Nimodipine-Dependent Protection of Schwann Cells, Astrocytes and Neuronal Cells from Osmotic, Oxidative and Heat Stress Is Associated with the Activation of AKT and CREB


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
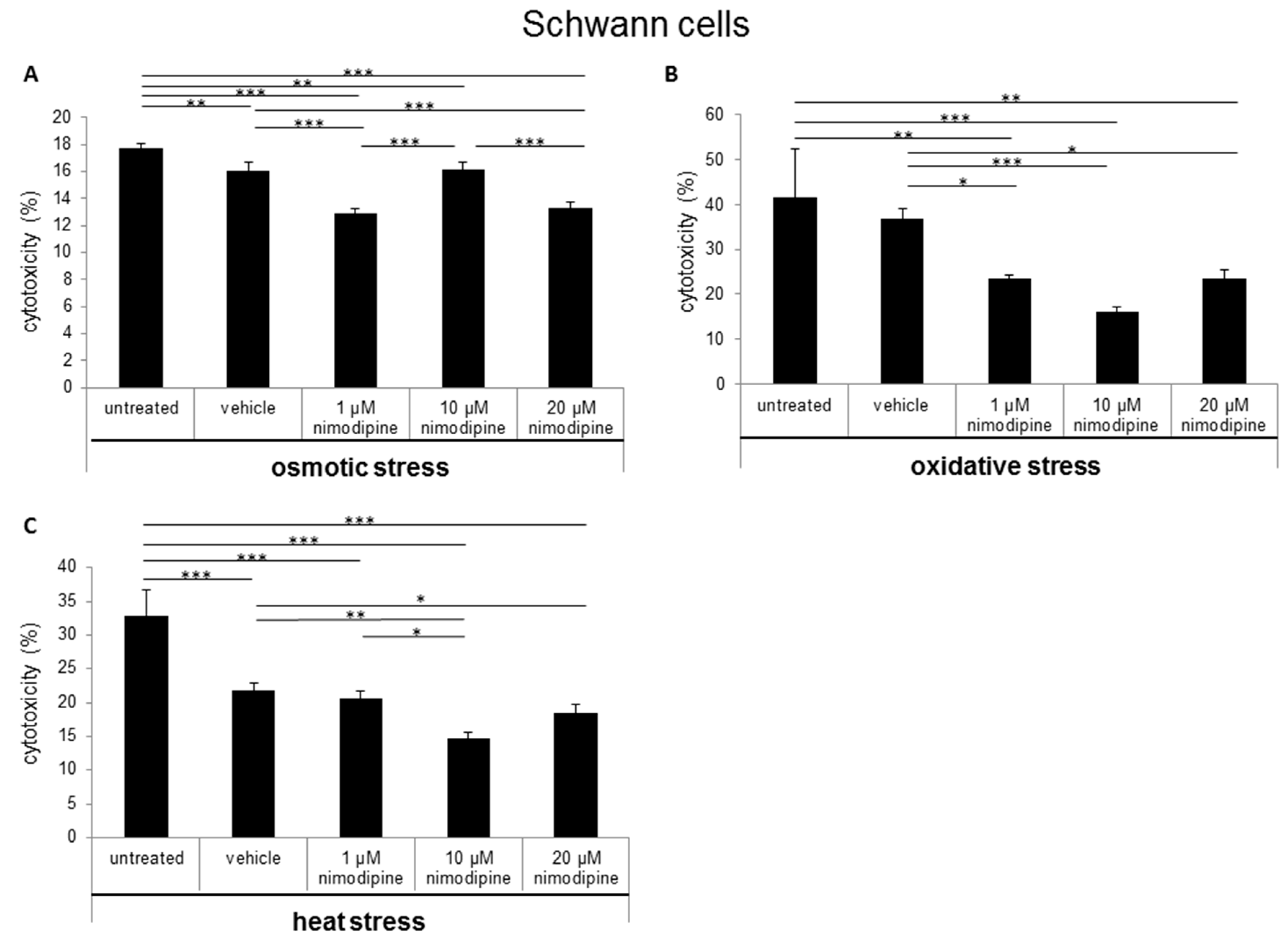
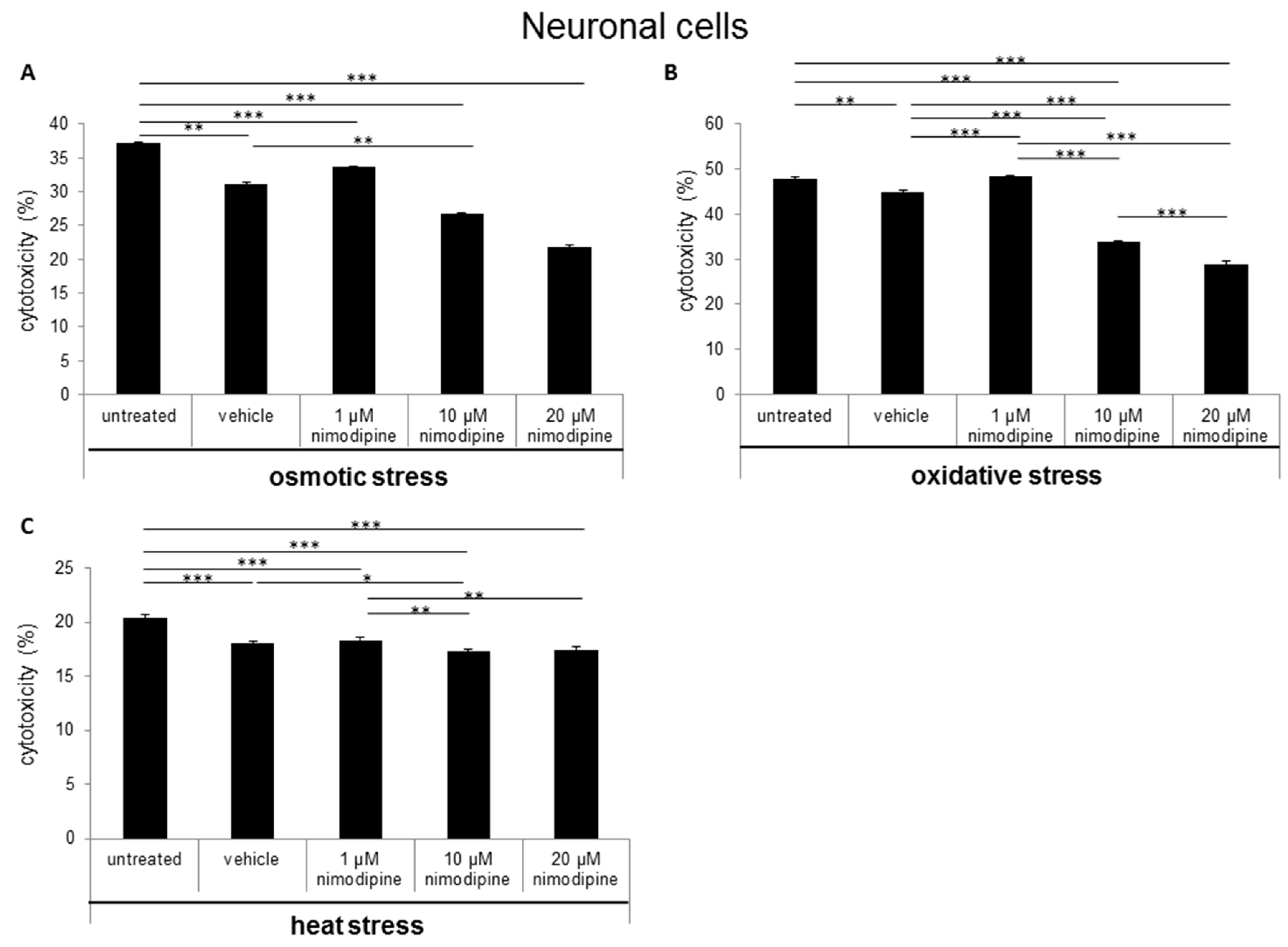
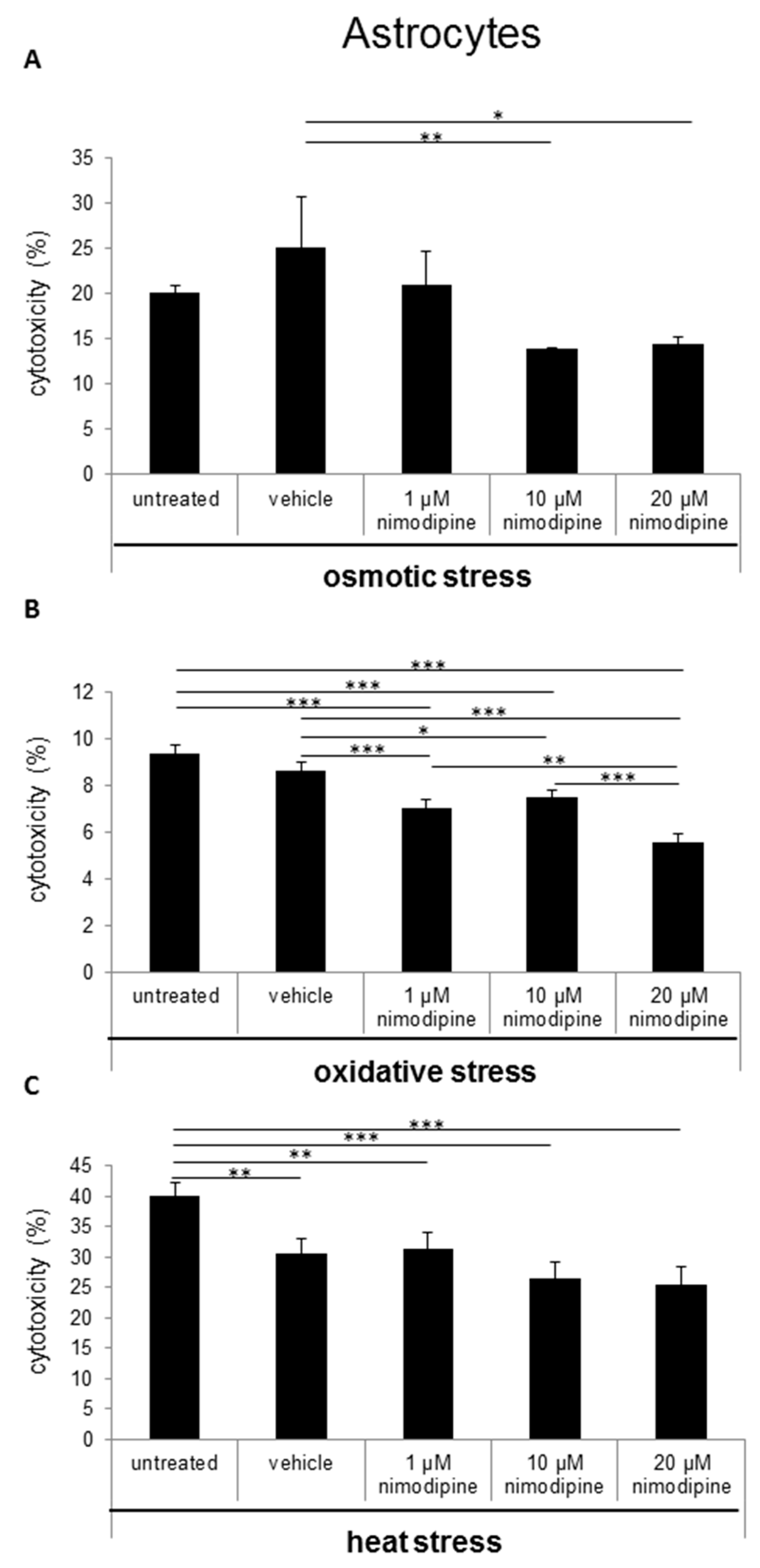
2.1. Nimodipine Decreases Cytotoxicity of Schwann Cells, Neuronal Cells and Astrocytes under Different Stress Conditions
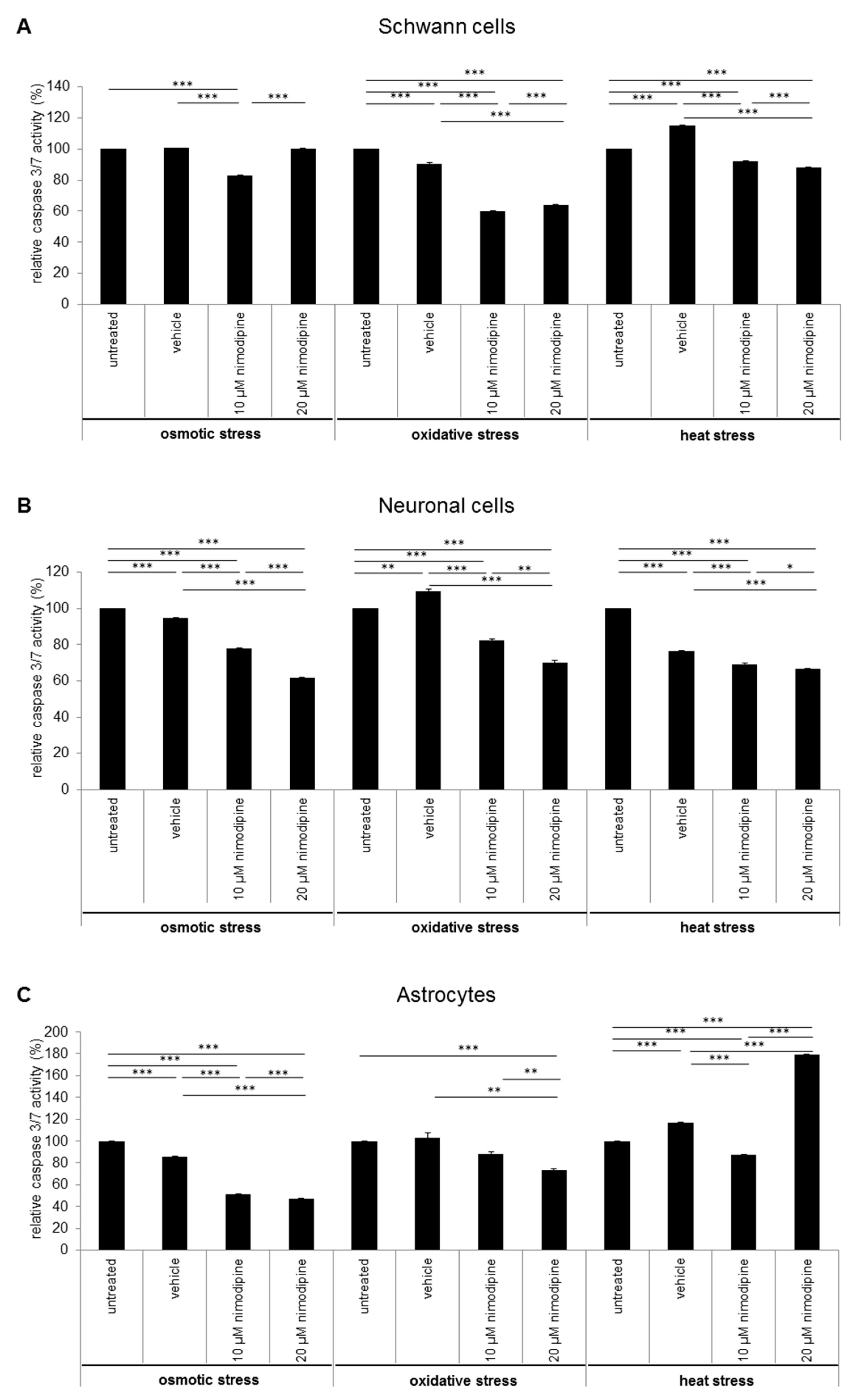
2.2. Caspase 3/7 Activity Analysis of Nimodipine Treated Schwann Cells, Neuronal Cells and Astrocytes
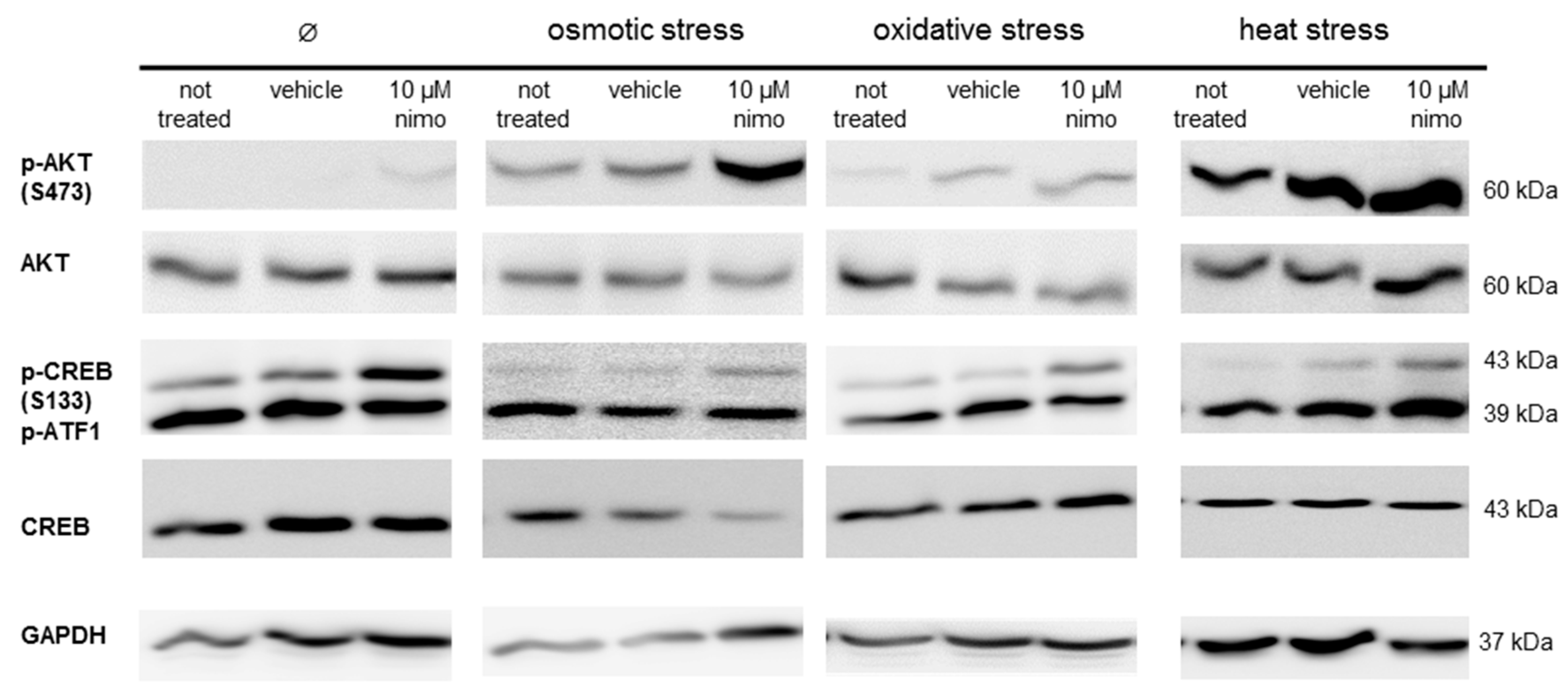
2.3. Neuroprotective Effect is Associated with Activation of AKT and CREB Signaling
3. Discussion
3.1. Nimodipine Pretreatment Leads to Cell Type-Independent Reduction of Cytotoxicity
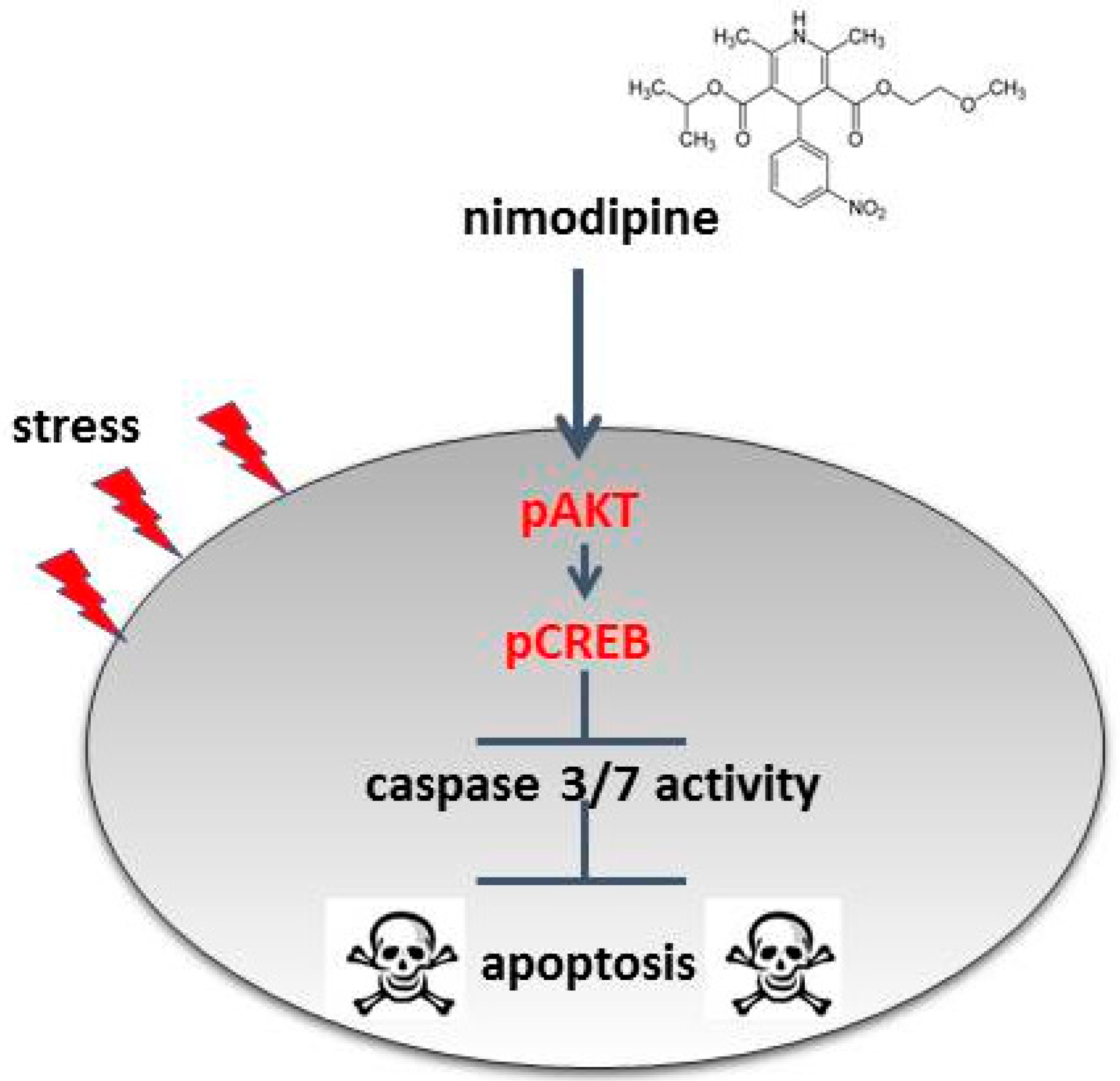
3.2. Nimodipine-Dependent Apoptosis Prevention Is Accompanied by Reduction of Caspase 3 and 7 Activity and Activation of CREB and AKT
4. Materials and Methods
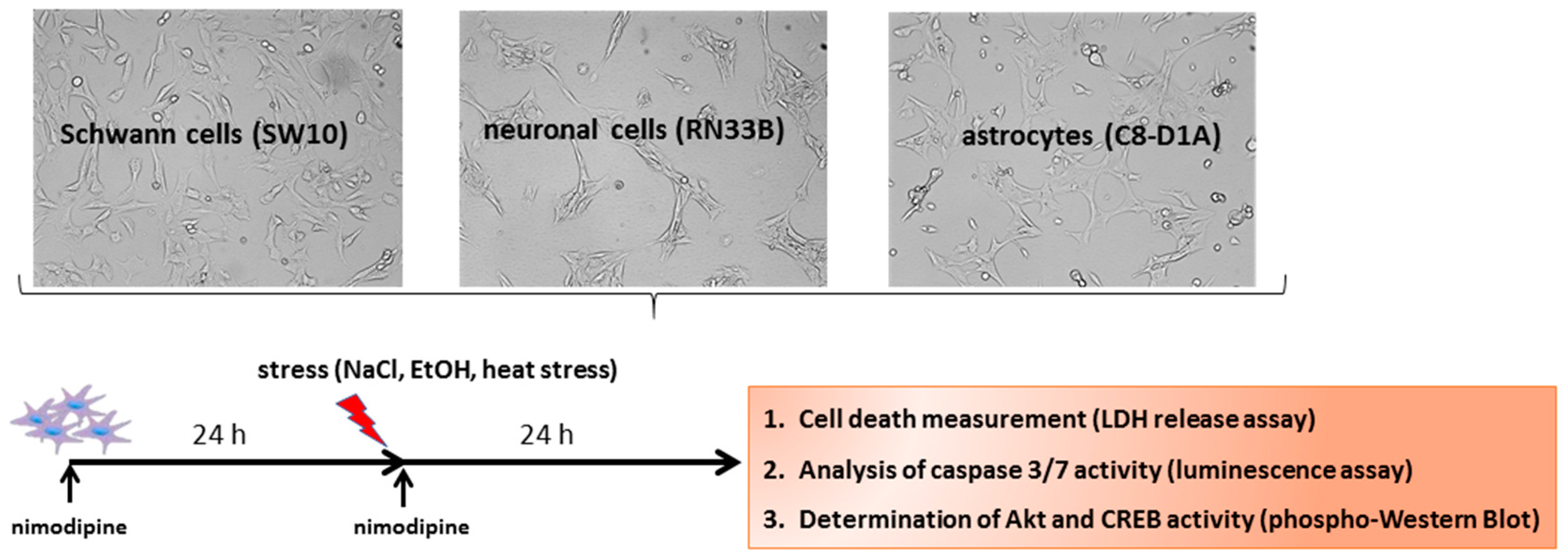
4.1. Cell Lines
4.2. Nimodipine Treatment
4.3. Stress Induction
4.4. Cytotoxicity Measurement
4.5. Western Blot Analysis
4.6. Caspase 3/7 Assay
4.7. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rabinstein, A.A.; Wijdicks, E.F. Cerebral Vasospasm in Subarachnoid Hemorrhage. Curr. Treat Options Neurol. 2005, 7, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Dorhout Mees, S.M.; Rinkel, G.J.; Feigin, V.L.; Algra, A.; van den Bergh, W.M.; Vermeulen, M.; van Gijn, J. Calcium antagonists for aneurysmal subarachnoid haemorrhage. Cochrane Database Syst. Rev. 2007. [Google Scholar] [CrossRef] [PubMed]
- Rabinstein, A.A.; Lanzino, G.; Wijdicks, E.F. Multidisciplinary management and emerging therapeutic strategies in aneurysmal subarachnoid haemorrhage. Lancet Neurol. 2010, 9, 504–519. [Google Scholar] [CrossRef]
- Scheller, K.; Scheller, C. Nimodipine promotes regeneration of peripheral facial nerve function after traumatic injury following maxillofacial surgery: An off label pilot-study. J. Craniomaxillofac. Surg. 2012, 40, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Scheller, C.; Vogel, A.S.; Simmermacher, S.; Rachinger, J.C.; Prell, J.; Strauss, C.; Reinsch, M.; Kunter, U.; Wienke, A.; Neumann, J.; et al. Prophylactic intravenous nimodipine treatment in skull base surgery: Pharmacokinetic aspects. J. Neurol. Surg. A Cent. Eur. Neurosurg. 2012, 73, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Scheller, K.; Scheller, C. Nimodipine for peripheral nerve recovery after maxillofacial and vestibular schwannoma surgery. Muscle Nerve 2014, 50, 1026–1027. [Google Scholar] [CrossRef]
- Hydman, J.; Remahl, S.; Björck, G.; Svensson, M.; Mattsson, P. Nimodipine improves reinnervation and neuromuscular function after injury to the recurrent laryngeal nerve in the rat. Ann. Otol. Rhinol. Laryngol. 2007, 116, 623–630. [Google Scholar] [CrossRef]
- Mattsson, P.; Björck, G.; Remahl, S.; Bäckdahl, M.; Hamberger, B.; Hydman, J.; Svensson, M. Nimodipine and microsurgery induced recovery of the vocal cord after recurrent laryngeal nerve resection. Laryngoscope 2005, 115, 1863–1865. [Google Scholar] [CrossRef]
- Mattsson, P.; Frostell, A.; Björck, G.; Persson, J.K.E.; Hakim, R.; Zedenius, J.; Svensson, M. Recovery of Voice After Reconstruction of the Recurrent Laryngeal Nerve and Adjuvant Nimodipine. World J. Surg. 2018, 42, 632–638. [Google Scholar] [CrossRef]
- Bork, K.; Wurm, F.; Haller, H.; Strauss, C.; Scheller, C.; Gnanapragassam, V.S.; Horstkorte, R. Neuroprotective and neuroregenerative effects of nimodipine in a model system of neuronal differentiation and neurite outgrowth. Molecules 2015, 20, 1003–1013. [Google Scholar] [CrossRef]
- Herzfeld, E.; Strauss, C.; Simmermacher, S.; Bork, K.; Horstkorte, R.; Dehghani, F.; Scheller, C. Investigation of the neuroprotective impact of nimodipine on Neuro2a cells by means of a surgery-like stress model. Int. J. Mol. Sci. 2014, 15, 18453–18465. [Google Scholar] [CrossRef] [PubMed]
- Herzfeld, E.; Speh, L.; Strauss, C.; Scheller, C. Nimodipine but Not Nifedipine Promotes Expression of Fatty Acid 2-Hydroxylase in a Surgical Stress Model Based on Neuro2a Cells. Int. J. Mol. Sci. 2017, 18, 964. [Google Scholar] [CrossRef] [PubMed]
- Koskimäki, J.; Matsui, N.; Umemori, J.; Rantamäki, T.; Castrén, E. Nimodipine activates TrkB neurotrophin receptors and induces neuroplastic and neuroprotective signaling events in the mouse hippocampus and prefrontal cortex. Cell Mol. Neurobiol. 2015, 35, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Castanares-Zapatero, D.; Hantson, P. Pharmacological treatment of delayed cerebral ischemia and vasospasm in subarachnoid hemorrhage. Ann. Intensive Care 2011, 1, 12. [Google Scholar] [CrossRef] [PubMed]
- Al-Mufti, F.; Amuluru, K.; Damodara, N.; El-Ghanem, M.; Nuoman, R.; Kamal, N.; Al-Marsoummi, S.; Morris, N.A.; Dangayach, N.S.; Mayer, S.A. Novel management strategies for medically-refractory vasospasm following aneurysmal subarachnoid hemorrhage. J. Neurol. Sci. 2018, 390, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Feigin, V.L.; Rinkel, G.J.; Algra, A.; Vermeulen, M.; van Gijn, J. Calcium antagonists in patients with aneurysmal subarachnoid hemorrhage: A systematic review. Neurology 1998, 50, 876–883. [Google Scholar] [CrossRef] [PubMed]
- Scheller, C.; Richter, H.P.; Engelhardt, M.; Köenig, R.; Antoniadis, G. The influence of prophylactic vasoactive treatment on cochlear and facial nerve functions after vestibular schwannoma surgery: A prospective and open-label randomized pilot study. Neurosurgery 2007, 61, 92–97, discussion 97-98. [Google Scholar] [CrossRef]
- Scheller, C.; Wienke, A.; Tatagiba, M.; Gharabaghi, A.; Ramina, K.F.; Ganslandt, O.; Bischoff, B.; Zenk, J.; Engelhorn, T.; Matthies, C.; et al. Prophylactic nimodipine treatment for cochlear and facial nerve preservation after vestibular schwannoma surgery: A randomized multicenter Phase III trial. J. Neurosurg. 2016, 124, 657–664. [Google Scholar] [CrossRef]
- Scheller, C.; Wienke, A.; Tatagiba, M.; Gharabaghi, A.; Ramina, K.F.; Ganslandt, O.; Bischoff, B.; Zenk, J.; Engelhorn, T.; Matthies, C.; et al. Prophylactic nimodipine treatment and improvement in hearing outcome after vestibular schwannoma surgery: A combined analysis of a randomized, multicenter, Phase III trial and its pilot study. J. Neurosurg. 2017, 127, 1376–1383. [Google Scholar] [CrossRef]
- Horn, J.; de Haan, R.J.; Vermeulen, M.; Limburg, M. Very Early Nimodipine Use in Stroke (VENUS): A randomized, double-blind, placebo-controlled trial. Stroke 2001, 32, 461–465. [Google Scholar] [CrossRef]
- Barker, F.G.; Ogilvy, C.S. Efficacy of prophylactic nimodipine for delayed ischemic deficit after subarachnoid hemorrhage: A metaanalysis. J. Neurosurg. 1996, 84, 405–414. [Google Scholar] [CrossRef]
- Pickard, J.D.; Murray, G.D.; Illingworth, R.; Shaw, M.D.; Teasdale, G.M.; Foy, P.M.; Humphrey, P.R.; Lang, D.A.; Nelson, R.; Richards, P. Effect of oral nimodipine on cerebral infarction and outcome after subarachnoid haemorrhage: British aneurysm nimodipine trial. BMJ 1989, 298, 636–642. [Google Scholar]
- Mattsson, P.; Aldskogius, H.; Svensson, M. Nimodipine-induced improved survival rate of facial motor neurons following intracranial transection of the facial nerve in the adult rat. J. Neurosurg. 1999, 90, 760–765. [Google Scholar] [CrossRef]
- Mattsson, P.; Janson, A.M.; Aldskogius, H.; Svensson, M. Nimodipine promotes regeneration and functional recovery after intracranial facial nerve crush. J. Comp. Neurol. 2001, 437, 106–117. [Google Scholar]
- Zhang, Q.; Li, Y.; Bao, Y.; Yin, C.; Xin, X.; Guo, Y.; Gao, F.; Huo, S.; Wang, X.; Wang, Q. Pretreatment with nimodipine reduces incidence of POCD by decreasing calcineurin mediated hippocampal neuroapoptosis in aged rats. BMC Anesthesiol. 2018, 18, 42. [Google Scholar] [CrossRef]
- Kurita, M.; Okazaki, M.; Ozaki, M.; Miyamoto, S.; Takushima, A.; Harii, K. Thermal effect of illumination on microsurgical transfer of free flaps: Experimental study and clinical implications. Scand. J. Plast. Reconstr. Surg. Hand Surg. 2008, 42, 58–66. [Google Scholar] [CrossRef]
- Comporti, M.; Signorini, C.; Leoncini, S.; Gardi, C.; Ciccoli, L.; Giardini, A.; Vecchio, D.; Arezzini, B. Ethanol-induced oxidative stress: Basic knowledge. Genes Nutr. 2010, 5, 101–109. [Google Scholar] [CrossRef]
- Schilling, T.; Eder, C. Stimulus-dependent requirement of ion channels for microglial NADPH oxidase-mediated production of reactive oxygen species. J. Neuroimmunol. 2010, 225, 190–194. [Google Scholar] [CrossRef]
- Clark, L.F.; Kodadek, T. The Immune System and Neuroinflammation as Potential Sources of Blood-Based Biomarkers for Alzheimer’s Disease, Parkinson’s Disease, and Huntington’s Disease. ACS Chem. Neurosci. 2016, 7, 520–527. [Google Scholar] [CrossRef]
- Bansal, L.R.; Zinkus, T. Osmotic Demyelination Syndrome in Children. Pediatr. Neurol. 2019, 97, 12–17. [Google Scholar] [CrossRef]
- Gankam-Kengne, F.; Couturier, B.S.; Soupart, A.; Brion, J.P.; Decaux, G. Osmotic Stress-Induced Defective Glial Proteostasis Contributes to Brain Demyelination after Hyponatremia Treatment. J. Am. Soc. Nephrol. 2017, 28, 1802–1813. [Google Scholar] [CrossRef]
- Schampel, A.; Volovitch, O.; Koeniger, T.; Scholz, C.J.; Jörg, S.; Linker, R.A.; Wischmeyer, E.; Wunsch, M.; Hell, J.W.; Ergün, S.; et al. Nimodipine fosters remyelination in a mouse model of multiple sclerosis and induces microglia-specific apoptosis. Proc. Natl. Acad. Sci. USA 2017, 114, E3295–E3304. [Google Scholar] [CrossRef] [Green Version]
- Ingwersen, J.; De Santi, L.; Wingerath, B.; Graf, J.; Koop, B.; Schneider, R.; Hecker, C.; Schröter, F.; Bayer, M.; Engelke, A.D.; et al. Nimodipine confers clinical improvement in two models of experimental autoimmune encephalomyelitis. J. Neurochem. 2018. [Google Scholar] [CrossRef]
- Sanz, J.M.; Chiozzi, P.; Colaianna, M.; Zotti, M.; Ferrari, D.; Trabace, L.; Zuliani, G.; Di Virgilio, F. Nimodipine inhibits IL-1β release stimulated by amyloid β from microglia. Br. J. Pharmacol. 2012, 167, 1702–1711. [Google Scholar] [CrossRef]
- Singh, A.; Verma, P.; Balaji, G.; Samantaray, S.; Mohanakumar, K.P. Nimodipine, an L-type calcium channel blocker attenuates mitochondrial dysfunctions to protect against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced Parkinsonism in mice. Neurochem. Int. 2016, 99, 221–232. [Google Scholar] [CrossRef]
- Ji, B.; Wang, M.; Gao, D.; Xing, S.; Li, L.; Liu, L.; Zhao, M.; Qi, X.; Dai, K. Combining nanoscale magnetic nimodipine liposomes with magnetic resonance image for Parkinson’s disease targeting therapy. Nanomedicine (Lond.) 2017, 12, 237–253. [Google Scholar] [CrossRef]
- Watters, D.; Waterhouse, N. Proteolytic targets in cell death. Results Probl. Cell. Differ. 1998, 24, 25–44. [Google Scholar]
- Kothakota, S.; Azuma, T.; Reinhard, C.; Klippel, A.; Tang, J.; Chu, K.; McGarry, T.J.; Kirschner, M.W.; Koths, K.; Kwiatkowski, D.J.; et al. Caspase-3-generated fragment of gelsolin: Effector of morphological change in apoptosis. Science 1997, 278, 294–298. [Google Scholar] [CrossRef]
- Jänicke, R.U.; Ng, P.; Sprengart, M.L.; Porter, A.G. Caspase-3 is required for alpha-fodrin cleavage but dispensable for cleavage of other death substrates in apoptosis. J. Biol. Chem. 1998, 273, 15540–15545. [Google Scholar]
- Jänicke, R.U.; Sprengart, M.L.; Wati, M.R.; Porter, A.G. Caspase-3 is required for DNA fragmentation and morphological changes associated with apoptosis. J. Biol. Chem. 1998, 273, 9357–9360. [Google Scholar]
- Stennicke, H.R.; Salvesen, G.S. Caspases—Controlling intracellular signals by protease zymogen activation. Biochim. Biophys. Acta 2000, 1477, 299–306. [Google Scholar] [CrossRef]
- Dudek, H.; Datta, S.R.; Franke, T.F.; Birnbaum, M.J.; Yao, R.; Cooper, G.M.; Segal, R.A.; Kaplan, D.R.; Greenberg, M.E. Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science 1997, 275, 661–665. [Google Scholar] [CrossRef]
- Franke, T.F.; Kaplan, D.R.; Cantley, L.C. PI3K: Downstream AKTion blocks apoptosis. Cell 1997, 88, 435–437. [Google Scholar] [CrossRef]
- Kulik, G.; Klippel, A.; Weber, M.J. Antiapoptotic signalling by the insulin-like growth factor I receptor, phosphatidylinositol 3-kinase, and Akt. Mol. Cell. Biol. 1997, 17, 1595–1606. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Hoell, P.; Ahlemeyer, B.; Sure, U.; Bertalanffy, H.; Krieglstein, J. Implication of PTEN in production of reactive oxygen species and neuronal death in in vitro models of stroke and Parkinson’s disease. Neurochem. Int. 2007, 50, 507–516. [Google Scholar] [CrossRef]
- Cheng, B.; Martinez, A.A.; Morado, J.; Scofield, V.; Roberts, J.L.; Maffi, S.K. Retinoic acid protects against proteasome inhibition associated cell death in SH-SY5Y cells via the AKT pathway. Neurochem. Int. 2013, 62, 31–42. [Google Scholar] [CrossRef]
- Ermak, G.; Hench, K.J.; Chang, K.T.; Sachdev, S.; Davies, K.J. Regulator of calcineurin (RCAN1-1L) is deficient in Huntington disease and protective against mutant huntingtin toxicity in vitro. J. Biol. Chem. 2009, 284, 11845–11853. [Google Scholar] [CrossRef]
- Walker, C.L.; Xu, X.M. PTEN inhibitor bisperoxovanadium protects oligodendrocytes and myelin and prevents neuronal atrophy in adult rats following cervical hemicontusive spinal cord injury. Neurosci. Lett. 2014, 573, 64–68. [Google Scholar] [CrossRef] [Green Version]
- Du, K.; Montminy, M. CREB is a regulatory target for the protein kinase Akt/PKB. J. Biol. Chem. 1998, 273, 32377–32379. [Google Scholar] [CrossRef]
- Steven, A.; Seliger, B. Control of CREB expression in tumors: From molecular mechanisms and signal transduction pathways to therapeutic target. Oncotarget 2016, 7, 35454–35465. [Google Scholar] [CrossRef]
- Pregi, N.; Belluscio, L.M.; Berardino, B.G.; Castillo, D.S.; Cánepa, E.T. Oxidative stress-induced CREB upregulation promotes DNA damage repair prior to neuronal cell death protection. Mol. Cell. Biochem. 2017, 425, 9–24. [Google Scholar] [CrossRef]
- Walton, M.R.; Dragunow, I. Is CREB a key to neuronal survival? Trends Neurosci. 2000, 23, 48–53. [Google Scholar] [CrossRef]
- Noriega, L.G.; Feige, J.N.; Canto, C.; Yamamoto, H.; Yu, J.; Herman, M.A.; Mataki, C.; Kahn, B.B.; Auwerx, J. CREB and ChREBP oppositely regulate SIRT1 expression in response to energy availability. EMBO Rep. 2011, 12, 1069–1076. [Google Scholar] [CrossRef] [Green Version]
- Martins, I.J. Heat Shock Gene Inactivation and Protein Aggregation with Links to Chronic Diseases. Diseases 2018, 6, 39. [Google Scholar] [CrossRef]
- Pillai, V.B.; Sundaresan, N.R.; Gupta, M.P. Regulation of Akt signaling by sirtuins: Its implication in cardiac hypertrophy and aging. Circ. Res. 2014, 114, 368–378. [Google Scholar] [CrossRef]
- Towart, R.; Kazda, S. The cellular mechanism of action of nimodipine (BAY e 9736), a new calcium antagonist [proceedings]. Br. J. Pharmacol. 1979, 67, 409P–410P. [Google Scholar]
- Meier, K.; Knepel, W.; Schöfl, C. Potassium depolarization elevates cytosolic free calcium concentration in rat anterior pituitary cells through 1,4-dihydropyridine-sensitive, omega-conotoxin-insensitive calcium channels. Endocrinology 1988, 122, 2764–2770. [Google Scholar] [CrossRef]
- Lecht, S.; Rotfeld, E.; Arien-Zakay, H.; Tabakman, R.; Matzner, H.; Yaka, R.; Lelkes, P.I.; Lazarovici, P. Neuroprotective effects of nimodipine and nifedipine in the NGF-differentiated PC12 cells exposed to oxygen-glucose deprivation or trophic withdrawal. Int. J. Dev. Neurosci. 2012, 30, 465–469. [Google Scholar] [CrossRef]
- Recktenwald, C.V.; Leisz, S.; Steven, A.; Mimura, K.; Müller, A.; Wulfänger, J.; Kiessling, R.; Seliger, B. HER-2/neu-mediated down-regulation of biglycan associated with altered growth properties. J. Biol. Chem. 2012, 287, 24320–24329. [Google Scholar] [CrossRef]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leisz, S.; Simmermacher, S.; Prell, J.; Strauss, C.; Scheller, C. Nimodipine-Dependent Protection of Schwann Cells, Astrocytes and Neuronal Cells from Osmotic, Oxidative and Heat Stress Is Associated with the Activation of AKT and CREB. Int. J. Mol. Sci. 2019, 20, 4578. https://doi.org/10.3390/ijms20184578
Leisz S, Simmermacher S, Prell J, Strauss C, Scheller C. Nimodipine-Dependent Protection of Schwann Cells, Astrocytes and Neuronal Cells from Osmotic, Oxidative and Heat Stress Is Associated with the Activation of AKT and CREB. International Journal of Molecular Sciences. 2019; 20(18):4578. https://doi.org/10.3390/ijms20184578
Chicago/Turabian StyleLeisz, Sandra, Sebastian Simmermacher, Julian Prell, Christian Strauss, and Christian Scheller. 2019. "Nimodipine-Dependent Protection of Schwann Cells, Astrocytes and Neuronal Cells from Osmotic, Oxidative and Heat Stress Is Associated with the Activation of AKT and CREB" International Journal of Molecular Sciences 20, no. 18: 4578. https://doi.org/10.3390/ijms20184578
APA StyleLeisz, S., Simmermacher, S., Prell, J., Strauss, C., & Scheller, C. (2019). Nimodipine-Dependent Protection of Schwann Cells, Astrocytes and Neuronal Cells from Osmotic, Oxidative and Heat Stress Is Associated with the Activation of AKT and CREB. International Journal of Molecular Sciences, 20(18), 4578. https://doi.org/10.3390/ijms20184578