1. Introduction
The conceptuses of sheep remain free floating within the uterine lumen as they elongate from spherical blastocysts to conceptuses with a filamentous morphology [
1]. Sheep embryos enter the uterus on day 3, develop to spherical blastocysts, and, subsequent to hatching from the zona pellucida, transform from spherical to tubular and filamentous conceptuses between days 12 and 15 of pregnancy. The conceptus extra-embryonic membranes extend into the contralateral uterine horn, relative to the ovary with a corpus luteum, between days 16 and 20 of pregnancy [
2]. During this period of rapid elongation and differentiation, the mononuclear trophoblast cells of ovine conceptuses secrete interferon tau as the pregnancy recognition signal and attachment of the trophoblast to the uterine LE is initiated [
3,
4,
5]. After firm attachment of the trophoblast to the uterine LE, ruminants, including sheep and cattle, develop synepitheliochorial placentae in which two morphologically and functionally distinct trophoblast cell types are present at the uterine–placental interface of placentomes: the mononucleated trophoblasts and the multinucleated syncytia. These trophoblast cells control the exchange of gases, nutrients, and other factors between the maternal and fetal circulations, protect the fetus against the maternal immune system, and are responsible for the production of many proteins, hormones, and growth factors [
1,
2].
Initially the mononucleate trophoblast cells constitute the majority of the trophoblast cells; however, between days 14 and 16 of gestation, binucleate trophoblast cells (BNCs) begin to differentiate from the mononuclear trophoblast cells by consecutive nuclear divisions without cytokinesis, also termed mitotic polyploidy [
6,
7,
8]. By day 18, they comprise 15–20% of the trophoblast cells that are apposed to the uterine LE at sites of conceptus implantation [
9,
10]. The BNCs pass through the tight junctions between adjacent mononucleate trophoblast cells and migrate to the uterine LE for syncytialization with LE cells [
7]. In addition, BNCs express placental lactogens and pregnancy-associated glycoproteins (PAGs). The measurement of PAGS in serum can be reliably utilized to assess BNC development, and diagnose pregnancy in both sheep and cattle [
11].
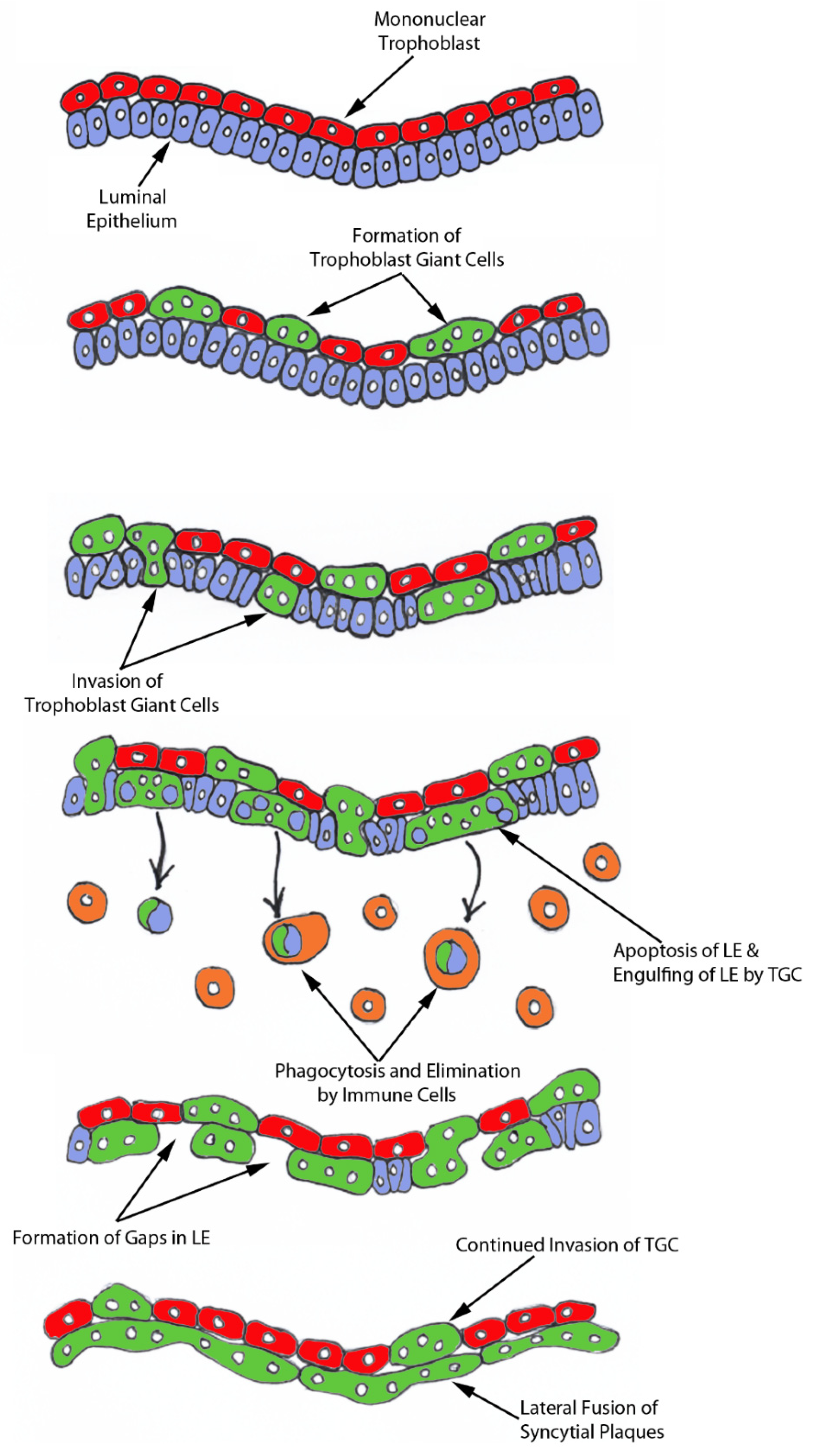
The process of syncytia formation in sheep is generally explained by Wooding’s hypothesis (
Figure S1) [
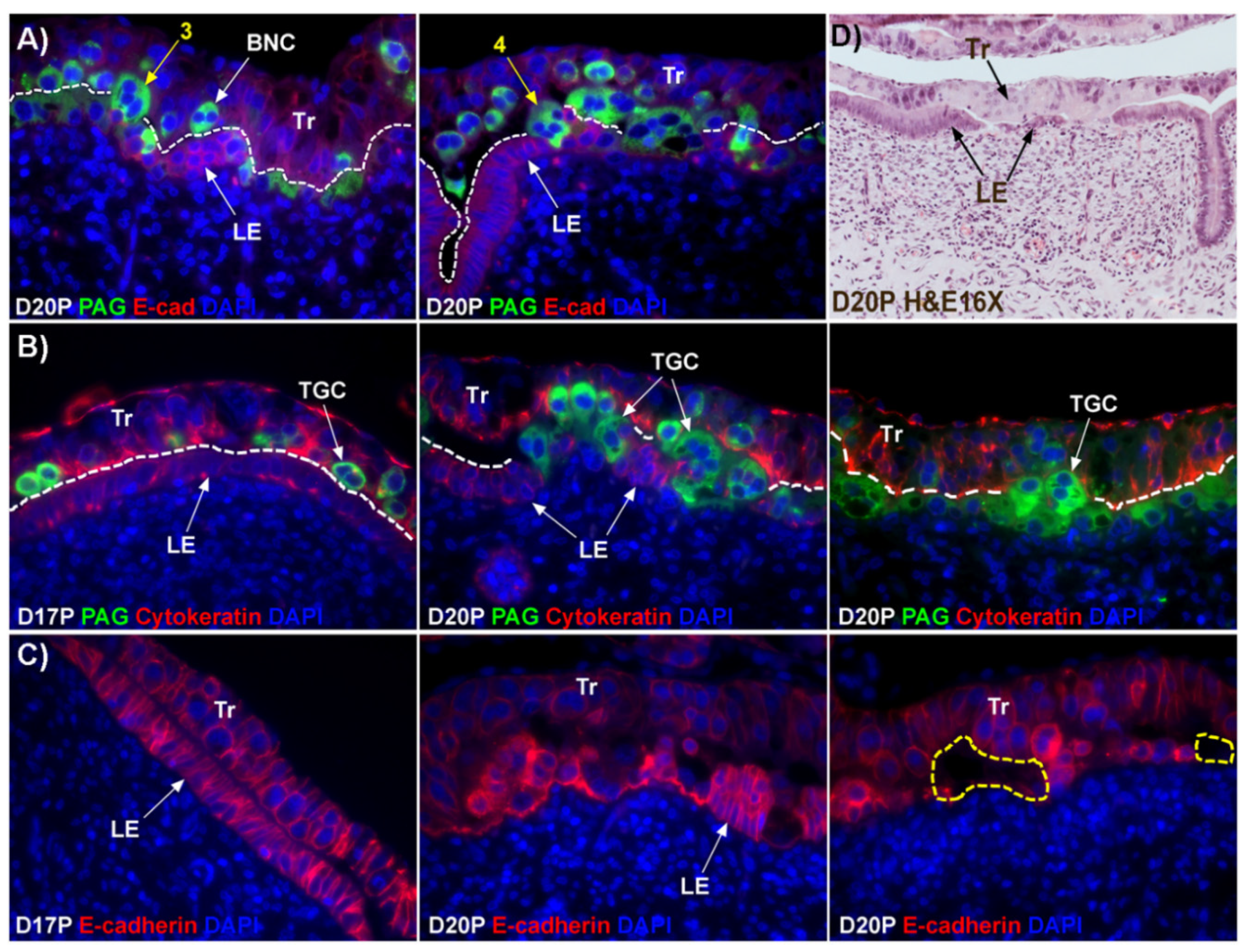
12]. This hypothesis states that BNCs differentiate from the mononuclear trophoblast cells and migrate and fuse with individual uterine LE cells to form trinucleate syncytial cells, thereby assimilating the LE cells. BNCs continue to develop and migrate to the LE layer and fuse with these growing trophoblast–LE syncytial cells to eventually form extensive syncytial plaques. Therefore, these syncytial plaques are conceptus–maternal hybrid cells that are composed of LE cells and BNCs, and they eventually cover the entire caruncular surface to form the epithelial interface between uterine caruncular and placental cotyledonary tissues within the placentome of sheep. However, this idea was based on electron microscopy studies, without the benefit of molecular markers of BNC and LE to support the conclusion. Therefore, the aim of this study was to perform immunohistochemical localization for molecular markers for BNCs and uterine LE cells, including PAGs, E-cadherin, cytokeratin, and caspase 3, as well as assess apoptosis (TUNEL staining) at the uterine–placental interface to better understand the mechanism for syncytialization during early placentation in sheep.
3. Discussion
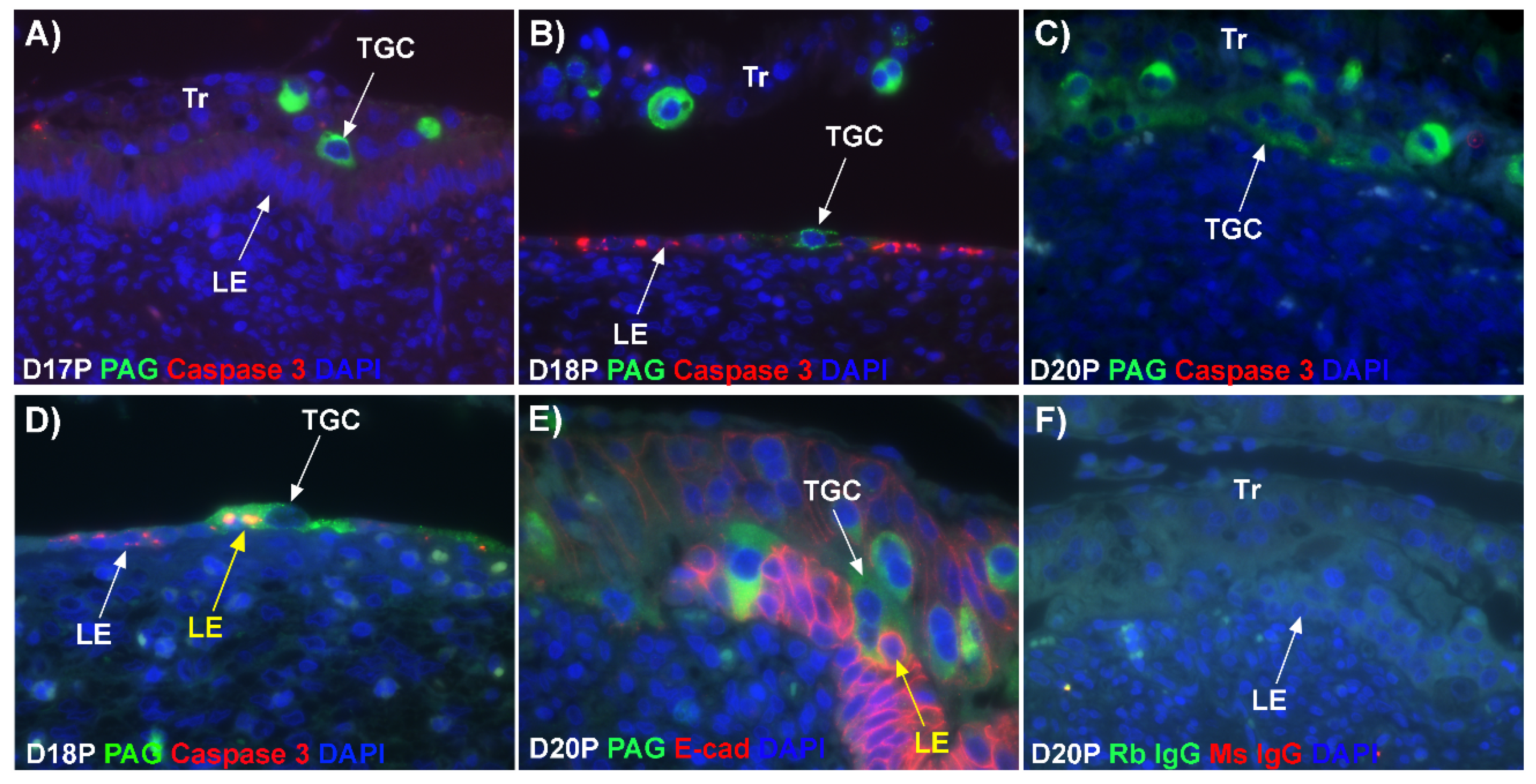
For over 20 years the scientific consensus has been that during trophoblast syncytialization in sheep, BNCs differentiate from the mononuclear trophoblast cells through mitotic polyploidy, individual BNCs fuse with individual LE cells to form trinucleate cells, and BNCs continue to migrate to the LE layer and fuse with these growing trophoblast–LE syncytial plaques. LE cells are incorporated into syncytial plaques, and there is no lateral fusion between syncytial plaques, rather all fusion events involve newly formed BNCs [
9,
11]. Results of the present study expand our understanding of the syncytialization process in sheep as they indicate that: 1) there are syncytial cells containing more than two nuclei within the trophoblast cell layer, suggesting that fusion between trophectoderm cells is not limited to the formation of BNCs, and that the term ‘trophoblast giant cell (TGC)’ may be appropriate; 2) depolarized uterine LE cells express caspase 3 and stain positively for TUNEL suggesting apoptosis of uterine LE during syncytialization; 3) engulfment of caspase 3-positive uterine LE cells by TGCs and empty spaces/voids within the uterine LE at sites of implantation indicating elimination of apoptotic LE cells by TGCs; 4) rapid enlargement of syncytial plaques suggests that fusion is not limited to the incorporation of new BNCs, but involves lateral fusion between growing syncytial plaques; and 5) E-cadherin- and TUNEL-positive LE cells and SHMT2-positive TGCs within the stroma underlying degenerating LE, coincident with the accumulation of CD45-positive immune cells at these sites suggesting that TGCs carry apoptotic LE cells away from the uterine–placental interface for elimination by immune cells within the uterine stroma.
The mechanism by which BNCs differentiate from mononuclear trophoblasts is poorly understood. BNCs may be generated from mononuclear trophoblast cells by consecutive nuclear divisions without cytokinesis, also termed mitotic polyploidy [
8]. Alternatively, the BNCs may form by fusion of mononuclear trophoblast cells [
13]. The current consensus for placental syncytialization was based on electron microscopic (EM) studies that observed BNCs in the trophoblast layer and trinucleate cells in the LE layer [
12]. With these observations, it is thought that only trophoblast BNCs develop in the trophoblast layer and migrate into the uterine LE, and trinucleate cells in the LE are formed by fusion between BNCs and LE cells (
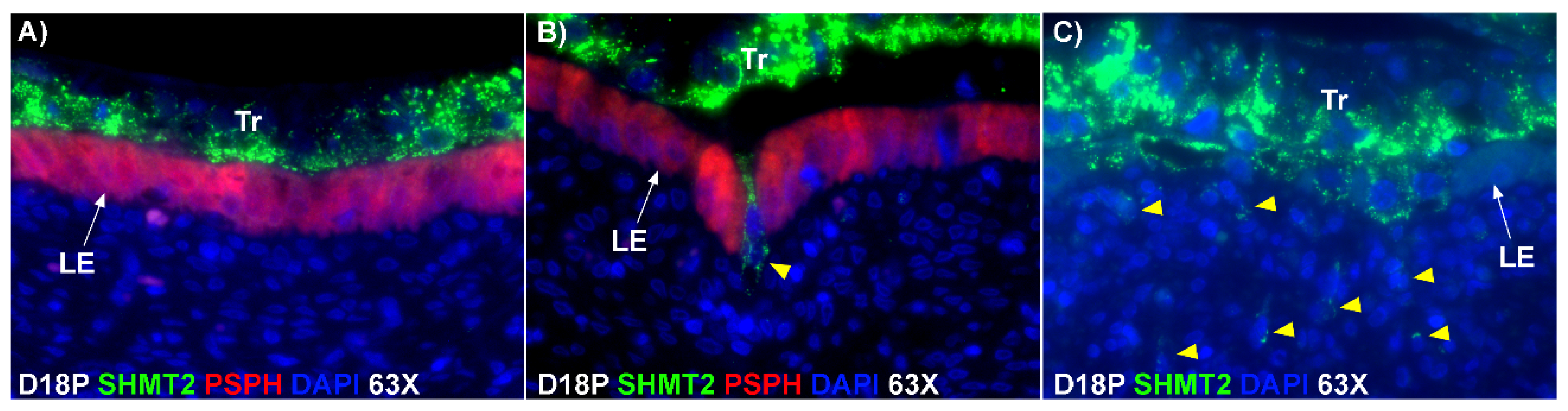
Figure 1D). If this were the case, all PAG-stained trophoblast cells in the trophoblast cell layer should be BNCs. However, results of the present study revealed that multinucleated trophoblast cells with three or four nuclei also migrate into the uterine LE. This suggests that differentiation of trophoblasts is not limited to the formation of BNCs. In addition, the presence of trophoblast cells with three nuclei, an odd number of nuclei that could not be generated by mitotic polyploidy, suggests that fusion of adjacent trophoblast cells may occur to generate multinucleated TGCs that have two or more nuclei. This raises the possibility that at least some of the trinucleate cells that were observed by EM in the LE layer could possibly be TGCs that have three nuclei, entirely of trophoblast origin, that have migrated from the trophoblast layer into the LE layer.
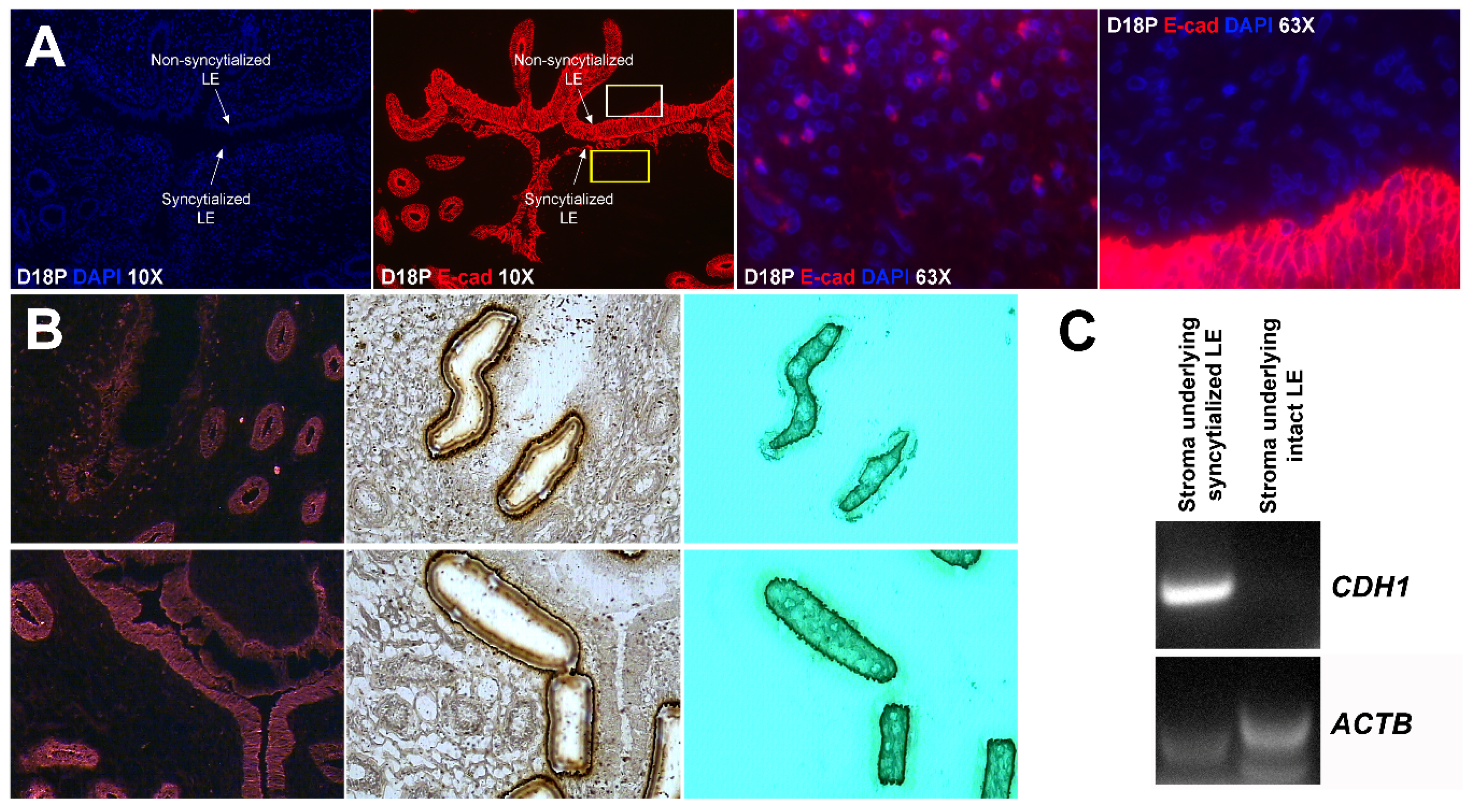
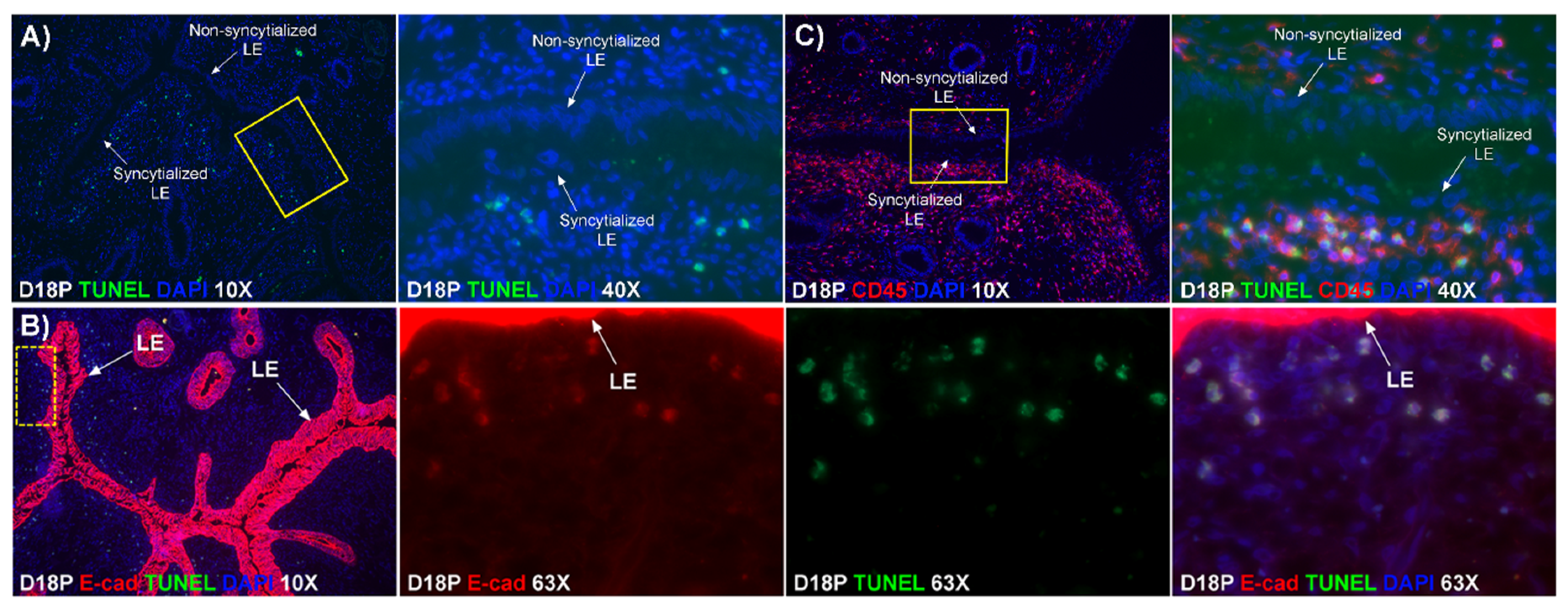
Our results show that extensive migration of TGCs into the LE layer and replacement of mononuclear LE cells by syncytial cells occurs between day 17 and day 20 of pregnancy. An important observation is that there were fewer nuclei in the syncytialized regions of the LE layer as compared to the number of nuclei observed in regions of intact and non-syncytialized LE, suggesting that LE cells may not be incorporated into the syncytial layer during syncytializations, rather there is loss of LE cells during syncytialization. In mice, the uterine LE cells are eliminated during implantation [
14]. Based on EM studies, it is hypothesized that degeneration of LE cells is intrinsic to the uterus and embryos play a minor role [
15,
16], whereas another hypothesis proposed that trophoblast cells trigger apoptosis of LE cells [
17]. Regardless of the mechanism involved, most studies found that uterine LE cells adjacent to the blastocyst exhibit characteristics of apoptosis, including cellular shrinkage and nuclear fragmentation, following attachment of blastocysts for implantation and that the apoptotic LE cells are phagocytized by trophoblast cells [
17,
18,
19]. Our data show that LE cells express caspase 3, an executor in the apoptosis cascade, during syncytialization. In addition, TGCs appeared to physically engulf, internalize or fuse with the caspase 3-positive LE cells. At present, the immunofluorescence staining does not discriminate between these three processes. However, taken together, these results suggest that LE cells undergo apoptosis and TGCs associate intimately with these apoptotic LE cells during syncytialization.
E-cadherin, an adherens junction molecule, is specifically expressed in epithelial cells and important for the integrity of epithelial cells. In the present study, E-cadherin-positive cells were detected within the uterine stroma underlying syncytialized regions of the LE layer compared to regions of intact LE. We hypothesize that the E-cadherin-positive cells were LE cells undergoing apoptosis because the localization of E-cadherin-positive cells was limited to the stroma just below the syncytialized LE wherein LE cells appear to undergo apoptosis. Indeed, TUNEL staining co-localized with E-cadherin in those cells. This supports our hypothesis that E-cadherin-positive cells within the stroma are apoptotic LE cells. Further, immunofluorescence staining for CD45, a common marker for leukocytes, showed the accumulation of CD45-positive cells closely associated with TUNEL-positive cells. We hypothesize that these immune cells eliminate the apoptotic LE cells within the stroma. The question arises, how do the LE cells move down to the stroma? We propose that TGCs carry apoptotic LE cells into the stroma, because TGCs appears to be able to engulf LE cells, TGCs are known to be inherently invasive [
7,
20], and SHMT2-positive trophoblast cells were detected within the uterine stroma underlying regions of active syncytialization. Therefore, it is possible that TGCs engulf or endocytose apoptotic LE cells and then invade the basement membrane cell barrier to carry the LE cells into the underlying stroma where they can be eliminated by immune cells. If the process described above is taking place, then gaps should develop in the LE cell layer where LE cells have been engulfed by TGCs. The results of this study show that gaps in the LE barrier are developed at implantation sites as syncytialization progresses. It is possible that these spaces are replaced by syncytia that likely arose through the lateral fusion of TGCs within the LE cell layer.
In light of our results, we hypothesize that mononuclear trophoblast cells fuse with one-another to become multinucleated TGCs, and a role for the endogenous betaretroviruses (enJSRV) in this process is likely [
13,
20]. Large numbers of TGCs migrate to insert themselves between the uterine LE cells that are simultaneously undergoing apoptosis. TGCs then engulf LE cells and carry them to the stroma for elimination by immune cells. The remaining TGCs then fuse with each other to form an extensive trophoblast syncytial layer that fills spaces left by removal of uterine LE cells and provides the direct interface between uterine and placental tissues within the placentomes of sheep (
Figure 6). Therefore, we suggest that the possibility exists that the composition of the syncytial layer at the uterine–placental interface of sheep is entirely of placental origin. In humans, neighboring cytotrophoblast cells begin to fuse to generate syncytiotrophoblasts at the time of adhesion of the blastocyst to the uterine LE that initiates implantation and this syncytiotrophoblast is the tissue that penetrates through the uterine LE during implantation of the human blastocyst [
21]. Our present study suggests that early placentation in sheep is more similar to early placentation in humans than has been understood in that both may develop mononucleated cytotrophoblast and multinucleated syncytiotrophoblast layers of entirely placental origin [
22,
23].
4. Materials and Methods
4.1. Animals and Tissue Preparation
Experimental procedures complied with the Guide for Care and Use of Agricultural Animals in Research and Teaching and were approved by the Texas A&M University Institutional Animal Care and Use Committee (AUP IACUC 2012-161). Mature western-range ewes of primarily Suffolk breeding were observed daily for estrous behavior. Following at least two estrous cycles of normal duration (16–18 days), ewes were mated to rams of proven fertility (day 0). Four ewes each were hysterectomized on days 17, 18, 20, and 30 of pregnancy. Several 1–1.5 cm sections of uterine wall from the middle of each horn from pregnant ewes were: (1) embedded in OCT compound, frozen in liquid nitrogen, and stored at −80 °C; and (2) fixed in 4% paraformaldehyde and paraffin-embedded.
4.2. Immunofluorescence Analyses
Immunoreactive PAG, E-cadherin, cytokeratin, caspase 3, SHMT2 (serine hydroxymethyltransferase 2), and PSPH (phosphoserine phosphatase) proteins were localized in paraffin-embedded samples from day 17–30 pregnant ewes using immuofluorescence microscopy. Antigen retrieval was performed using either boiling citrate (E-cadherin, cytokeratin, caspase 3, SHMT2, and PSPH) or protease (PAG, E-cadherin, cytokeratin, and caspase 3). Sections were then blocked in 10% normal goat serum for 1 h at room temperature. These sections were incubated overnight at 4 °C with the following primary antibodies: rabbit anti-PAG polyclonal antibody (kindly provided by Jonathan A. Green, University of Missouri-Columbia, Columbia, MO; 1:100), rabbit anti-SHMT2 polyclonal antibody (Sigma-Aldrich, St. Louis, MO, USA; HPA020543; 1:100), rabbit anti-PSPH polyclonal antibody (Lifespan Biosciences, Seattle, WA, USA; LS-B2935; 1:100), mouse anti-E-cadherin monoclonal antibody (BD Biosciences; San Jose, CA, USA; 610182; 1:200), mouse anti-cytokeratin monoclonal antibody (Sigma-Aldrich; C-6909; 1:500), and mouse anti-caspase 3 monoclonal antibody (Cell Signaling Technology; Danvers, MA, USA; 9668; 1:100). Each antibody was used at a dilution optimized for that antibody. Normal rabbit (EMD Millipore; Billerica, MA, USA; 12-370) or mouse (EMD Millipore; 12-371) IgG, at a concentration equal to that for the primary IgG, was used as the negative control. Immunoreactive proteins were detected using the appropriate Alexa Fluor 488- or Alexa Fluor 594-conjugated secondary antibodies (Life Technologies, Grand Island, NY, USA) for 1 h at room temperature at a dilution of 1:250. Tissue sections were then washed three times for 5 min/wash in PBS. Slides were counterstained with Prolong Gold Antifade reagent containing DAPI (Life Technologies, Carlsbad, CA, USA) and coverslipped. Images were taken using an Axioplan 2 microscope (Carl Zeiss, Thornwood, NY, USA) interfaced with an Axioplan HR digital camera.
Frozen sections of uterine–placental interface (8 μm) from day 18 of pregnancy were used to localize CD45- or E-cadherin-positive cells using immunofluorescence microscopy as described previously [
24]. Sections were fixed in methanol at −20 °C and washed in PBS. These sections were then blocked in 10% normal goat serum diluted in antibody dilution buffer for 1 h at room temperature. Mouse anti-CD45 monoclonal antibody (Pierce Biotechnology, Rockford, IL, USA; MA1-81267; 1:500) or anti-E-cadherin antibody (1:200) were added at a dilution optimized for each antibody and incubated overnight at 4 °C in a humidified chamber. Tissue sections were then washed three times for 5 min/wash in PBS. Goat anti-mouse IgG Alexa 594 (Life Technologies; 1:250) was added to tissue sections and incubated for 1 h at room temperature. Tissue sections were then washed three times for 5 min/wash in PBS. Slides were processed for counterstaining and imaging as described previously.
For dual immunofluorescence staining (PAG + E-cadherin, PAG + cytokeratin, PAG + caspase 3, SHMT2 + PSPH), we followed the same procedures as described for normal immunofluorescence staining except that we added the two primary antibodies simultaneously on the first day and added the two secondary antibodies (goat anti-rabbit-Alexa Fluor 488-conjugated and goat anti-mouse-Alexa Fluor 594-conjugated) simultaneously on the second day.
4.3. TUNEL Apoptosis Assay
Apoptosis was assessed using a terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick-end labeling (TUNEL) assay (Promega) according to the manufacturer’s instructions for frozen tissue. Briefly, sections were fixed in 4% methanol free paraformaldehyde (PFA) in PBS for 25 min at 4 °C. The tissue was washed with PBS, and then slides were covered with equilibration buffer for 10 min at room temperature followed by incubation with TdT incubation buffer (containing TdT and nucleotide mix) for 1 h at 37 °C in a humidified chamber. The reaction was terminated by submersion of slides in 2× SSC for 15 min at room temperature. The sections were then washed with PBS, counterstained with Prolong Gold Antifade reagent containing DAPI (Life Technologies), and coverslipped. Images were taken using an Axioplan 2 microscope (Carl Zeiss, Thornwood, NY, USA) interfaced with an Axioplan HR digital camera.
For double staining for TUNEL and E-cadherin, mouse anti-E-cadherin antibody was added to TUNEL-stained sections and incubated overnight at 4 °C in a humidified chamber in the dark. Tissue sections were then washed three times for 5 min/wash in PBS. Goat anti-mouse IgG Alexa 594 (Life Technologies; 1:250) was added and incubated for 1 h at room temperature. Tissue sections were then washed three times for 5 min per wash in PBS and processed for imaging as described previously.
4.4. Rapid Immunofluorescent Staining and Laser Capture Microdissection (LCM)
Endometrial tissues frozen in OCT were sectioned at 8 μm using a cryostat. Sections were placed on polyethylene naphthalate (PEN) membrane slides and stored at −80 °C until analyzed. Immunofluorescent staining was performed as previously described with slight modifications to maintain mRNA integrity [
25]. Briefly, the sections were fixed in ice-cold 100% methanol for 3 min, washed briefly in cold PBS, and incubated with mouse anti-E-cadherin antibody (1:50) for 5 min, and followed by four brief rinses in cold PBS. These sections were then incubated with anti-mouse secondary antibody conjugated to Alexa Fluor 594 for 5 min. Sections were washed four times in cold PBS and dehydrated (1 min each in 75%, 95%, and 2 × 1 min in 100% EtOH), followed by 5 min in xylene. After air-drying for 10 min, sections were visualized using a Veritas™ Laser Microdissection System (Arcturus Bioscience, Mountain View, CA, USA). Stroma containing no E-cadherin-fluorescence-positive cells and stroma containing E-cadherin-fluorescence-positive cells were collected on the CapSure Macro LCM Caps (Thermo Fisher Scientific, Waltham, MA, USA).
4.5. Reverse Transcription–Polymerase Chain Reaction (RT–PCR)
Total RNA was prepared from captured cells using a PicoPure RNA Isolation Kit (Thermo Fisher Scientific), and cDNA was synthesized using a Superscript III First Strand Kit (Life Technologies, Carlsbad, CA, USA) according to the manufacturer’s instructions. The cDNA templates were amplified by PCR using Taq polymerase and specific primers based on mRNA sequences of sheep CDH1 (GenBank accession number XM_004015093.3; forward, 5′-TAC TAT GAT GAA GAA GGA GGT GGA G-3′; reverse, 5′-CAA TAA AGT TTC CAA TTT CAT CAG G-3′) or ACTB (GenBank accession number NM_001009784.1; forward, 5′-ATT CAC GAA ACT ACC TTC AAT TCC-3′; reverse, 5′-ATG ATC TTG ATC TTC ATC GTG CT-3′), which amplify cDNAs of 175 bp and 170 bp, respectively. PCR conditions for both CDH1 and ACTB were 45 cycles of 94 °C for 45 s, 60 °C for 45 s, and 72 °C for 1 min. PCR products were separated on 2% agarose gels and visualized following ethidium bromide staining.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





