Protective Effects and Mechanisms of Vaccarin on Vascular Endothelial Dysfunction in Diabetic Angiopathy


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents and Chemicals
2.2. Cell Culture and Treatments
2.3. NO Production
2.4. Transfection of miRNA.
2.5. Real-Time PCR
2.6. Western Blotting
2.7. Animal Models and Treatments
2.8. Insulin Tolerance Test (ITT) and Glucose Tolerance Test (GTT)
2.9. In Vitro Vasorelaxation Assay
2.10. Statistical Analysis
3. Results
3.1. In Vivo Verification of VAC Effects on Vascular Endothelial Dysfunction in T2DM Mice
3.2. VAC Promotes eNOS Phosphorylation in HG-Exposed HMEC-1 Cells
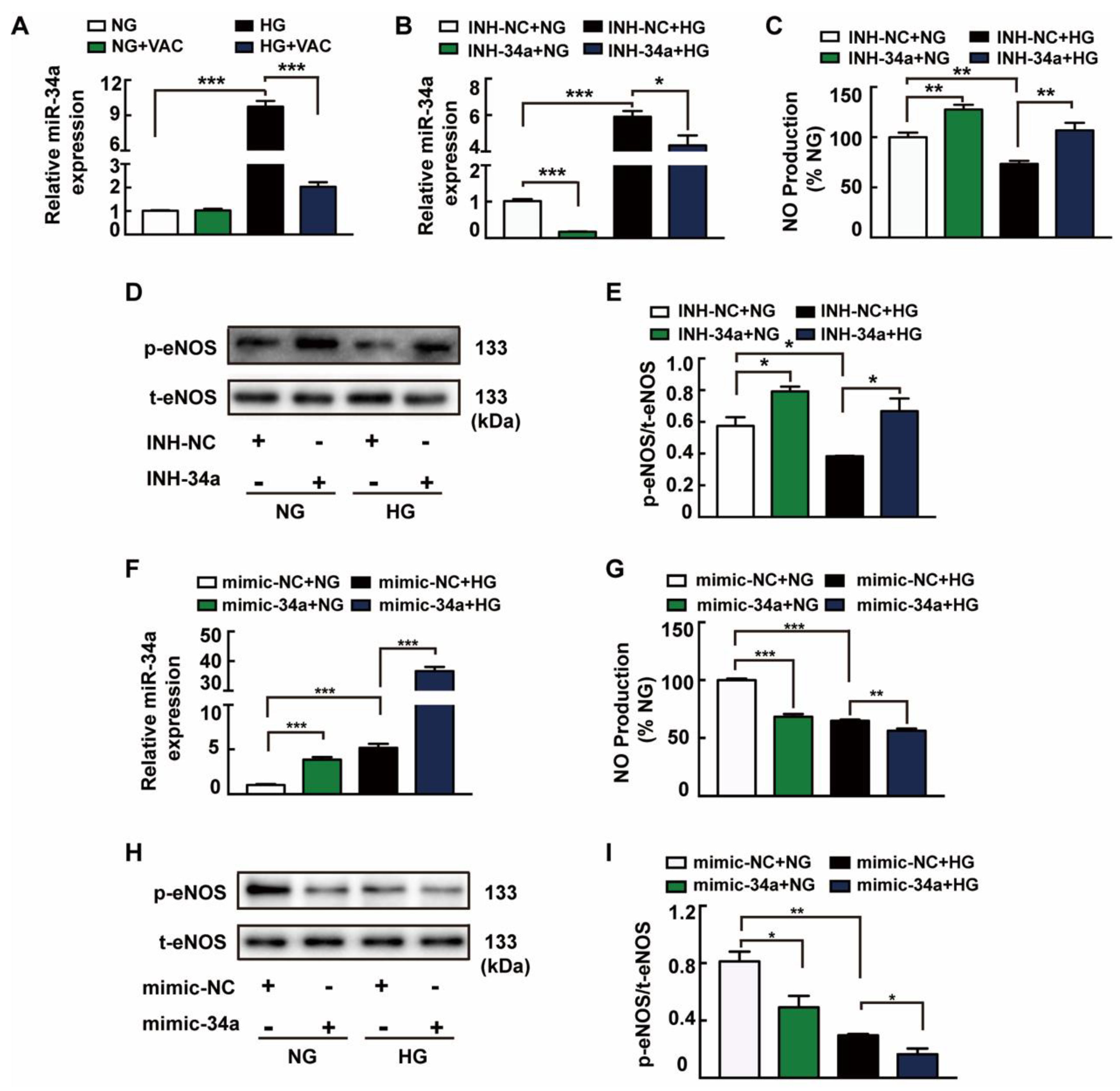
3.3. The Role of miRNA-34a in VAC-Promoting NO Production
3.4. VAC Stimulated AMPK to Improve NO Production and Attenuate miRNA-34a Expression
3.5. ROS Were Responsible for HG-Induced Endothelial Dysfunction
3.6. VAC Repressed Endothelial Dysfunction and Suppressed ROS/AMPK/miRNA-34a/eNOS Signaling Cascade In Vivo
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Kannel, W.B.; McGee, D.L. Diabetes and cardiovascular disease. The Framingham study. JAMA 1979, 241, 2035–2038. [Google Scholar] [CrossRef] [PubMed]
- Rahman, S.; Ismail, A.A.; Rahman, A.R. Treatment of diabetic vasculopathy with rosiglitazone and ramipril: Hype or hope? Int. J. Diabetes Dev. Ctries 2009, 29, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Dhananjayan, R.; Koundinya, K.S.; Malati, T.; Kutala, V.K. Endothelial Dysfunction in Type 2 Diabetes Mellitus. Indian J. Clin. Biochem. 2016, 31, 372–379. [Google Scholar] [CrossRef] [PubMed]
- Vanhoutte, P.M.; Shimokawa, H.; Feletou, M.; Tang, E.H. Endothelial dysfunction and vascular disease—A 30th anniversary update. Acta Physiol. 2017, 219, 22–96. [Google Scholar] [CrossRef] [PubMed]
- Cooke, J.P. The endothelium: A new target for therapy. Vasc. Med. 2000, 5, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Chrysohoou, C.; Kollia, N.; Tousoulis, D. The link between depression and atherosclerosis through the pathways of inflammation and endothelium dysfunction. Maturitas 2018, 109, 5. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, K.; Bessho, N.; Hasegawa, M.; Narimatsu, H.; Matsumoto, T.; Kobayashi, T. Co-treatment with clonidine and a GRK2 inhibitor prevented rebound hypertension and endothelial dysfunction after withdrawal in diabetes. Hypertens Res. 2018, 41, 263–274. [Google Scholar] [CrossRef]
- Yang, G.; Lucas, R.; Caldwell, R.; Yao, L.; Romero, M.J.; Caldwell, R.W. Novel mechanisms of endothelial dysfunction in diabetes. J. Cardiovasc. Dis. Res. 2010, 1, 59–63. [Google Scholar] [CrossRef] [Green Version]
- Vecoli, C.; Novelli, M.; Pippa, A.; Giacopelli, D.; Beffy, P.; Masiello, P.; L’Abbate, A.; Neglia, D. Partial deletion of eNOS gene causes hyperinsulinemic state, unbalance of cardiac insulin signaling pathways and coronary dysfunction independently of high fat diet. PLoS ONE 2014, 9, e104156. [Google Scholar] [CrossRef]
- Blazevic, T.; Schwaiberger, A.V.; Schreiner, C.E.; Schachner, D.; Schaible, A.M.; Grojer, C.S.; Atanasov, A.G.; Werz, O.; Dirsch, V.M.; Heiss, E.H. 12/15-lipoxygenase contributes to platelet-derived growth factor-induced activation of signal transducer and activator of transcription 3. J. Biol. Chem. 2013, 288, 35592–35603. [Google Scholar] [CrossRef]
- Clempus, R.E.; Griendling, K.K. Reactive oxygen species signaling in vascular smooth muscle cells. Cardiovasc. Res. 2006, 71, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Xie, F.; Feng, L.; Cai, W.; Qiu, Y.; Liu, Y.; Li, Y.; Du, B.; Qiu, L. Vaccarin promotes endothelial cell proliferation in association with neovascularization in vitro and in vivo. Mol. Med. Rep. 2015, 12, 1131–1136. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.J.; Cai, W.W.; Gong, L.L.; Wang, X.; Zhu, X.X.; Wan, M.Y.; Wang, P.Y.; Qiu, L.Y. FGF-2-mediated FGFR1 signaling in human microvascular endothelial cells is activated by vaccarin to promote angiogenesis. Biomed. Pharm. 2017, 95, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Du, B.; Xie, F.; Cai, W.; Liu, Y.; Li, Y.; Feng, L.; Qiu, L. Vaccarin attenuates high glucose-induced human EA*hy926 endothelial cell injury through inhibition of Notch signaling. Mol. Med. Rep. 2016, 13, 2143–2150. [Google Scholar] [CrossRef] [PubMed]
- Xie, F.; Cai, W.; Liu, Y.; Li, Y.; Du, B.; Feng, L.; Qiu, L. Vaccarin attenuates the human EA.hy926 endothelial cell oxidative stress injury through inhibition of Notch signaling. Int. J. Mol. Med. 2015, 35, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Zhou, Z.; Zhang, Q.; Cai, W.; Zhou, Y.; Sun, H.; Qiu, L. Vaccarin administration ameliorates hypertension and cardiovascular remodeling in renovascular hypertensive rats. J. Cell. Biochem. 2018, 119, 926–937. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.X.; Ou, H.; Shen, Y.H.; Wang, J.; Wang, J.; Coselli, J.; Wang, X.L. Regulation of endothelial nitric oxide synthase by small RNA. Proc. Natl. Acad. Sci. USA 2005, 102, 16967–16972. [Google Scholar] [CrossRef] [Green Version]
- Vinayagam, R.; Jayachandran, M.; Chung, S.S.M.; Xu, B. Guava leaf inhibits hepatic gluconeogenesis and increases glycogen synthesis via AMPK/ACC signaling pathways in streptozotocin-induced diabetic rats. Biomed. Pharm. 2018, 103, 1012–1017. [Google Scholar] [CrossRef]
- Guo, M.; Ding, J.; Li, J.; Wang, J.; Zhang, T.; Liu, C.; Huang, W.; Long, Y.; Gao, C.; Xu, Y. SGLT2 inhibitors and risk of stroke in patients with type 2 diabetes: A systematic review and meta-analysis. Diabetes Obes. Metab. 2018, 20, 1977–1982. [Google Scholar] [CrossRef]
- Piwowar, A.; Knapik-Kordecka, M.; Warwas, M. Oxidative stress and endothelium dysfunction in diabetes mellitus type 2. Polski merkuriusz lekarski Organ Polskiego Towarzystwa Lekarskiego 2008, 25, 120–123. [Google Scholar]
- Fancher, I.S.; Ahn, S.J.; Adamos, C.; Osborn, C.; Oh, M.J.; Fang, Y.; Reardon, C.A.; Getz, G.S.; Phillips, S.A.; Levitan, I. Hypercholesterolemia-Induced Loss of Flow-Induced Vasodilation and Lesion Formation in Apolipoprotein E-Deficient Mice Critically Depend on Inwardly Rectifying K(+) Channels. J. Am. Heart Assoc. 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Mann, G.E. Vascular NAD(P)H oxidase activation in diabetes: A double-edged sword in redox signalling. Cardiovasc. Res. 2009, 82, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Potdar, S.; Kavdia, M. NO/peroxynitrite dynamics of high glucose-exposed HUVECs: Chemiluminescent measurement and computational model. Microvasc. Res. 2009, 78, 191–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Wang, J.; Zhao, A.; Li, J. SIRT1 activation inhibits hyperglycemia-induced apoptosis by reducing oxidative stress and mitochondrial dysfunction in human endothelial cells. Mol. Med. Rep. 2017, 16, 3331–3338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schober, A.; Nazari-Jahantigh, M.; Weber, C. MicroRNA-mediated mechanisms of the cellular stress response in atherosclerosis. Nat. Rev. Cardiol. 2015, 12, 361–374. [Google Scholar] [CrossRef]
- Liu, J. Screening and Functional Study of AMPK-related MicroRNAs in Mouse Hepatocytes. Ph.D. Thesis, Tianjin Medical University, Tianjin, China, 2013. [Google Scholar]
- Arunachalam, G.; Lakshmanan, A.P.; Samuel, S.M.; Triggle, C.R.; Ding, H. Molecular Interplay between microRNA-34a and Sirtuin1 in Hyperglycemia-Mediated Impaired Angiogenesis in Endothelial Cells: Effects of Metformin. J. Pharm. Exp. Ther. 2016, 356, 314–323. [Google Scholar] [CrossRef]
- Raitoharju, E.; Lyytikainen, L.P.; Levula, M.; Oksala, N.; Mennander, A.; Tarkka, M.; Klopp, N.; Illig, T.; Kahonen, M.; Karhunen, P.J.; et al. miR-21, miR-210, miR-34a, and miR-146a/b are up-regulated in human atherosclerotic plaques in the Tampere Vascular Study. Atherosclerosis 2011, 219, 211–217. [Google Scholar] [CrossRef]
- Umezawa, S.; Higurashi, T.; Nakajima, A. AMPK: Therapeutic Target for Diabetes and Cancer Prevention. Curr. Pharm. Des. 2017, 23, 3629–3644. [Google Scholar] [CrossRef]
- Zhang, B.B.; Zhou, G.; Li, C. AMPK: An emerging drug target for diabetes and the metabolic syndrome. Cell Metab. 2009, 9, 407–416. [Google Scholar] [CrossRef]
- Ikeda, Y.; Aihara, K.; Yoshida, S.; Iwase, T.; Tajima, S.; Izawa-Ishizawa, Y.; Kihira, Y.; Ishizawa, K.; Tomita, S.; Tsuchiya, K.; et al. Heparin cofactor II, a serine protease inhibitor, promotes angiogenesis via activation of the AMP-activated protein kinase-endothelial nitric-oxide synthase signaling pathway. J. Biol. Chem. 2012, 287, 34256–34263. [Google Scholar] [CrossRef]
- Chen, M.B.; Wei, M.X.; Han, J.Y.; Wu, X.Y.; Li, C.; Wang, J.; Shen, W.; Lu, P.H. MicroRNA-451 regulates AMPK/mTORC1 signaling and fascin1 expression in HT-29 colorectal cancer. Cell. Signal. 2014, 26, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Nan, Y.; Zhen, Y.; Zhang, Y.; Guo, L.; Yu, K.; Huang, Q.; Zhong, Y. miRNA-451 inhibits glioma cell proliferation and invasion by downregulating glucose transporter 1. Tumour Biol. 2016, 37, 13751–13761. [Google Scholar] [CrossRef] [PubMed]
- Qu, J.; Lu, D.; Guo, H.; Miao, W.; Wu, G.; Zhou, M. MicroRNA-9 regulates osteoblast differentiation and angiogenesis via the AMPK signaling pathway. Mol. Cell. Biochem. 2016, 411, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.W.; Zhu, Y.R.; Zhou, X.Z.; Zhuo, B.B.; Wang, X.D. microRNA-135b expression silences Ppm1e to provoke AMPK activation and inhibit osteoblastoma cell proliferation. Oncotarget 2017, 8, 26424–26433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, X.L.; Edelstein, D.; Dimmeler, S.; Ju, Q.; Sui, C.; Brownlee, M. Hyperglycemia inhibits endothelial nitric oxide synthase activity by posttranslational modification at the Akt site. J. Clin. Investig. 2001, 108, 1341–1348. [Google Scholar] [CrossRef]
- Qin, W.; Ren, B.; Wang, S.; Liang, S.; He, B.; Shi, X.; Wang, L.; Liang, J.; Wu, F. Apigenin and naringenin ameliorate PKCbetaII-associated endothelial dysfunction via regulating ROS/caspase-3 and NO pathway in endothelial cells exposed to high glucose. Vasc. Pharm. 2016, 85, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Wu, M.; Lu, Y.; Xian, T.; Wang, Y.; Huang, B.; Zeng, G.; Huang, Q. Protective effects of 6-Gingerol on vascular endothelial cell injury induced by high glucose via activation of PI3K-AKT-eNOS pathway in human umbilical vein endothelial cells. Biomed. Pharm. 2017, 93, 788–795. [Google Scholar] [CrossRef]
- Liu, J.; Yang, J. Uncarboxylated osteocalcin inhibits high glucose-induced ROS production and stimulates osteoblastic differentiation by preventing the activation of PI3K/Akt in MC3T3-E1 cells. Int. J. Mol. Med. 2016, 37, 173–181. [Google Scholar] [CrossRef]
- Yang, L.; Wu, L.; Du, S.; Hu, Y.; Fan, Y.; Ma, J. 1,25(OH)2D3 inhibits high glucose-induced apoptosis and ROS production in human peritoneal mesothelial cells via the MAPK/P38 pathway. Mol. Med. Rep. 2016, 14, 839–844. [Google Scholar] [CrossRef] [Green Version]
- Yan, X.; Xun, M.; Li, J.; Wu, L.; Dou, X.; Zheng, J. Activation of Na+/K+-ATPase attenuates high glucose-induced H9c2 cell apoptosis via suppressing ROS accumulation and MAPKs activities by DRm217. Acta Biochim. Biophys. Sin. (Shanghai) 2016, 48, 883–893. [Google Scholar] [CrossRef] [Green Version]
- Qi, P.; Fan, M.; Li, Z.; Chen, M.; Sun, Z.; Wu, B.; Huang, C. In vivo metabolism study of vaccarin in rat using HPLC-LTQ-MSn. Biomed. Chromatogr. 2013, 27, 96–101. [Google Scholar] [CrossRef] [PubMed]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, F.; Liu, Y.; Zhu, X.; Li, S.; Shi, X.; Li, Z.; Ai, M.; Sun, J.; Hou, B.; Cai, W.; et al. Protective Effects and Mechanisms of Vaccarin on Vascular Endothelial Dysfunction in Diabetic Angiopathy. Int. J. Mol. Sci. 2019, 20, 4587. https://doi.org/10.3390/ijms20184587
Xu F, Liu Y, Zhu X, Li S, Shi X, Li Z, Ai M, Sun J, Hou B, Cai W, et al. Protective Effects and Mechanisms of Vaccarin on Vascular Endothelial Dysfunction in Diabetic Angiopathy. International Journal of Molecular Sciences. 2019; 20(18):4587. https://doi.org/10.3390/ijms20184587
Chicago/Turabian StyleXu, Fei, Yixiao Liu, Xuexue Zhu, Shuangshuang Li, Xuelin Shi, Zhongjie Li, Min Ai, Jiangnan Sun, Bao Hou, Weiwei Cai, and et al. 2019. "Protective Effects and Mechanisms of Vaccarin on Vascular Endothelial Dysfunction in Diabetic Angiopathy" International Journal of Molecular Sciences 20, no. 18: 4587. https://doi.org/10.3390/ijms20184587
APA StyleXu, F., Liu, Y., Zhu, X., Li, S., Shi, X., Li, Z., Ai, M., Sun, J., Hou, B., Cai, W., Sun, H., Ni, L., Zhou, Y., & Qiu, L. (2019). Protective Effects and Mechanisms of Vaccarin on Vascular Endothelial Dysfunction in Diabetic Angiopathy. International Journal of Molecular Sciences, 20(18), 4587. https://doi.org/10.3390/ijms20184587