef] [Green Version]
- Levy, J.M.M.; Towers, C.G.; Thorburn, A. Targeting autophagy in cancer. Nat. Rev. Cancer 2017, 17, 528. [Google Scholar] [CrossRef]
- Galluzzi, L.; Pietrocola, F.; Bravo-San Pedro, J.M.; Amaravadi, R.K.; Baehrecke, E.H.; Cecconi, F.; Codogno, P.; Debnath, J.; Gewirtz, D.A.; Karantza, V.; et al. Autophagy in malignant transformation and cancer progression. EMBO J. 2015, 34, 856–880. [Google Scholar] [CrossRef]
- Jin, J.; Britschgi, A.; Schläfli, A.M.; Humbert, M.; Shan-Krauer, D.; Batliner, J.; Federzoni, E.A.; Ernst, M.; Torbett, B.E.; Yousefi, S.; et al. Low Autophagy (ATG) Gene Expression Is Associated with an Immature AML Blast Cell Phenotype and Can Be Restored during AML Differentiation Therapy. Oxid. Med. Cell. Longev. 2018, 2018, 1482795. [Google Scholar] [CrossRef]
- Ségal-Bendirdjian, E.; Tschan, M.P.; Reiffers, J.; Djavaheri-Mergny, M. Pro-survival role of p62 during granulocytic differentiation of acute myeloid leukemia cells. Mol. Cell. Oncol. 2014, 1, e970066. [Google Scholar] [CrossRef] [Green Version]
- Trocoli, A.; Bensadoun, P.; Richard, E.; Labrunie, G.; Merhi, F.; Schläfli, A.M.; Brigger, D.; Souquere, S.; Pierron, G.; Pasquet, J.M.; et al. p62/SQSTM1 upregulation constitutes a survival mechanism that occurs during granulocytic differentiation of acute myeloid leukemia cells. Cell Death Differ. 2014, 21, 1852. [Google Scholar] [CrossRef]
- Meenhuis, A.; van Veelen, P.A.; de Looper, H.; van Boxtel, N.; van den Berge, I.J.; Sun, S.M.; Taskesen, E.; Stern, P.; de Ru, A.H.; van Adrichem, A.J.; et al. MiR-17/20/93/106 promote hematopoietic cell expansion by targeting sequestosome 1-regulated pathways in mice. Blood 2011, 118, 916–925. [Google Scholar] [CrossRef]
- Nguyen, T.D.; Shaid, S.; Vakhrusheva, O.; Koschade, S.E.; Klann, K.; Thölken, M.; Baker, F.; Zhang, J.; Oellerich, T.; Sürün, D.; et al. Loss of the selective autophagy receptor p62 impairs murine myeloid leukemia progression and mitophagy. Blood 2019, 133, 168–179. [Google Scholar] [CrossRef] [Green Version]
- Riffelmacher, T.; Clarke, A.; Richter, F.C.; Stranks, A.; Pandey, S.; Danielli, S.; Hublitz, P.; Yu, Z.; Johnson, E.; Schwerd, T.; et al. Autophagy-Dependent Generation of Free Fatty Acids Is Critical for Normal Neutrophil Differentiation. Immunity 2017, 47, 466–480. [Google Scholar] [CrossRef]
- Brigger, D.; Proikas-Cezanne, T.; Tschan, M.P. WIPI-dependent autophagy during neutrophil differentiation of NB4 acute promyelocytic leukemia cells. Cell Death Dis. 2014, 5, e1315. [Google Scholar] [CrossRef]
- Huang, Y.; Hou, J.K.; Chen, T.T.; Zhao, X.Y.; Yan, Z.W.; Zhang, J.; Yang, J.; Kogan, S.C.; Chen, G.Q. PML-RARα enhances constitutive autophagic activity through inhibiting the Akt/mTOR pathway. Autophagy 2011, 7, 1132–1144. [Google Scholar] [CrossRef] [PubMed]
- Trocoli, A.; Mathieu, J.; Priault, M.; Reiffers, J.; Souquere, S.; Pierron, G.; Besançon, F.; Djavaheri-Mergny, M. ATRA-induced upregulation of Beclin 1 prolongs the life span of differentiated acute promyelocytic leukemia cells. Autophagy 2011, 7, 1108. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Chai, W.; Wang, Y.; Cao, L.; Xie, M.; Yang, M.; Kang, R.; Yu, Y. Reactive oxygen species regulate the differentiation of acute promyelocytic leukemia cells through HMGB1-mediated autophagy. Am. J. Cancer Res. 2015, 5, 714–725. [Google Scholar] [PubMed]
- Wu, D.; Shao, K.; Zhou, Q.; Sun, J.; Wang, Z.; Yan, F.; Liu, T.; Wu, X.; Ye, B.; Huang, H.; et al. Autophagy and Ubiquitin-Mediated Proteolytic Degradation of PML/Rarα Fusion Protein in Matrine-Induced Differentiation Sensitivity Recovery of ATRA-Resistant APL (NB4-LR1) Cells: In Vitro and in Vivo Studies. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2018, 48, 2286–2301. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Wang, H.; Shen, Q.; Feng, L.; Jin, H. Long non-coding RNAs involved in autophagy regulation. Cell Death Dis. 2017, 8, e3073. [Google Scholar] [CrossRef] [PubMed]
- Zeng, C.W.; Chen, Z.H.; Zhang, X.J.; Han, B.W.; Lin, K.Y.; Li, X.J.; Wei, P.P.; Zhang, H.; Li, Y.; Chen, Y.Q. MIR125B1 represses the degradation of the PML-RARA oncoprotein by an autophagy-lysosomal pathway in acute promyelocytic leukemia. Autophagy 2014, 10, 1726–1737. [Google Scholar] [CrossRef]
- Wang, Z.; Cao, L.; Kang, R.; Yang, M.; Liu, L.; Zhao, Y.; Yu, Y.; Xie, M.; Yin, X.; Livesey, K.M.; et al. Autophagy regulates myeloid cell differentiation by p62/SQSTM1-mediated degradation of PML-RARα oncoprotein. Autophagy 2011, 7, 401–411. [Google Scholar] [CrossRef]
- Schläfli, A.M.; Isakson, P.; Garattini, E.; Simonsen, A.; Tschan, M.P. The autophagy scaffold protein ALFY is critical for the granulocytic differentiation of AML cells. Sci. Rep. 2017, 7, 12980. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Wan, J.; Kong, Y.; Zhang, Y.; Wan, L.; Zhang, Z. Inhibition of CRL-NEDD8 pathway as a new approach to enhance ATRA-induced differentiation of acute promyelocytic leukemia cells. Int. J. Med. Sci. 2018, 15, 674–681. [Google Scholar] [CrossRef] [Green Version]
- Qian, W.; Liu, J.; Jin, J.; Ni, W.; Xu, W. Arsenic trioxide induces not only apoptosis but also autophagic cell death in leukemia cell lines via up-regulation of Beclin-1. Leuk. Res. 2007, 31, 329–339. [Google Scholar] [CrossRef]
- Ganesan, S.; Alex, A.A.; Chendamarai, E.; Balasundaram, N.; Palani, H.K.; David, S.; Kulkarni, U.; Aiyaz, M.; Mugasimangalam, R.; Korula, A.; et al. Rationale and efficacy of proteasome inhibitor combined with arsenic trioxide in the treatment of acute promyelocytic leukemia. Leukemia 2016, 30, 2169–2178. [Google Scholar] [CrossRef]
- Goussetis, D.J.; Altman, J.K.; Glaser, H.; McNeer, J.L.; Tallman, M.S.; Platanias, L.C. Autophagy is a critical mechanism for the induction of the antileukemic effects of arsenic trioxide. J. Biol. Chem. 2010, 285, 29989–29997. [Google Scholar] [CrossRef]
- Ma, R.; Li, T.; Cao, M.; Si, Y.; Wu, X.; Zhao, L.; Yao, Z.; Zhang, Y.; Fang, S.; Deng, R.; et al. Extracellular DNA traps released by acute promyelocytic leukemia cells through autophagy. Cell Death Dis. 2016, 7, e2283. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Liang, Z.; Gao, B.; Jia, Y.; Qin, Z. Dynamic effects of autophagy on arsenic trioxide-induced death of human leukemia cell line HL60 cells. Acta Pharmacol. Sin. 2008, 29, 123. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, S.; Kumar Palani, H.; Lakshmanan, V.; Balasundaram, N.; Alex, A.A.; David, S.; Venkatraman, A.; Kulkarni, U.P.; George, B.; Srivastava, A. Stromal-Cells Downregulate MiR-23a-5p Levels in Myeloid Leukemic Cells to Activate Protective-Autophagy Against Chemotherapeutic Agents. Blood 2017, 130, 3780. [Google Scholar]
- Fakhimahmadi, A.; Nazmi, F.; Rahmati, M.; Bonab, N.M.; Hashemi, M.; Moosavi, M.A. Nucleostemin silencing induces differentiation and potentiates all-trans-retinoic acid effects in human acute promyelocytic leukemia NB4 cells via autophagy. Leuk. Res. 2017, 63, 15–21. [Google Scholar] [CrossRef] [PubMed]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moosavi, M.A.; Djavaheri-Mergny, M. Autophagy: New Insights into Mechanisms of Action and Resistance of Treatment in Acute Promyelocytic leukemia. Int. J. Mol. Sci. 2019, 20, 3559. https://doi.org/10.3390/ijms20143559
Moosavi MA, Djavaheri-Mergny M. Autophagy: New Insights into Mechanisms of Action and Resistance of Treatment in Acute Promyelocytic leukemia. International Journal of Molecular Sciences. 2019; 20(14):3559. https://doi.org/10.3390/ijms20143559
Chicago/Turabian StyleMoosavi, Mohammad Amin, and Mojgan Djavaheri-Mergny. 2019. "Autophagy: New Insights into Mechanisms of Action and Resistance of Treatment in Acute Promyelocytic leukemia" International Journal of Molecular Sciences 20, no. 14: 3559. https://doi.org/10.3390/ijms20143559
APA StyleMoosavi, M. A., & Djavaheri-Mergny, M. (2019). Autophagy: New Insights into Mechanisms of Action and Resistance of Treatment in Acute Promyelocytic leukemia. International Journal of Molecular Sciences, 20(14), 3559. https://doi.org/10.3390/ijms20143559