Detailed Clinical Features of Deafness Caused by a Claudin-14 Variant



Abstract
:1. Introduction
1.1. Hearing Loss
1.2. Tight Junctions in the Inner Ear
1.3. CLDN14 Variants in Deafness
2. Results
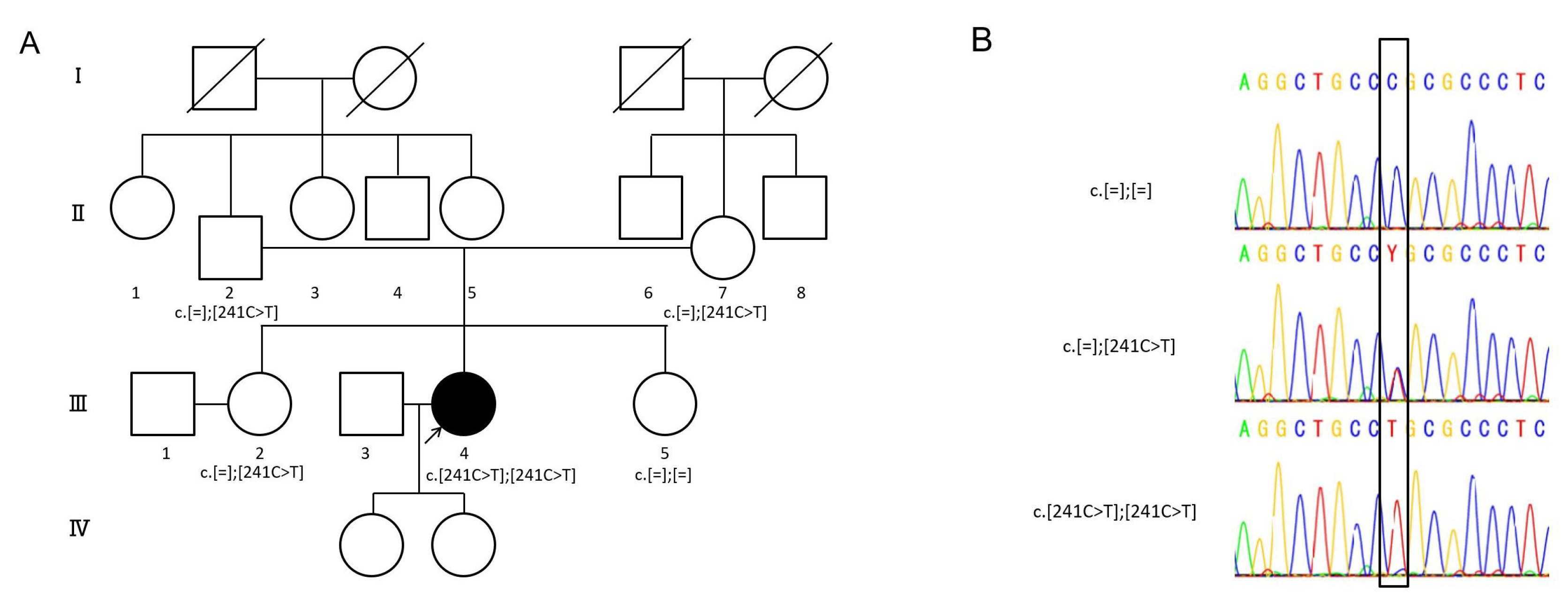
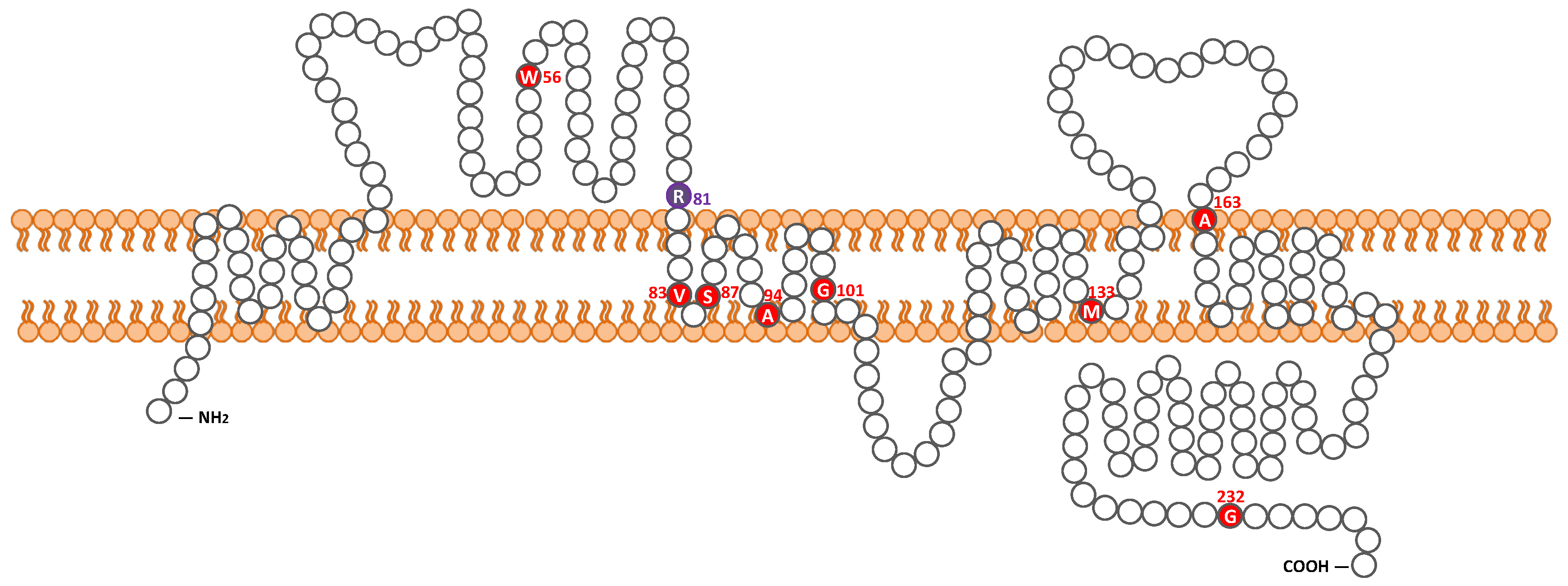
2.1. Detected Variant
2.2. Clinical Findings
3. Discussion
3.1. Frequency of CLDN14-Associated HL in the Japanese Population
3.2. Clinical Characteristics
3.2.1. Progression and Onset of DFNB29
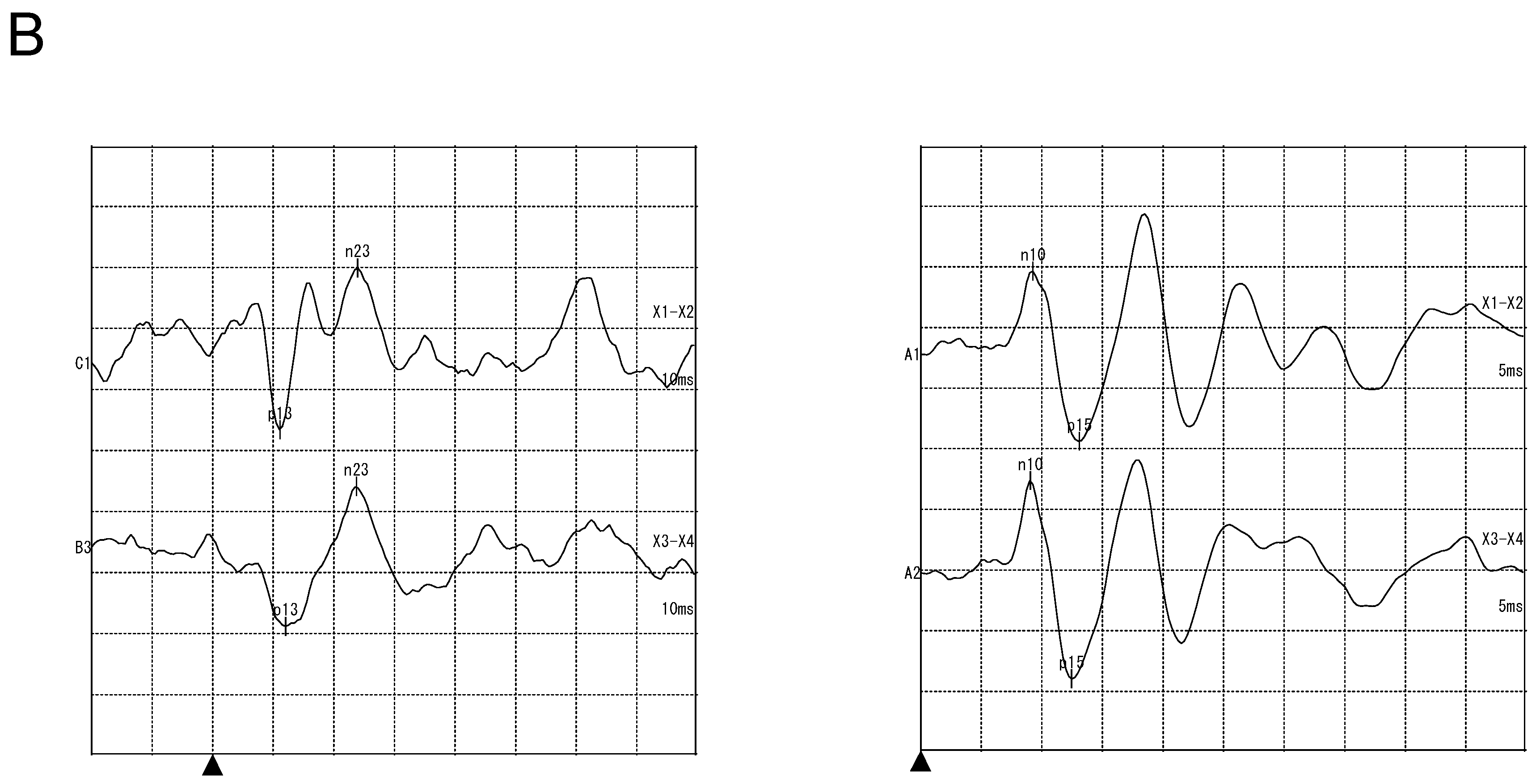
3.2.2. Vestibular Examination
3.2.3. Outcome of Cochlear Implantation
4. Materials and Methods
4.1. Subjects
4.2. Variant Analysis
4.3. Clinical Evaluations
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Data Availability
Abbreviations
| HL | Hearing loss |
| ARNSHL | Autosomal recessive nonsyndromic hereditary hearing loss |
| EP | Endocochlear potential |
| TJ | Tight junction |
| CI | Cochlear implantation |
| MPS | Massively parallel DNA sequencing |
| PTA | Pure-tone average |
| cVEMPs | Cervical vestibular evoked myogenic potentials |
| oVEMPs | Ocular vestibular evoked myogenic potentials |
| SNHL | Sensorineural hearing loss |
| dBSPL | Decibel sound pressure level |
| dBHL | Decibel hearing level |
References
- Morton, C.C.; Nance, W.E. Newborn Hearing Screening—A Silent Revolution. N. Engl. J. Med. 2006, 354, 2151–2164. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.J.H.; Bale, J.F.; White, K.R. Sensorineural hearing loss in children. Lancet 2005, 365, 879–890. [Google Scholar] [CrossRef]
- Hilgert, N.; Smith, R.J.H.; Van Camp, G. Forty-six genes causing nonsyndromic hearing impairment: Which ones should be analyzed in DNA diagnostics? Mutat. Res. 2009, 681, 189–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hereditary Hearing Loss Homepage. Available online: https://hereditaryhearingloss.org/ (accessed on 13 May 2019).
- Richardson, G.P.; de Monvel, J.B.; Petit, C. How the Genetics of Deafness Illuminates Auditory Physiology. Annu. Rev. Physiol. 2011, 73, 311–334. [Google Scholar] [CrossRef] [PubMed]
- Wangemann, P.; Schacht, J. Homeostatic Mechanisms in the Cochlea. In The Cochlea; Dallos, P., Popper, A.N., Fay, R.R., Eds.; Springer: New York, NY, USA, 1996; Volume 8, pp. 130–185. [Google Scholar]
- Ferrary, E.; Sterkers, O. Mechanisms of endolymph secretion. Kidney Int. 1998, 65, S98–S103. [Google Scholar]
- Sterkers, O.; Ferrary, E.; Amiel, C. Production of inner ear fluids. Phys. Rev. 1988, 68, 1083–1128. [Google Scholar] [CrossRef] [PubMed]
- Tasaki, I.; Spyroupoulos, C.S. Stria vascularis as source of endocochlear potential. J. Neurophysiol. 1959, 22, 149–155. [Google Scholar] [CrossRef]
- Konishi, T.; Hamrick, P.E.; Walsh, P.J. Ion transport in guinea pig cochlea. I. Potassium and sodium transport. Acta Otolaryngol. 1978, 86, 22–34. [Google Scholar] [CrossRef]
- Hudspeth, A.J. How the ear’s works work. Nature 1989, 341, 397–404. [Google Scholar] [CrossRef]
- Nunes, F.D.; Lopez, L.N.; Lin, H.W.; Davies, C.; Azevedo, R.B.; Gow, A.; Kachar, B. Distinctsubdomain organization and molecular composition of a tight junction with adherens junction features. J. Cell Sci. 2006, 119, 4819–4827. [Google Scholar] [CrossRef]
- Wilcox, E.R.; Burton, Q.L.; Naz, S.; Riazuddin, S.; Smith, T.N.; Ploplis, B.; Belyantseva, I.; Ben-Yosef, T.; Liburd, N.A.; Morell, R.J.; et al. Mutations in the gene encoding tight junction claudin-14 cause autosomal recessive deafness DFNB29. Cell 2001, 104, 165–172. [Google Scholar] [CrossRef]
- Riazuddin, S.; Ahmed, Z.M.; Fanning, A.S.; Lagziel, A.; Kitajiri, S.; Ramzan, K.; Khan, S.N.; Chattaraj, P.; Friedman, P.L.; Anderson, J.M.; et al. Tricellulin Is a Tight-Junction Protein Necessary for Hearing. Am. J. Hum. Genet. 2006, 79, 1040–1051. [Google Scholar] [CrossRef] [Green Version]
- Borck, G.; Rehman, A.U.; Lee, K.; Pogoda, H.; Kakar, N.; von Ameln, S.; Grillet, N.; Hildebrand, M.S.; Ansar, M.; Basit, S.; et al. Loss-of-Function Mutations of ILDR1 Cause Autosomal-Recessive Hearing Impairment DFNB42. Am. J. Hum. Genet. 2011, 88, 127–137. [Google Scholar] [CrossRef] [Green Version]
- Furuse, M.; Fujita, K.; Hiiragi, T.; Fujimoto, K.; Tsukita, S. Claudin-1 and -2: Novel integral membrane proteins localizing at tight junctions with no sequence similarity to occludin. J. Cell Biol. 1998, 141, 1539–1550. [Google Scholar] [CrossRef] [PubMed]
- Elkouby-Naor, L.; Ben-Yosef, T. Functions of claudin tight junction proteins and their complex interactions in various physiologicalsystems. Int. Rev. Cell Mol. Biol. 2010, 279, 1–32. [Google Scholar] [PubMed]
- Gunzel, D.; Fromm, M. Claudins and other tight junction proteins. Compr. Physiol. 2012, 2, 1819–1852. [Google Scholar] [PubMed]
- Kitajiri, S.-I.; Furuse, M.; Morita, K. Expression patterns of claudins, tight junction adhesion molecules, in the inner ear. Hear. Res. 2004, 187, 25–34. [Google Scholar] [CrossRef]
- Gow, A.; Davies, C.; Southwood, C.M.; Frolenkov, G.; Chrustowski, M.; Ng, L.; Yamauchi, D.; Marcus, D.C.; Kachar, B. Deafness in Claudin 11-null mice reveals the critical contribution of basal cell tight junctions to stria vascularis function. J. Neurosci. 2004, 24, 7051–7062. [Google Scholar] [CrossRef] [PubMed]
- Kitajiri, S.; Miyamoto, T.; Mineharuetal, A. Compartmentalization established by claudin-11-based tight junctions in stria vascularis is required for hearing through generation of endocochlear potential. J. Cell Sci. 2004, 117, 5087–5096. [Google Scholar] [CrossRef] [Green Version]
- Ben-Yosef, T.; Belyantseva, I.A.; Saunders, T.L.; Hughes, E.D.; Kawamoto, K.; Van Itallie, C.M.; Beyer, L.A.; Halsey, K.; Gardner, D.J.; Wilcox, E.R.; et al. Claudin 14 knockout mice, a model for autosomal recessive deafness DFNB29, are deaf due to cochlear hair cell degeneration. Hum. Mol. Genet. 2003, 12, 2049–2061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakano, Y.; Kim, S.H.; Kim, H.M.; Sanneman, J.D.; Zhang, Y.; Smith, R.J.; Marcus, D.C.; Wangemann, P.; Nessler, R.A.; Banfi, B. A claudin-9—Based ion permeability barrier is essential for hearing. PLoS Genet. 2009, 5, e1000610. [Google Scholar] [CrossRef] [PubMed]
- Matter, K.; Balda, M.S. Signalling to and from tight junctions. Nat. Rev. Mol. Cell Biol. 2003, 4, 225–237. [Google Scholar] [CrossRef]
- Schneeberger, E.E.; Lynch, R.D. The tight junction: A multifunctional complex. Am. J. Physiol. Cell Physiol. 2004, 286, C1213–C1228. [Google Scholar] [CrossRef] [PubMed]
- Wattenhofer, M.; Reymond, A.; Falciola, V.; Charollais, A.; Caille, D.; Borel, C.; Lyle, R.; Estivill, X.; Petersen, M.B.; Meda, P.; et al. Different mechanisms preclude mutant CLDN14 proteins from forming tight junctions in vitro. Hum. Mutat. 2005, 25, 543–549. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Ansar, M.; Andrade, P.B.; Khan, B.; Santos-Cortez, R.L.P.; Ahmad, W.; Leal, S.M. Novel CLDN14 mutations in Pakistani families with autosomal recessive non-syndromic hearing loss. Am. J. Med. Genet. A 2012, 158, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Bashir, Z.-E.-H.; Latief, N.; Belyantseva, I.A.; Iqbal, F.; Amer Riazuddin, S.; Khan, S.N.; Friedman, T.B.; Riazuddin, S.; Riazuddin, S. Phenotypic variability of CLDN14 mutations causing DFNB29 hearing loss in the Pakistani population HHS Public Access. J. Hum. Genet. 2013, 58143, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Pater, J.A.; Benteau, T.; Griffin, A.; Penney, C.; Stanton, S.G.; Predham, S.; Kielley, B.; Squires, J.; Zhou, J.; Li, Q.; et al. A common variant in CLDN14 causes precipitous, prelingual sensorineural hearing loss in multiple families due to founder effect. Hum. Genet. 2017, 136, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.-A.; Kim, Y.-R.; Sagong, B.; Cho, H.-J.; Bae, J.W.; Kim, J.; Lee, J.; Park, H.-J.; Choi, J.Y.; Lee, K.-Y.; et al. Genetic Analysis of Genes Related to Tight Junction Function in the Korean Population with Non-Syndromic Hearing Loss. PLoS ONE 2014, 9, e95646. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Yao, J.; Wei, Q.; Xu, J.; Xing, G.; Cao, X. Genetic analysis of CLDN14 in the Chinese population affected with non-syndromic hearing loss. Int. J. Pediatr. Otorhinolaryngol. 2018, 105, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Roche, J.P.; City, I.; Hansen, M.R. On the Horizon: Cochlear Implant Technology. Otolaryngol. Clin. North Am. 2016, 48, 1097–1116. [Google Scholar] [CrossRef] [PubMed]
- Wilson, B.S. Getting a decent (but sparse) signal to the brain for users of cochlear implants. Hear. Res. 2014, 322, 24–38. [Google Scholar] [CrossRef] [PubMed]
- Deep, N.L.; Dowling, E.M.; Jethanamest, D.; Carlson, M.L. Cochlear Implantation: An Overview. J. Neurol. Surg. Part B 2019, 80, 169–177. [Google Scholar] [CrossRef]
- Nishio, S.; Takumi, Y.; Usami, S. Laser-capture micro dissection combined with next-generation sequencing analysis of cell type-specific deafness gene expression in the mouse cochlea. Hear. Res. 2017, 348, 87–97. [Google Scholar] [CrossRef]
- Nishio, S.Y.; Usami, S.I. Deafness Gene Variations in a 1120 Nonsyndromic Hearing Loss Cohort: Molecular Epidemiology and Deafness Mutation Spectrum of Patients in Japan. Ann. Otol. Rhinol. Laryngol. 2015, 124, 49S–60S. [Google Scholar] [CrossRef] [PubMed]
- Kitano, T.; Miyagawa, M.; Nishio, S.-Y.; Moteki, H.; Oda, K.; Ohyama, K.; Miyazaki, H.; Hidaka, H.; Nakamura, K.-I.; Murata, T.; et al. POU4F3 mutation screening in Japanese hearing loss patients: Massively parallel DNA sequencing-based analysis identified novel variants associated with autosomal dominant hearing loss. PLoS ONE 2017, 12, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Miyagawa, M.; Nishio, S.Y.; Ikeda, T.; Fukushima, K.; Usami, S.I. Massively Parallel DNA Sequencing Successfully Identifies New Causative Mutations in Deafness Genes in Patients with Cochlear Implantation and EAS. PLoS ONE 2013, 8, e75793. [Google Scholar] [CrossRef] [PubMed]
- Nishio, S.Y.; Moteki, H.; Usami, S.I. Simple and efficient germline copy number variant visualization method for the Ion AmpliSeqTM custom panel. Mol. Genet. Genomic Med. 2018, 6, 678–686. [Google Scholar] [CrossRef] [PubMed]
- Chang, X.; Wang, K. wANNOVAR: Annotating genetic variants for personal genomes via the web. J. Med. Genet. 2012, 49, 433–436. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Abboud, H.E.; Abecasis, G.; Aguilar-Salinas, C.A.; Arellano-Campos, O.; Atzmon, G.; Aukrust, I.; Barr, C.L.; Bell, G.I.; Bergen, S.; Bjørkhaug, L.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [Green Version]
- The Exome Aggregation Consortium Database (ExAC). Available online: http://exac.broadinstitute.org/ (accessed on 13 May 2019).
- The Genome Aggregation Database (gnomAD). Available online: https://gnomad.broadinstitute.org/ (accessed on 13 May 2019).
- Integrative Japanese Genome Variation Database (3.5KJPN). Available online: https://ijgvd.megabank.tohoku.ac.jp/statistics/statistics-3.5kjpn-all (accessed on 13 May 2019).
- Narahara, M.; Higasa, K.; Nakamura, S.; Tabara, Y.; Kawaguchi, T.; Ishii, M.; Matsubara, K.; Matsuda, F.; Yamada, R. Large-scale East-Asian eQTL mapping reveals novel candidate genes for LD mapping and the genomic landscape of transcriptional effects of sequence variants. PLoS ONE 2014, 9, e100924. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009, 4, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chun, S.; Fay, J.C. Identification of deleterious mutations within three human genomes. Genome Res. 2009, 19, 1553–1561. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, J.M.; Rödelsperger, C.; Schuelke, M.; Seelow, D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods 2010, 7, 575–576. [Google Scholar] [CrossRef]
- Reva, B.; Antipin, Y.; Sander, C. Predicting the functional impact of protein mutations: Application to cancer genomics. Nucleic Acids Res. 2011, 39, 37–43. [Google Scholar] [CrossRef]
- Ioannidis, N.M.; Rothstein, J.H.; Pejaver, V.; Middha, S.; McDonnell, S.K.; Baheti, S.; Musolf, A.; Li, Q.; Holzinger, E.; Karyadi, D.; et al. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am. J. Hum. Genet. 2016, 99, 877–885. [Google Scholar] [CrossRef]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef] [Green Version]
- The Human Gene Mutation Database Professional (HGMD). Available online: http://www.hgmd.cf.ac.uk/ (accessed on 13 May 2019).
- Mazzoli, M.; Van Camp, G.; Newton, V.; Giarbini, N.; Declau, F.; Parving, A. Recommendations for the Description of Genetic and Audiological Data for Families with Nonsyndromic Hereditary Hearing Impairment. Audiol. Med. 2003, 1, 148–150. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Prediction Score | Allele Frequency in Controls | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Nucleotide Change | Amino Acid Change | SIFT * | PolyPhen2_HVAR * | LRT * | Mut_Taster * | Mut_Assessor * | REVEL * | CADD | Evolutional Conservation ** | ExAC | gnomAD | 3.5kJPN |
| c.241C > T | p.R81C | 0.78 | 0.88 | 0.84 | 0.81 | 0.98 | 0.89 | 27.6 | Yes (100%) | 0.000042 | 0.000037 | 0 |
| HL | Pure-Tone Audiometry | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Nucleotide Change | Genotype | Amino Acid Change | Onset | Progression | Severity of HL | Audiometric Configuration | Consanguineous Marriage | Vestibuler Function | Hearing Intervention | Family Origin | Reference |
| c.167G > A | homozygote | p.Trp56 * | NA | No | severe to profound | gently sloping | Yes | NA | NA | Pakistan | Lee et al. 2011 [27] |
| c.241C > T | homozygote | p.Arg81Cys | 9 y.o. | Yes | Severe | steeply sloping | No | Normal | CI | Japan | This study |
| c.242G > A | homozygote | p.Arg81His | NA | No | severe to profound | gently sloping | Yes | NA | NA | Pakistan | Lee et al. 2011 [27] |
| c.254T > A | homozygote | p.Val85Asp | prelingual | No | moderate to profound | HF | Yes | NA | NA | Pakistan | Wilcox et al. 2001 [13], Bashir et al. 2013 [28] |
| c.259_260TC > AT | homozygote | p.Ser87Ile | prelingual | No | severe to profound | flat | Yes | NA | NA | Pakistan | Bashir et al. 2013 [28] |
| c.281C > T | homozygote | p.Ala94Val | NA | No | moderate to profound | flat, DE | Yes | NA | NA | Pakistan | Bashir et al. 2013 [28] |
| c.301G > A | homozygote | p.Gly101Arg | NA | NA | NA | NA | NA | NA | NA | Greece | Wattenhofer et al. 2005 [26] |
| c.398delT | homozygote | p.Met133Argfs *24 | congenital | NA | moderate to profound | gently sloping | Yes | NA | NA | Pakistan | Wilcox et al. 2001 [13], Bashir et al. 2013 [28] |
| c.488C > T | homozygote | p.Ala163Val | 5–7 y.o. | Yes | mild to severe | steeply sloping | Yes | Normal | HA | Canada | Pater et al. 2016 [29] |
| c.694G > A | homozygote | p.Gly232Arg | NA | No | severe to profound | gently sloping | Yes | NA | NA | Pakistan | Lee et al. 2011 [27] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kitano, T.; Kitajiri, S.-i.; Nishio, S.-y.; Usami, S.-i. Detailed Clinical Features of Deafness Caused by a Claudin-14 Variant. Int. J. Mol. Sci. 2019, 20, 4579. https://doi.org/10.3390/ijms20184579
Kitano T, Kitajiri S-i, Nishio S-y, Usami S-i. Detailed Clinical Features of Deafness Caused by a Claudin-14 Variant. International Journal of Molecular Sciences. 2019; 20(18):4579. https://doi.org/10.3390/ijms20184579
Chicago/Turabian StyleKitano, Tomohiro, Shin-ichiro Kitajiri, Shin-ya Nishio, and Shin-ichi Usami. 2019. "Detailed Clinical Features of Deafness Caused by a Claudin-14 Variant" International Journal of Molecular Sciences 20, no. 18: 4579. https://doi.org/10.3390/ijms20184579
APA StyleKitano, T., Kitajiri, S.-i., Nishio, S.-y., & Usami, S.-i. (2019). Detailed Clinical Features of Deafness Caused by a Claudin-14 Variant. International Journal of Molecular Sciences, 20(18), 4579. https://doi.org/10.3390/ijms20184579