Melatonin Enhances Mitophagy by Upregulating Expression of Heat Shock 70 kDa Protein 1L in Human Mesenchymal Stem Cells under Oxidative Stress


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
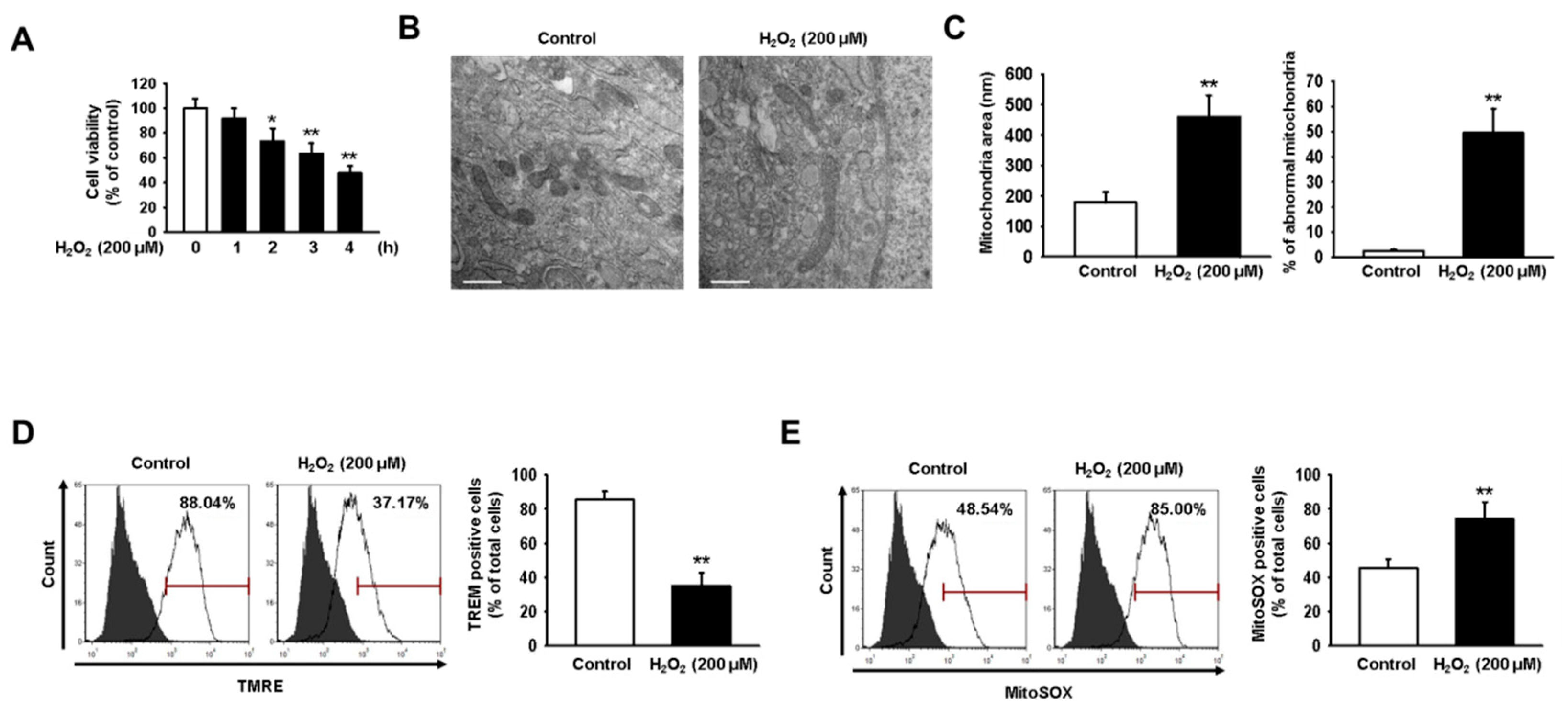
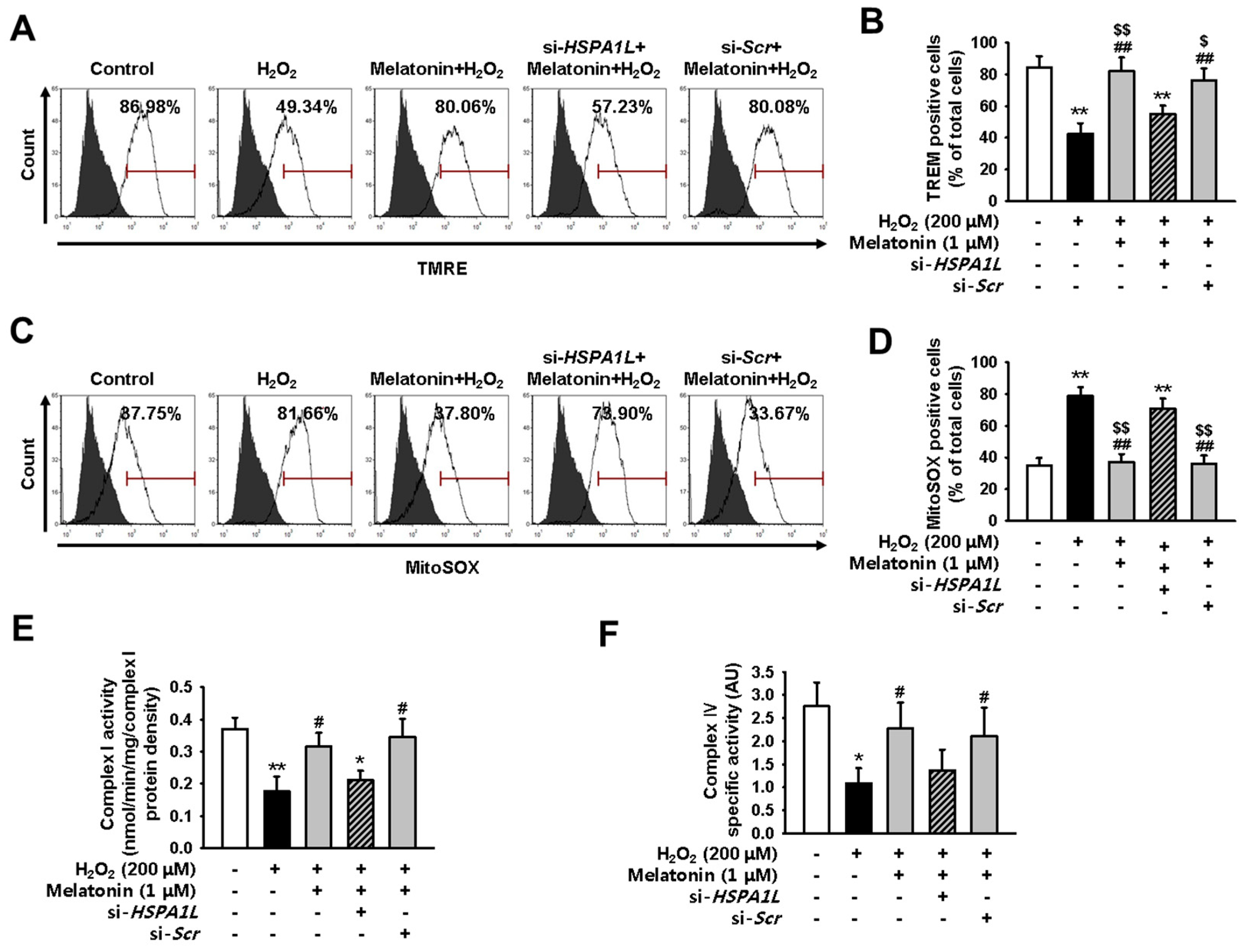
2.1. Oxidative Stress Impairs Mitochondrial Function in hMSCs
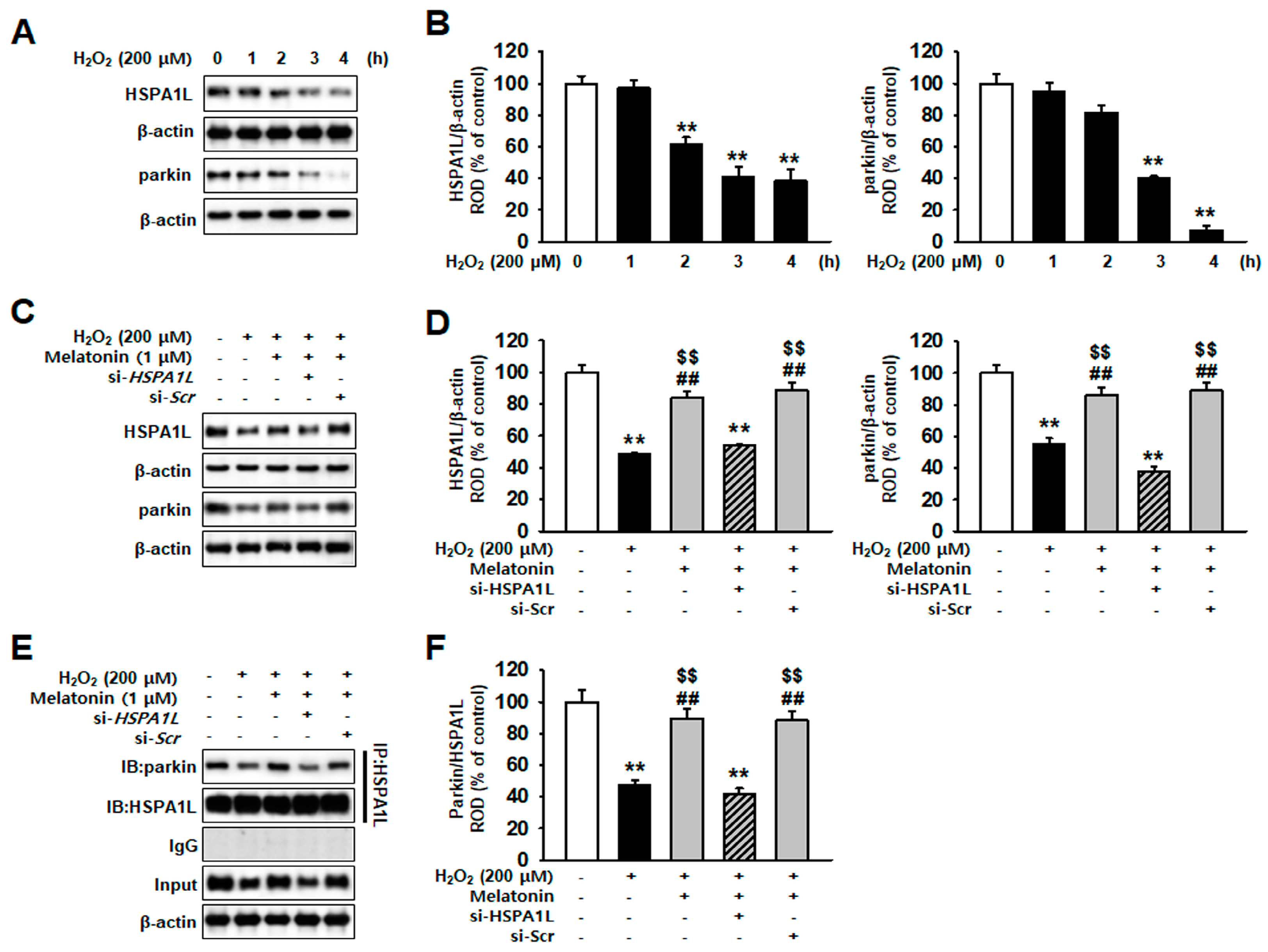
2.2. Melatonin-Treated hMSCs Show Increased HSPA1L Expression and Parkin Stability under Conditions of Oxidative Stress
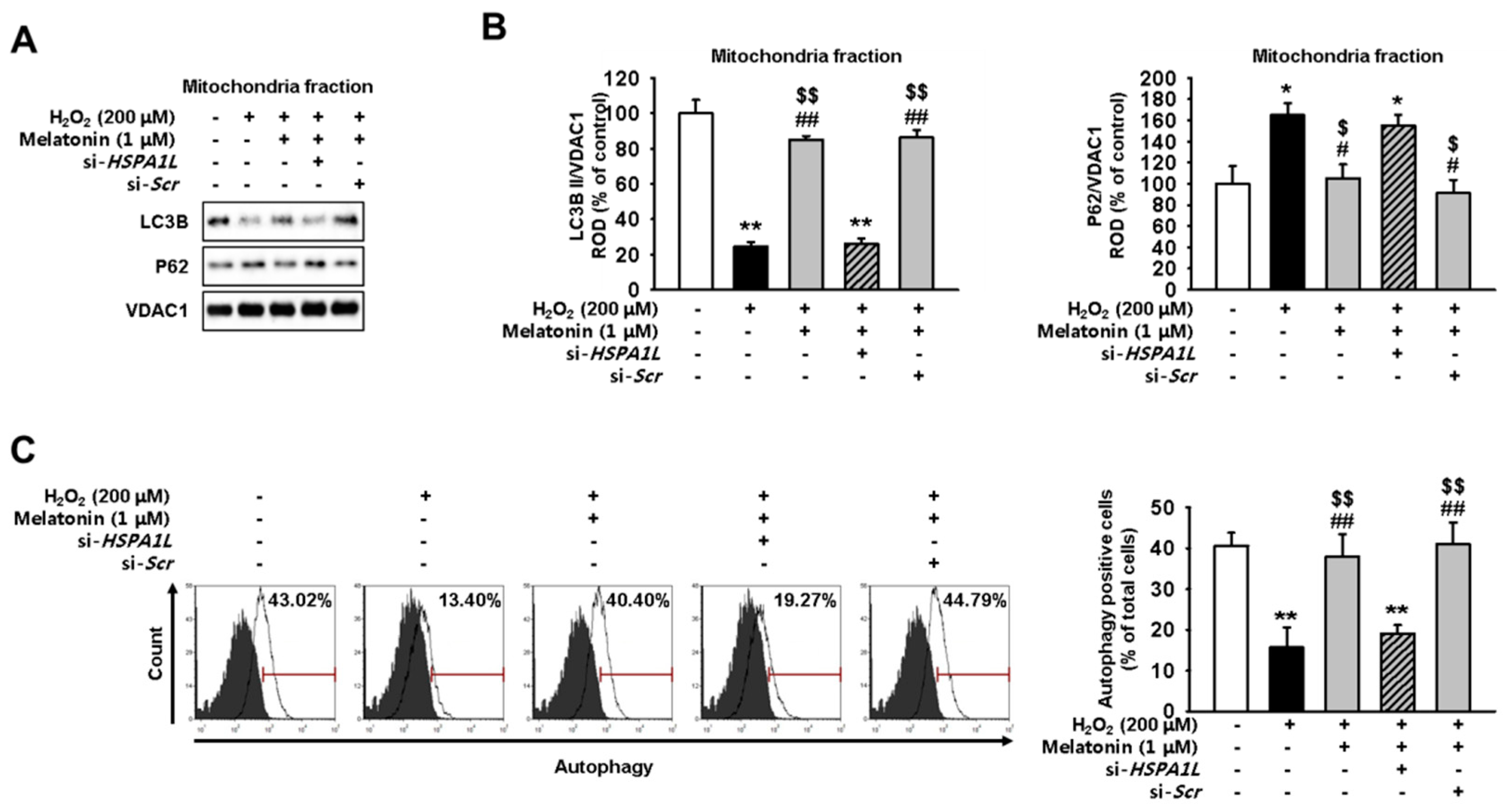
2.3. Mitophagy Pathway Is Enhanced via Increased Expression of HSPA1L in Melatonin-Treated hMSCs Subjected to Oxidative Stress
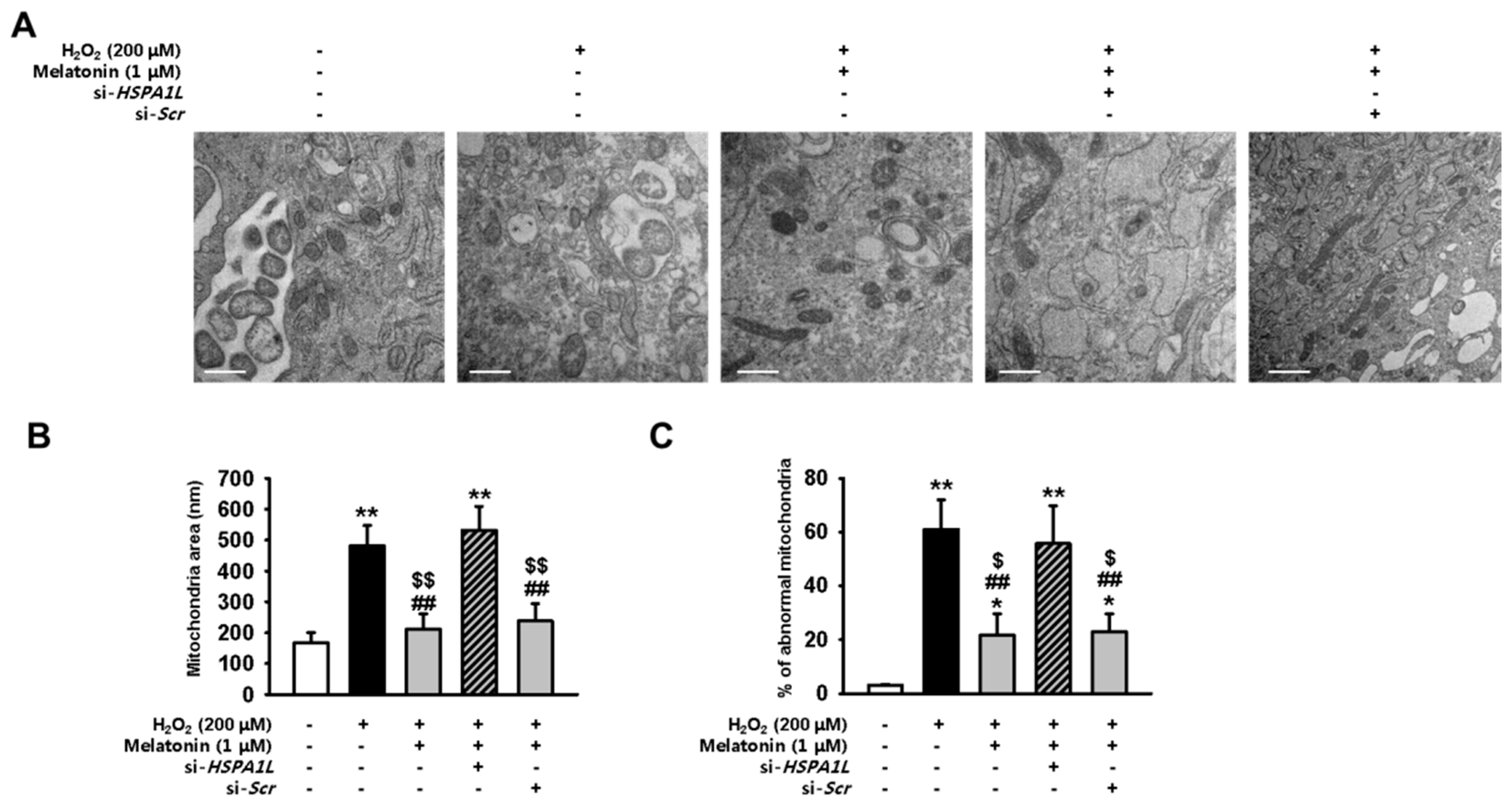
2.4. Melatonin-Treated hMSCs Show Enhanced Mitochondrial Function under Conditions of Oxidative Stress
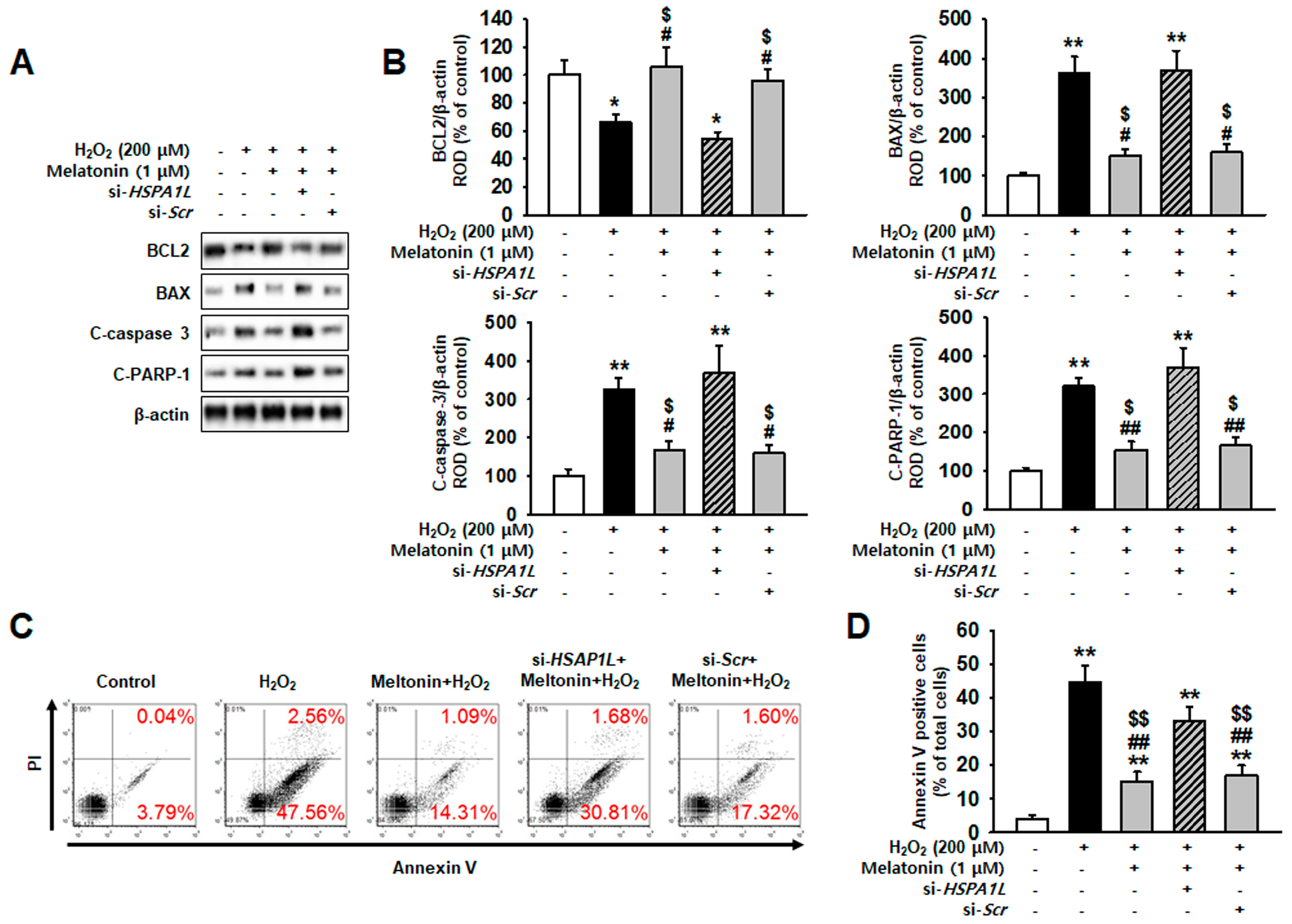
2.5. Melatonin Protects hMSCs against Oxidative Stress by Augmenting Mitophagy
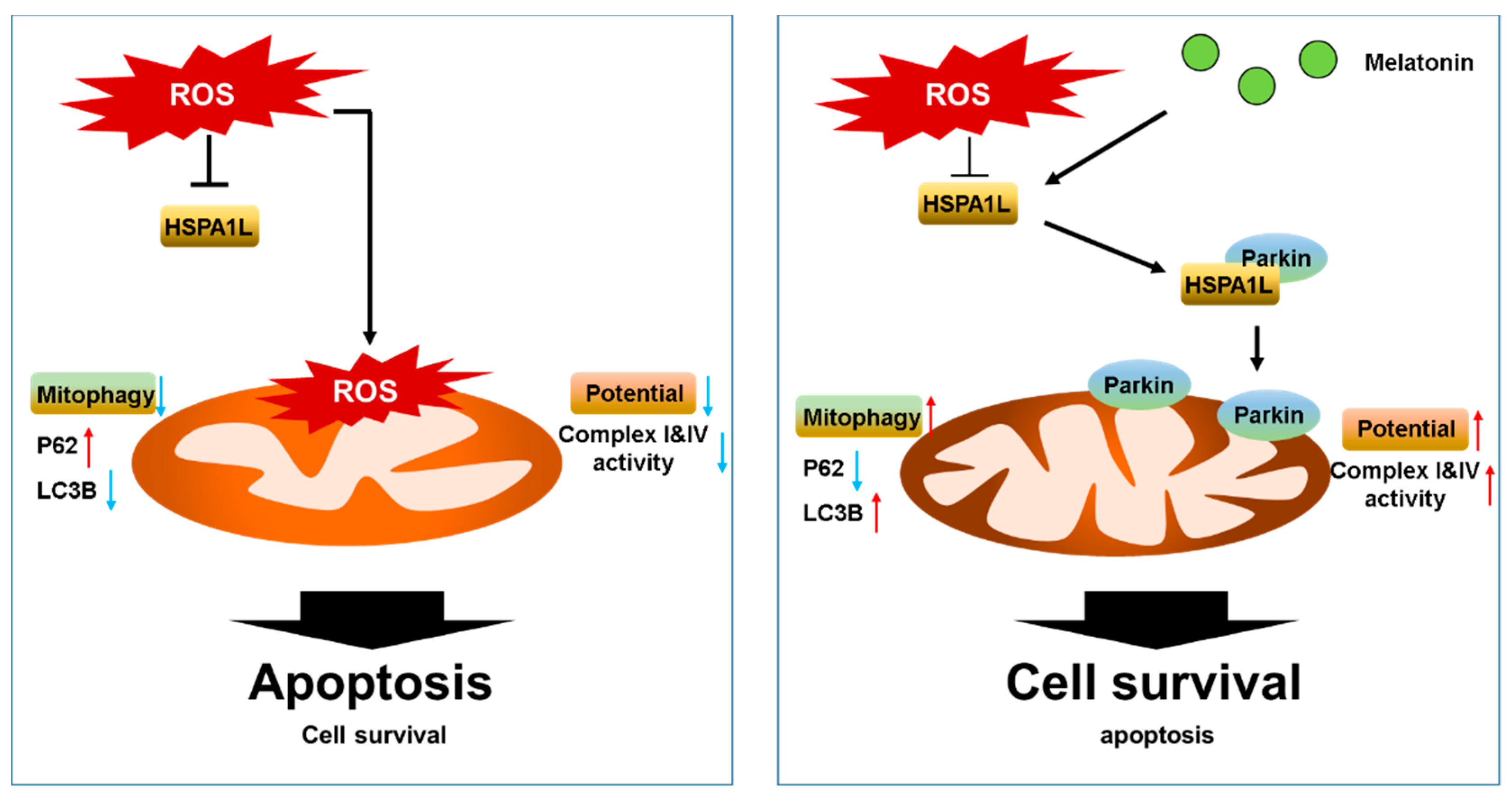
3. Discussion
4. Materials and Methods
4.1. Human MSC Cultures
4.2. Treatments Administered to hMSCs
4.3. Inhibition of HSPA1L Expression by RNA Interference
4.4. Electron Microscopy
4.5. Measurement of Mitochondrial Membrane Potential
4.6. Measurement of Mitochondrial Superoxide (O2•−) Generation
4.7. Autophagy Assessment
4.8. PI/Annexin V Flow Cytometric Analysis
4.9. Western Blotting
4.10. Immunoprecipitation
4.11. Evaluation of Electron Transport Chain Complex I Activity
4.12. Evaluation of Electron Transport Chain Complex IV Activity
4.13. Statistical Analysis
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Phinney, D.G.; Pittenger, M.F. Concise review: Msc-derived exosomes for cell-free therapy. Stem Cells 2017, 35, 851–858. [Google Scholar] [CrossRef] [PubMed]
- Bian, S.; Zhang, L.; Duan, L.; Wang, X.; Min, Y.; Yu, H. Extracellular vesicles derived from human bone marrow mesenchymal stem cells promote angiogenesis in a rat myocardial infarction model. J. Mol. Med. 2014, 92, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Teng, X.; Chen, L.; Chen, W.; Yang, J.; Yang, Z.; Shen, Z. Mesenchymal stem cell-derived exosomes improve the microenvironment of infarcted myocardium contributing to angiogenesis and anti-inflammation. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2015, 37, 2415–2424. [Google Scholar] [CrossRef] [PubMed]
- Yoon, Y.M.; Kim, S.; Han, Y.S.; Yun, C.W.; Lee, J.H.; Noh, H.; Lee, S.H. Tudca-treated chronic kidney disease-derived hmscs improve therapeutic efficacy in ischemic disease via prp(c). Redox Biol. 2019, 22, 101144. [Google Scholar] [CrossRef] [PubMed]
- Yoon, Y.M.; Lee, J.H.; Yun, S.P.; Han, Y.S.; Yun, C.W.; Lee, H.J.; Noh, H.; Lee, S.J.; Han, H.J.; Lee, S.H. Tauroursodeoxycholic acid reduces er stress by regulating of akt-dependent cellular prion protein. Sci. Rep. 2016, 6, 39838. [Google Scholar] [CrossRef] [PubMed]
- Yun, S.P.; Yoon, Y.M.; Lee, J.H.; Kook, M.; Han, Y.S.; Jung, S.K.; Lee, S.H. Tauroursodeoxycholic acid protects against the effects of p-cresol-induced reactive oxygen species via the expression of cellular prion protein. Int. J. Mol. Sci. 2018, 19, 352. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.S.; Lee, J.H.; Jung, J.S.; Noh, H.; Baek, M.J.; Ryu, J.M.; Yoon, Y.M.; Han, H.J.; Lee, S.H. Fucoidan protects mesenchymal stem cells against oxidative stress and enhances vascular regeneration in a murine hindlimb ischemia model. Int. J. Cardiol. 2015, 198, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Yoon, Y.M.; Lee, J.H.; Yun, C.W.; Lee, S.H. Pioglitazone improves the function of human mesenchymal stem cells in chronic kidney disease patients. Int. J. Mol. Sci. 2019, 20, 2314. [Google Scholar] [CrossRef]
- Ott, M.; Gogvadze, V.; Orrenius, S.; Zhivotovsky, B. Mitochondria, oxidative stress and cell death. Apoptosis Int. J. Program. Cell Death 2007, 12, 913–922. [Google Scholar] [CrossRef]
- Ott, M.; Robertson, J.D.; Gogvadze, V.; Zhivotovsky, B.; Orrenius, S. Cytochrome c release from mitochondria proceeds by a two-step process. Proc. Natl. Acad. Sci. USA 2002, 99, 1259–1263. [Google Scholar] [CrossRef]
- Fan, P.; Xie, X.H.; Chen, C.H.; Peng, X.; Zhang, P.; Yang, C.; Wang, Y.T. Molecular regulation mechanisms and interactions between reactive oxygen species and mitophagy. DNA Cell Biol. 2019, 38, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Rahal, A.; Kumar, A.; Singh, V.; Yadav, B.; Tiwari, R.; Chakraborty, S.; Dhama, K. Oxidative stress, prooxidants, and antioxidants: The interplay. BioMed Res. Int. 2014, 2014, 761264. [Google Scholar] [CrossRef] [PubMed]
- Sergi, D.; Naumovski, N.; Heilbronn, L.K.; Abeywardena, M.; O’Callaghan, N.; Lionetti, L.; Luscombe-Marsh, N. Mitochondrial (dys)function and insulin resistance: From pathophysiological molecular mechanisms to the impact of diet. Front. Physiol. 2019, 10, 532. [Google Scholar] [CrossRef] [PubMed]
- Thannickal, V.J.; Fanburg, B.L. Reactive oxygen species in cell signaling. Am. J. Physiol. Lung Cell. Mol. Physiol. 2000, 279, L1005–L1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Truban, D.; Hou, X.; Caulfield, T.R.; Fiesel, F.C.; Springer, W. Pink1, parkin, and mitochondrial quality control: What can we learn about parkinson’s disease pathobiology? J. Park. Dis. 2017, 7, 13–29. [Google Scholar] [CrossRef] [PubMed]
- Ashrafi, G.; Schwarz, T.L. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2013, 20, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Narendra, D.; Kane, L.A.; Hauser, D.N.; Fearnley, I.M.; Youle, R.J. P62/sqstm1 is required for parkin-induced mitochondrial clustering but not mitophagy; vdac1 is dispensable for both. Autophagy 2010, 6, 1090–1106. [Google Scholar] [CrossRef]
- MacVicar, T. Mitophagy. Essays Biochem. 2013, 55, 93–104. [Google Scholar] [CrossRef]
- Lerner, A.B.; Case, J.D.; Takahashi, Y.; Lee, T.H.; Mori, W. Isolation of melatonin, the pineal gland factor that lightens melanocytes. J. Am. Chem. Soc. 1958, 80, 2587. [Google Scholar] [CrossRef]
- Reiter, R.J.; Rosales-Corral, S.; Tan, D.X.; Jou, M.J.; Galano, A.; Xu, B. Melatonin as a mitochondria-targeted antioxidant: One of evolution’s best ideas. Cell. Mol. Life Sci. CMLS 2017, 74, 3863–3881. [Google Scholar] [CrossRef]
- Lee, J.H.; Yoon, Y.M.; Han, Y.S.; Jung, S.K.; Lee, S.H. Melatonin protects mesenchymal stem cells from autophagy-mediated death under ischaemic er-stress conditions by increasing prion protein expression. Cell Prolif. 2019, 52, e12545. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.S.; Kim, S.M.; Lee, J.H.; Jung, S.K.; Noh, H.; Lee, S.H. Melatonin protects chronic kidney disease mesenchymal stem cells against senescence via prp(c)—dependent enhancement of the mitochondrial function. J. Pineal Res. 2019, 66, e12535. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Han, Y.S.; Lee, S.H. Potentiation of biological effects of mesenchymal stem cells in ischemic conditions by melatonin via upregulation of cellular prion protein expression. J. Pineal Res. 2017, 62, e12385. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Yoon, Y.M.; Han, Y.S.; Yun, C.W.; Lee, S.H. Melatonin promotes apoptosis of oxaliplatin-resistant colorectal cancer cells through inhibition of cellular prion protein. Anticancer Res. 2018, 38, 1993–2000. [Google Scholar] [PubMed]
- Han, Y.S.; Kim, S.M.; Lee, J.H.; Lee, S.H. Co-administration of melatonin effectively enhances the therapeutic effects of pioglitazone on mesenchymal stem cells undergoing indoxyl sulfate-induced senescence through modulation of cellular prion protein expression. Int. J. Mol. Sci. 2018, 19, 1367. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Yang, J.; Yang, C.; Zhu, M.; Jin, Y.; McNutt, M.A.; Yin, Y. Ptenalpha regulates mitophagy and maintains mitochondrial quality control. Autophagy 2018, 14, 1742–1760. [Google Scholar] [CrossRef]
- Weber, H.; Huhns, S.; Jonas, L.; Sparmann, G.; Bastian, M.; Schuff-Werner, P. Hydrogen peroxide-induced activation of defense mechanisms against oxidative stress in rat pancreatic acinar ar42j cells. Free Radic. Biol. Med. 2007, 42, 830–841. [Google Scholar] [CrossRef]
- Hasson, S.A.; Kane, L.A.; Yamano, K.; Huang, C.H.; Sliter, D.A.; Buehler, E.; Wang, C.; Heman-Ackah, S.M.; Hessa, T.; Guha, R.; et al. High-content genome-wide rnai screens identify regulators of parkin upstream of mitophagy. Nature 2013, 504, 291–295. [Google Scholar] [CrossRef]
- Matsuda, N.; Sato, S.; Shiba, K.; Okatsu, K.; Saisho, K.; Gautier, C.A.; Sou, Y.S.; Saiki, S.; Kawajiri, S.; Sato, F.; et al. Pink1 stabilized by mitochondrial depolarization recruits parkin to damaged mitochondria and activates latent parkin for mitophagy. J. Cell Biol. 2010, 189, 211–221. [Google Scholar] [CrossRef]
- Colmegna, I.; Stochaj, U. Msc—targets for atherosclerosis therapy. Aging 2018, 11, 285–286. [Google Scholar] [CrossRef]
- Bari, E.; Ferrarotti, I.; Torre, M.L.; Corsico, A.G.; Perteghella, S. Mesenchymal stem/stromal cell secretome for lung regeneration: The long way through “pharmaceuticalization” for the best formulation. J. Control. Release Off. J. Control. Release Soc. 2019, 309, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Soria-Juan, B.; Escacena, N.; Capilla-Gonzalez, V.; Aguilera, Y.; Llanos, L.; Tejedo, J.R.; Bedoya, F.J.; Juan, V.; De la Cuesta, A.; Ruiz-Salmeron, R.; et al. Cost-effective, safe, and personalized cell therapy for critical limb ischemia in type 2 diabetes mellitus. Front. Immunol. 2019, 10, 1151. [Google Scholar] [CrossRef] [PubMed]
- Klinkhammer, B.M.; Kramann, R.; Mallau, M.; Makowska, A.; van Roeyen, C.R.; Rong, S.; Buecher, E.B.; Boor, P.; Kovacova, K.; Zok, S.; et al. Mesenchymal stem cells from rats with chronic kidney disease exhibit premature senescence and loss of regenerative potential. PLoS ONE 2014, 9, e92115. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Jung, Y.H.; Oh, S.Y.; Yun, S.P.; Han, H.J. Melatonin enhances the human mesenchymal stem cells motility via melatonin receptor 2 coupling with galphaq in skin wound healing. J. Pineal Res. 2014, 57, 393–407. [Google Scholar] [CrossRef] [PubMed]
- Hardeland, R. Melatonin and the pathologies of weakened or dysregulated circadian oscillators. J. Pineal Res. 2017, 62, e12377. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Cai, B.; Yuan, F.; He, X.; Lin, X.; Wang, J.; Wang, Y.; Yang, G.Y. Melatonin pretreatment improves the survival and function of transplanted mesenchymal stem cells after focal cerebral ischemia. Cell Transplant. 2014, 23, 1279–1291. [Google Scholar] [CrossRef] [PubMed]
- Mortezaee, K.; Khanlarkhani, N.; Sabbaghziarani, F.; Nekoonam, S.; Majidpoor, J.; Hosseini, A.; Pasbakhsh, P.; Kashani, I.R.; Zendedel, A. Preconditioning with melatonin improves therapeutic outcomes of bone marrow-derived mesenchymal stem cells in targeting liver fibrosis induced by ccl4. Cell Tissue Res. 2017, 369, 303–312. [Google Scholar] [CrossRef]
- Langrzyk, A.; Nowak, W.N.; Stepniewski, J.; Jazwa, A.; Florczyk-Soluch, U.; Jozkowicz, A.; Dulak, J. Critical view on mesenchymal stromal cells in regenerative medicine. Antioxid. Redox Signal. 2018, 29, 169–190. [Google Scholar] [CrossRef]
- Munro, D.; Treberg, J.R. A radical shift in perspective: Mitochondria as regulators of reactive oxygen species. J. Exp. Biol. 2017, 220, 1170–1180. [Google Scholar] [CrossRef]
- Trub, A.G.; Hirschey, M.D. Reactive acyl-coa species modify proteins and induce carbon stress. Trends Biochem. Sci. 2018, 43, 369–379. [Google Scholar] [CrossRef]
- van der Reest, J.; Lilla, S.; Zheng, L.; Zanivan, S.; Gottlieb, E. Proteome-wide analysis of cysteine oxidation reveals metabolic sensitivity to redox stress. Nat. Commun. 2018, 9, 1581. [Google Scholar] [CrossRef] [PubMed]
- Shutt, T.; Geoffrion, M.; Milne, R.; McBride, H.M. The intracellular redox state is a core determinant of mitochondrial fusion. EMBO Rep. 2012, 13, 909–915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bueno, M.; Lai, Y.C.; Romero, Y.; Brands, J.; St Croix, C.M.; Kamga, C.; Corey, C.; Herazo-Maya, J.D.; Sembrat, J.; Lee, J.S.; et al. Pink1 deficiency impairs mitochondrial homeostasis and promotes lung fibrosis. J. Clin. Investig. 2015, 125, 521–538. [Google Scholar] [CrossRef] [PubMed]
- Padman, B.S.; Bach, M.; Lucarelli, G.; Prescott, M.; Ramm, G. The protonophore cccp interferes with lysosomal degradation of autophagic cargo in yeast and mammalian cells. Autophagy 2013, 9, 1862–1875. [Google Scholar] [CrossRef] [PubMed]
- Rahmani, M.; Nkwocha, J.; Hawkins, E.; Pei, X.; Parker, R.E.; Kmieciak, M.; Leverson, J.D.; Sampath, D.; Ferreira-Gonzalez, A.; Grant, S. Cotargeting bcl-2 and pi3k induces bax-dependent mitochondrial apoptosis in aml cells. Cancer Res. 2018, 78, 3075–3086. [Google Scholar] [CrossRef] [PubMed]
- Sinha, K.; Das, J.; Pal, P.B.; Sil, P.C. Oxidative stress: The mitochondria-dependent and mitochondria-independent pathways of apoptosis. Arch. Toxicol. 2013, 87, 1157–1180. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S.; Andreoletti, G.; Chen, R.; Munehira, Y.; Batra, A.; Afzal, N.A.; Beattie, R.M.; Bernstein, J.A.; Ennis, S.; Snyder, M. De novo and rare mutations in the hspa1l heat shock gene associated with inflammatory bowel disease. Genome Med. 2017, 9, 8. [Google Scholar] [CrossRef] [PubMed]
- Huusko, J.M.; Karjalainen, M.K.; Graham, B.E.; Zhang, G.; Farrow, E.G.; Miller, N.A.; Jacobsson, B.; Eidem, H.R.; Murray, J.C.; Bedell, B.; et al. Whole exome sequencing reveals hspa1l as a genetic risk factor for spontaneous preterm birth. PLoS Genet. 2018, 14, e1007394. [Google Scholar]
- Jin, S.M.; Lazarou, M.; Wang, C.; Kane, L.A.; Narendra, D.P.; Youle, R.J. Mitochondrial membrane potential regulates pink1 import and proteolytic destabilization by parl. J. Cell Biol. 2010, 191, 933–942. [Google Scholar] [CrossRef]
- Lazarou, M.; Jin, S.M.; Kane, L.A.; Youle, R.J. Role of pink1 binding to the tom complex and alternate intracellular membranes in recruitment and activation of the e3 ligase parkin. Dev. Cell 2012, 22, 320–333. [Google Scholar] [CrossRef]
- Nguyen, T.N.; Padman, B.S.; Lazarou, M. Deciphering the molecular signals of pink1/parkin mitophagy. Trends Cell Biol. 2016, 26, 733–744. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.S.; Lee, J.H.; Yoon, Y.M.; Yun, C.W.; Noh, H.; Lee, S.H. Hypoxia-induced expression of cellular prion protein improves the therapeutic potential of mesenchymal stem cells. Cell Death Dis. 2016, 7, e2395. [Google Scholar] [CrossRef] [PubMed]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yoon, Y.M.; Kim, H.J.; Lee, J.H.; Lee, S.H. Melatonin Enhances Mitophagy by Upregulating Expression of Heat Shock 70 kDa Protein 1L in Human Mesenchymal Stem Cells under Oxidative Stress. Int. J. Mol. Sci. 2019, 20, 4545. https://doi.org/10.3390/ijms20184545
Yoon YM, Kim HJ, Lee JH, Lee SH. Melatonin Enhances Mitophagy by Upregulating Expression of Heat Shock 70 kDa Protein 1L in Human Mesenchymal Stem Cells under Oxidative Stress. International Journal of Molecular Sciences. 2019; 20(18):4545. https://doi.org/10.3390/ijms20184545
Chicago/Turabian StyleYoon, Yeo Min, Hyung Joo Kim, Jun Hee Lee, and Sang Hun Lee. 2019. "Melatonin Enhances Mitophagy by Upregulating Expression of Heat Shock 70 kDa Protein 1L in Human Mesenchymal Stem Cells under Oxidative Stress" International Journal of Molecular Sciences 20, no. 18: 4545. https://doi.org/10.3390/ijms20184545
APA StyleYoon, Y. M., Kim, H. J., Lee, J. H., & Lee, S. H. (2019). Melatonin Enhances Mitophagy by Upregulating Expression of Heat Shock 70 kDa Protein 1L in Human Mesenchymal Stem Cells under Oxidative Stress. International Journal of Molecular Sciences, 20(18), 4545. https://doi.org/10.3390/ijms20184545