Beneficial and Deleterious Effects of Female Sex Hormones, Oral Contraceptives, and Phytoestrogens by Immunomodulation on the Liver

 ,
, 
Abstract
:1. Introduction
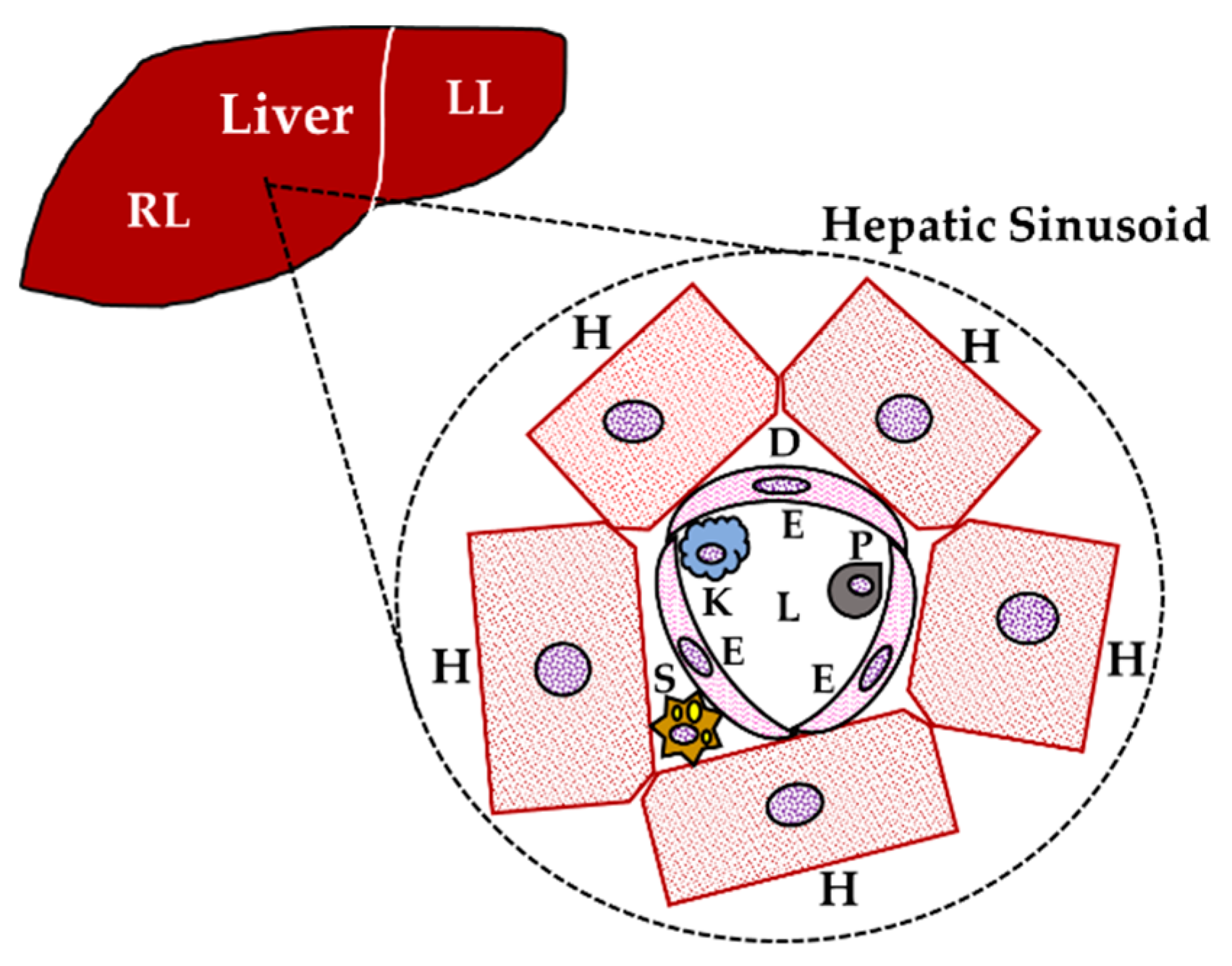
2. Liver and the Immune System: Immunomodulation
2.1. Cytokines and the Th1/Th2 Theory
2.2. Other Sets of Cytokines: Th9, Th17, and Treg cells in the Liver
2.3. Hepatic Cytokines
2.4. Cell Signaling Pathways and Effects of Hepatic IL-6
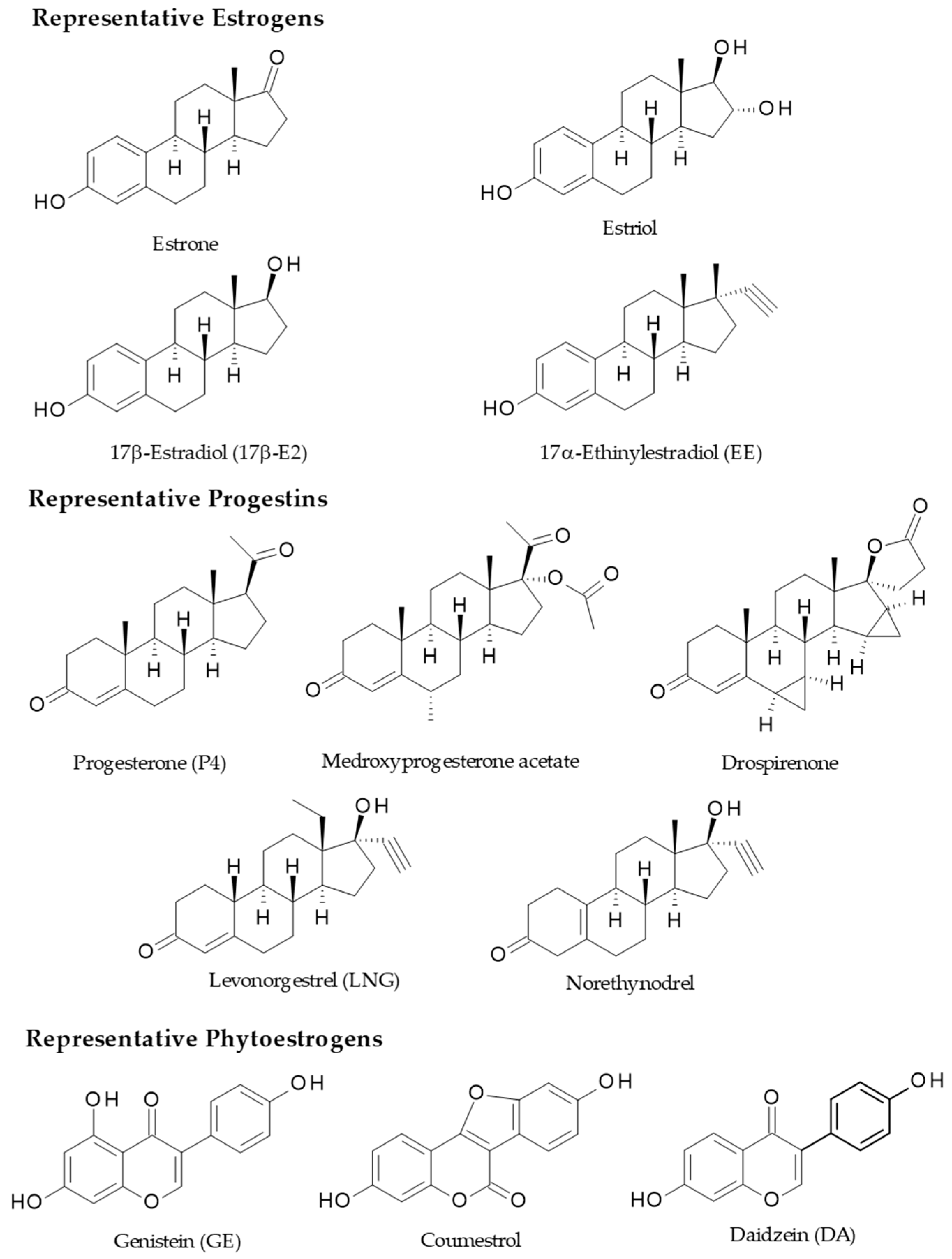
3. Female Sex Hormones, Oral Contraceptives, and Phytoestrogens
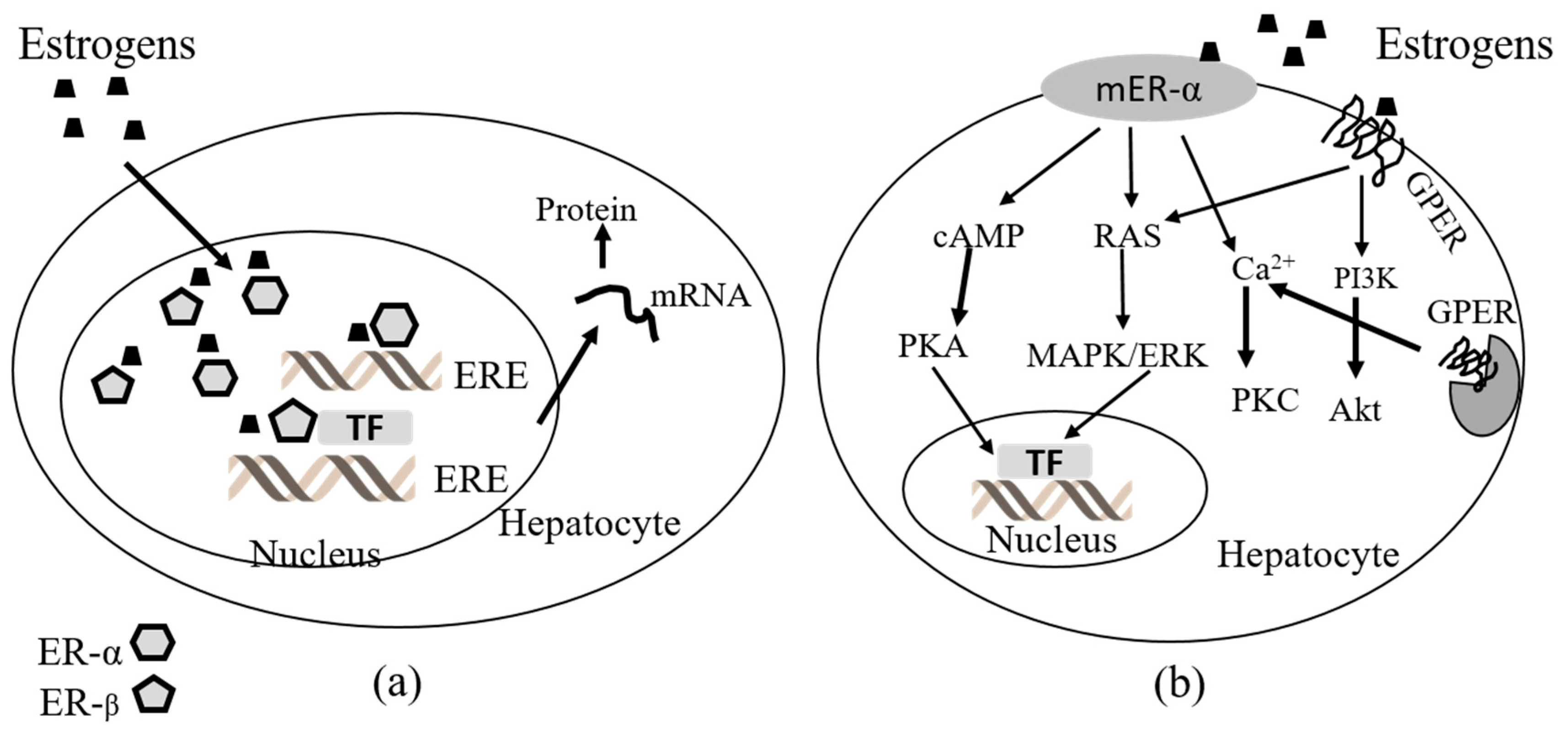
3.1. Estrogen Receptors in the Liver
3.2. Estrogen Receptor-Signaling in the Liver
3.3. Sex Hormone Receptors in HCC
3.4. Immunomodulatory Effects of Female Sex Hormones, Oral Contraceptives, and Phytoestrogens in the Liver
4. Effects of Female Sex Hormones, Oral Contraceptives, and Phytoestrogens on the Liver by Immunomodulation
4.1. Deleterious Effects by Cholestasis
4.2. Bidirectional Effects on Hepatic Oxidative Stress and Metabolism Regulation
4.3. Beneficial Effects on Hepatic Fibrosis
5. Preclinical and Clinical Studies on the Effects of Sex Hormones or their Analogs in Specific Liver Diseases
5.1. HBV and HCV Chronic Infections: Oxidative Stress and Fibrosis as a Consequence
5.2. Cholangitis and Cholestasis
5.3. Non-Alcoholic Fatty Liver Disease (NAFLD) and Non-Alcoholic Steatohepatitis (NASH): Gender Differences
5.4. Hepatocellular Carcinoma (HCC)
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AAF | 2-Acetylaminofluorene |
| Ab | Antibody |
| AHR | Aryl hydrocarbon receptor |
| AKT | Kinase/protein kinase B |
| AMH | Anti-müllerian hormone |
| AMPK | AMP-Activated protein kinase |
| AP-1 | Activator protein-1 |
| APC | Antigen-presenting cell |
| APR | Acute phase reactants |
| ATP | Adenosine triphosphate |
| ATP5a | ATP synthase mitochondria F1 complex α subunit 1 |
| BA | Bile acid(s) |
| BCA | Biochanin |
| BSEP | Bile salt export pump |
| BSF-2 | B cell stimulatory factor 2 |
| cAMP | Cyclic adenosine monophosphate |
| CBG | Corticosteroid-binding globulin |
| CCL20 | C-C Chemokine ligand 20 |
| CCND1 | Cyclin D1 |
| CCR4 | C-C Chemokine receptor type 4 |
| CCR6 | C-C Chemokine receptor type 6 |
| CD | Cluster of differentiation |
| CD4+ | CD4+ T lymphocytes |
| CD8+ | CD8+ T lymphocytes |
| Cdk | Cyclin-dependent kinase |
| CEBP-β | CCAAT/enhancer-binding protein-β |
| cGMP | Cyclic guanosine monophosphate |
| CNTF | Cilliary neurotrophic factor |
| COX | Cyclooxygenase |
| CRP | C-reactive protein |
| CSF | Colony-stimulating factor |
| CTLA-4 | Cytotoxic T-lymphocyte-associated antigen-4 |
| CU | Coumestrol |
| Cxcl | Chemokine C-X-C motif ligand |
| DA | Daidzein |
| DEN | Diethynylnitrosamine |
| DNA | Deoxyribonucleic acid |
| E1 | Estrone |
| E2-17G | Estradiol-17β-glucuronide |
| E2V | E2 valerate |
| ECM | Extracellular matrix |
| EE | 17α-ethinylestradiol |
| EGF | Epidermal growth factor |
| EGFR | Epidermal growth factor receptor |
| ENL | Enteronolactone |
| ER | Estrogen receptor(s) |
| ERE | Estrogen response element |
| ERK | Extracellular signal-regulated kinase |
| FAS | Fatty acid synthase |
| FAT | Fatty acid influx |
| FDA | Food and Drug Administration |
| FGF | Fibroblast growth factor |
| FoxP3 | Forkhead transcription factor |
| FSH | Follicle-stimulating hormone |
| GE | Genistein |
| GF | Growth factor |
| GITR | Glucocorticoid-induced TNF-alpha receptor |
| GnRH | Gonadotropin-releasing hormone |
| GPCR | G protein coupled receptor |
| GPER/GPR 30 | G protein-coupled estrogenic receptor |
| GR | Glucocorticoid receptor |
| Grb2 | Growth-factor-receptor-bound protein 2 |
| GSH | Reduced glutathione |
| GTF | General transcription Factor |
| HB-EGF | Heparin-binding-EGF |
| HBV-LF | Hepatitis B virus-related liver fibrosis |
| HCC | Hepatocellular carcinoma |
| HCV | Hepatitis C virus |
| HDL | High-density lipoproteins |
| HGF | Hepatocyte growth factor |
| HIV | Human immunodeficiency viruses |
| HRE | Hormone response element |
| HSC | Hepatic stellate cells |
| HSP | Heat shock protein |
| ICP | Intrahepatic cholestasis of pregnancy |
| IFN | Interferon(s) |
| IGF-1 | Insulin-like growth factor 1 |
| IL | Interleukin(s) |
| iNOS | Inducible nitric oxide synthase |
| JAK | Janus kinase |
| LBD | Ligand-binding-domain |
| LDA | Ligand dependent pathway of activation |
| LDL | Low density lipoproteins |
| LH | Luteinizing hormone |
| LIF | Leukemia inhibitory factor |
| LNG | Levonorgestrel |
| LP | Lipid peroxidation |
| M | Macrophage(s) |
| MAP | Mitogen-activated protein |
| MAPK | Mitogen activated protein kinase |
| MCP | Macrophage chemotactic protein |
| MetS | Metabolic syndrome |
| mPR | Membrane localized progestin receptor |
| MRP | Multidrug resistance-associated protein |
| MyD88 | Myeloid differentiation primary response protein 88 |
| NAFLD | Non-alcoholic fatty liver disease |
| NASH | Non-alcoholic steatohepatitis |
| NDUFA9 | NADH dehydrogenase 1α subcomplex |
| NF | Nuclear factor |
| βNGF | β-Nerve growth factor |
| NHR | Nuclear hormone receptors |
| NK | Natural killer |
| NLRP | NOD-like receptor |
| NO | Nitric oxide |
| Nrf2 | Nuclear factor erythroid 2 |
| NT | Neurotransmitter |
| NTCP | Na+-taurocholate cotransporter protein |
| OATP | Organic anion transporting protein |
| OC | Oral contraceptive(s) |
| O-DMA | O-demethylangiotensin |
| OVX | Ovariectomy |
| P4 | Progesterone |
| PDGF-BB | Platelet-Derived Growth Factor-BB |
| PE | Phytoestrogen(s) |
| PELP1/MNAR | Proline-, glutamic acid-, and leucine-rich protein 1, modulator of non-genomic activity of ERs |
| p-ERK1 | Proline-rich receptor-like protein kinase |
| PI3K | Phosphoinositide 3-kinase |
| PKA | Protein kinase A |
| PKC | Protein kinase C |
| PON | Paraoxonase |
| PPAR-γ | Peroxisome proliferator-activated receptor-gamma |
| 8PN | 8-Prenylnaringenin |
| PR | Progestin receptor |
| PRL | Prolactin |
| Ras | Small GTPase named by abbreviating rat sarcoma |
| ROS | Reactive oxygen species |
| SCF | Stem cell factor |
| SCGF-β | Stem cell growth factor beta |
| SDHA | Succinate dehydrogenase complex subunit A |
| Sgk | Serum and glucocorticoid regulated kinase |
| SGK | Glucocorticoid induced kinase |
| SHBG | Sex hormone-binding globulin |
| SHP | Protein tyrosine phosphatase |
| sIL-6R | Soluble derivative of the IL-6 receptor |
| SIRT | Silent mating type information regulation 2 homolog (Sirtuin) NAD+-dependent protein deacetylase |
| SOCS | Suppressor of cytokine signaling |
| SOD | Superoxide dismutase |
| Sos | Protein son-of-sevenless |
| Src | Schmidt-Ruppin A-2 viral oncogene tyrosine-protein kinase |
| SREBP-1c | Sterol regulatory element binding protein-1c |
| STAT3 | Signal transducer and activator of transcription factor-3 |
| TCR | T cell receptor |
| TF | Transcription factor |
| TGF | Transforming growth factor |
| Th | T helper cell(s) |
| TLR | Toll-like receptor |
| TNF | Tumor necrosis factor |
| Treg | T regulatory cell(s) |
| TX | Tromboxane |
| TYK | Phosphorylated tyrosine Y |
| UQCRC2 | Ubiquinol cytochrome c reductase core protein |
| VEGF | Vascular endothelial growth factor |
| VLDL | Very low density lipoprotein |
| XN | Xanthohumol |
References
- Kuntz, E.; Kuntz, H.D. Hepatology: Principles and Practice, 2nd ed.; Springer Medizin Verlag: Berlin/Heidelberg, Germany, 2006; p. 906. [Google Scholar]
- Baruch, Y. The liver: A large endocrine gland. J. Hepatol. 2000, 32, 505–507. [Google Scholar] [CrossRef]
- Adeva-Andany, M.M.; Perez-Felpete, N.; Fernandez-Fernandez, C.; Donapetry-Garcia, C.; Pazos-Garcia, C. Liver glucose metabolism in humans. Biosci. Rep. 2016, 36, e00416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, H.R.; Wang, J.L.; Ren, H.Z.; Shi, X.L. Lipometabolism and glycometabolism in liver diseases. Biomed. Res. Int. 2018, 2018, 1287127. [Google Scholar] [CrossRef] [PubMed]
- Mittal, S.; El-Serag, H.B. Epidemiology of hepatocellular carcinoma: Consider the population. J. Clin. Gastroenterol. 2013, 47 (Suppl. 1), S2–S6. [Google Scholar] [CrossRef]
- Kabir, E.R.; Rahman, M.S.; Rahman, I. A review on endocrine disruptors and their possible impacts on human health. Environ. Toxicol. Pharmacol. 2015, 40, 241–258. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Cao, M.; You, D.; Qin, G.; Liu, Z. Research progress on the animal models of drug-induced liver injury: Current status and further perspectives. Biomed. Res. Int. 2019, 2019, 1283824. [Google Scholar] [CrossRef]
- Heymann, F.; Tacke, F. Immunology in the liver--from homeostasis to disease. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 88–110. [Google Scholar] [CrossRef] [PubMed]
- Verthelyi, D. Sex hormones as immunomodulators in health and disease. Int. Immunopharmacol. 2001, 1, 983–993. [Google Scholar] [CrossRef]
- Bouman, A.; Heineman, M.J.; Faas, M.M. Sex hormones and the immune response in humans. Hum. Reprod. Update 2005, 11, 411–423. [Google Scholar] [CrossRef] [Green Version]
- Rubinow, K.B. An intracrine view of sex steroids, immunity, and metabolic regulation. Mol. Metab. 2018, 15, 92–103. [Google Scholar] [CrossRef]
- Fernández-Martínez, E.; Pérez-Soto, E.; González-Hernández, C.; Ortiz, M.I.; Pérez-Álvarez, V.; Muriel, P.; Shibayama, M. Immunomodulatory effects by oral contraceptives in normal and cholestatic female rats: Role of cytokines. Int. Immunopharmacol. 2014, 21, 10–19. [Google Scholar] [CrossRef]
- Van Duursen, M.B.M. Modulation of estrogen synthesis and metabolism by phytoestrogens in vitro and the implications for women’s health. Toxicol. Res. 2017, 6, 772–794. [Google Scholar] [CrossRef]
- Kuiper, G.G.; Lemmen, J.G.; Carlsson, B.; Corton, J.C.; Safe, S.H.; van der Saag, P.T.; van der Burg, B.; Gustafsson, J.A. Interaction of estrogenic chemicals and phytoestrogens with estrogen receptor beta. Endocrinology 1998, 139, 4252–4263. [Google Scholar] [CrossRef]
- Lecomte, S.; Demay, F.; Ferriere, F.; Pakdel, F. Phytochemicals targeting estrogen receptors: Beneficial rather than adverse effects? Int. J. Mol. Sci. 2017, 18, 1381. [Google Scholar] [CrossRef]
- Moutsatsou, P. The spectrum of phytoestrogens in nature: Our knowledge is expanding. Hormones 2007, 6, 173–193. [Google Scholar]
- Vacchio, M.S.; Bosselut, R. What happens in the thymus does not stay in the thymus: How T cells recycle the CD4+-CD8+ lineage commitment transcriptional circuitry to control their function. J. Immunol. 2016, 196, 4848–4856. [Google Scholar] [CrossRef]
- Berger, A. Th1 and Th2 responses: What are they? Br. Med. J. 2000, 321, 424. [Google Scholar] [CrossRef]
- Kidd, P. Th1/Th2 balance: The hypothesis, its limitations, and implications for health and disease. Altern. Med. Rev. 2003, 8, 223–246. [Google Scholar]
- Lafdil, F.; Miller, A.M.; Ki, S.H.; Gao, B. Th17 cells and their associated cytokines in liver diseases. Cell. Mol. Immunol. 2010, 7, 250–254. [Google Scholar] [CrossRef] [Green Version]
- Farrar, J.D.; Asnagli, H.; Murphy, K.M. T helper subset development: Roles of instruction, selection, and transcription. J. Clin. Investig. 2002, 109, 431–435. [Google Scholar] [CrossRef]
- Seki, S.; Habu, Y.; Kawamura, T.; Takeda, K.; Dobashi, H.; Ohkawa, T.; Hiraide, H. The liver as a crucial organ in the first line of host defense: The roles of Kupffer cells, natural killer (NK) cells and NK1.1 Ag+ T cells in T helper 1 immune responses. Immunol. Rev. 2000, 174, 35–46. [Google Scholar] [CrossRef]
- Kern, L.; Mittenbuhler, M.J.; Vesting, A.J.; Ostermann, A.L.; Wunderlich, C.M.; Wunderlich, F.T. Obesity-induced TNFalpha and IL-6 signaling: The missing link between obesity and inflammation-driven liver and colorectal cancers. Cancers 2019, 11, 24. [Google Scholar] [CrossRef]
- Fernández-Martínez, E.; Pérez-Álvarez, V.; Tsutsumi, V.; Shibayama, M.; Muriel, P. Chronic bile duct obstruction induces changes in plasma and hepatic levels of cytokines and nitric oxide in the rat. Exp. Toxicol. Pathol. 2006, 58, 49–58. [Google Scholar] [CrossRef]
- Pinheiro, D.; Leiros, L.; Dau, J.B.T.; Stumbo, A.C.; Thole, A.A.; Cortez, E.A.C.; Mandarim-de-Lacerda, C.A.; Carvalho, L.; Carvalho, S.N. Cytokines, hepatic cell profiling and cell interactions during bone marrow cell therapy for liver fibrosis in cholestatic mice. PLoS ONE 2017, 12, e0187970. [Google Scholar] [CrossRef]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 2014, 6, 1–13. [Google Scholar] [CrossRef]
- Luo, W.; Xu, Q.; Wang, Q.; Wu, H.; Hua, J. Effect of modulation of PPAR-gamma activity on Kupffer cells M1/M2 polarization in the development of non-alcoholic fatty liver disease. Sci. Rep. 2017, 7, 44612. [Google Scholar] [CrossRef]
- Toniolo, A.; Fadini, G.P.; Tedesco, S.; Cappellari, R.; Vegeto, E.; Maggi, A.; Avogaro, A.; Bolego, C.; Cignarella, A. Alternative activation of human macrophages is rescued by estrogen treatment in vitro and impaired by menopausal status. J. Clin. Endocrinol. Metab. 2015, 100, E50–E58. [Google Scholar] [CrossRef]
- Kiziltas, S. Toll-like receptors in pathophysiology of liver diseases. World J. Hepatol. 2016, 8, 1354–1369. [Google Scholar] [CrossRef]
- Zheng, S.G. Regulatory T cells vs Th17: Differentiation of Th17 versus Treg, are the mutually exclusive? Am. J. Clin. Exp. Immunol. 2013, 2, 94–106. [Google Scholar]
- Oo, Y.H.; Adams, D.H. The role of chemokines in the recruitment of lymphocytes to the liver. J. Autoimmun. 2010, 34, 45–54. [Google Scholar] [CrossRef] [Green Version]
- Muranski, P.; Restifo, N.P. Essentials of Th17 cell commitment and plasticity. Blood 2013, 121, 2402–2414. [Google Scholar] [CrossRef]
- Quintana, F.J. The aryl hydrocarbon receptor: A molecular pathway for the environmental control of the immune response. Immunology 2013, 138, 183–189. [Google Scholar] [CrossRef]
- Qin, S.Y.; Lu, D.H.; Guo, X.Y.; Luo, W.; Hu, B.L.; Huang, X.L.; Chen, M.; Wang, J.X.; Ma, S.J.; Yang, X.W.; et al. A deleterious role for Th9/IL-9 in hepatic fibrogenesis. Sci. Rep. 2016, 6, 18694. [Google Scholar] [CrossRef]
- Vignali, D.A.; Collison, L.W.; Workman, C.J. How regulatory T cells work. Nat. Rev. Immunol. 2008, 8, 523–532. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Su, Y.; Hua, X.; Xie, C.; Liu, J.; Huang, Y.; Zhou, L.; Zhang, M.; Li, X.; Gao, Z. Levels of hepatic Th17 cells and regulatory T cells upregulated by hepatic stellate cells in advanced HBV-related liver fibrosis. J. Transl. Med. 2017, 15, 75. [Google Scholar] [CrossRef]
- Vyas, S.P.; Goswami, R. A Decade of Th9 Cells: Role of Th9 Cells in Inflammatory Bowel Disease. Front. Immunol. 2018, 9, 1139. [Google Scholar] [CrossRef] [Green Version]
- Malik, S.; Awasthi, A. Transcriptional Control of Th9 Cells: Role of Foxo1 in Interleukin-9 Induction. Front. Immunol. 2018, 9, 995. [Google Scholar] [CrossRef] [Green Version]
- Nedoszytko, B.; Lange, M.; Sokolowska-Wojdylo, M.; Renke, J.; Trzonkowski, P.; Sobjanek, M.; Szczerkowska-Dobosz, A.; Niedoszytko, M.; Gorska, A.; Romantowski, J.; et al. The role of regulatory T cells and genes involved in their differentiation in pathogenesis of selected inflammatory and neoplastic skin diseases. Part II: The Treg role in skin diseases pathogenesis. Postepy Dermatol. Alergol. 2017, 34, 405–417. [Google Scholar] [CrossRef] [Green Version]
- Crome, S.Q.; Wang, A.Y.; Levings, M.K. Translational mini-review series on Th17 cells: Function and regulation of human T helper 17 cells in health and disease. Clin. Exp. Immunol. 2010, 159, 109–119. [Google Scholar] [CrossRef]
- Wang, X.J.; Leveson-Gower, D.; Golab, K.; Wang, L.J.; Marek-Trzonkowska, N.; Krzystyniak, A.; Wardowska, A.; Millis, J.M.; Trzonkowski, P.; Witkowski, P. Influence of pharmacological immunomodulatory agents on CD4(+)CD25(high)FoxP3(+) T regulatory cells in humans. Int. Immunopharmacol. 2013, 16, 364–370. [Google Scholar] [CrossRef]
- Niedzwiecki, M.; Budzilo, O.; Adamkiewicz-Drozynska, E.; Pawlik-Gwozdecka, D.; Zielinski, M.; Maciejka-Kemblowska, L.; Szczepanski, T.; Trzonkowski, P. CD4(+)CD25(high)CD127(low/-)FoxP3 (+) Regulatory T-Cell Population in Acute Leukemias: A Review of the Literature. J. Immunol. Res. 2019, 2019, 2816498. [Google Scholar] [CrossRef]
- Wawman, R.E.; Bartlett, H.; Oo, Y.H. Regulatory T Cell Metabolism in the Hepatic Microenvironment. Front. Immunol. 2017, 8, 1889. [Google Scholar] [CrossRef]
- Racanelli, V.; Rehermann, B. The liver as an immunological organ. Hepatology 2006, 43, S54–S62. [Google Scholar] [CrossRef]
- Zhou, Z.; Xu, M.J.; Gao, B. Hepatocytes: A key cell type for innate immunity. Cell. Mol. Immunol. 2016, 13, 301–315. [Google Scholar] [CrossRef]
- Bode, J.G.; Albrecht, U.; Haussinger, D.; Heinrich, P.C.; Schaper, F. Hepatic acute phase proteins--regulation by IL-6- and IL-1-type cytokines involving STAT3 and its crosstalk with NF-kappaB-dependent signaling. Eur. J. Cell Biol. 2012, 91, 496–505. [Google Scholar] [CrossRef]
- Fabbi, M.; Carbotti, G.; Ferrini, S. Dual roles of IL-27 in cancer biology and immunotherapy. Mediat. Inflamm. 2017, 2017, 3958069. [Google Scholar] [CrossRef]
- Strazzabosco, M.; Fiorotto, R.; Cadamuro, M.; Spirli, C.; Mariotti, V.; Kaffe, E.; Scirpo, R.; Fabris, L. Pathophysiologic implications of innate immunity and autoinflammation in the biliary epithelium. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1374–1379. [Google Scholar] [CrossRef]
- Seidensticker, M.; Powerski, M.; Seidensticker, R.; Damm, R.; Mohnike, K.; Garlipp, B.; Klopffleisch, M.; Amthauer, H.; Ricke, J.; Pech, M. Cytokines and (90)Y-radioembolization: Relation to liver function and overall survival. Cardiovasc. Interv. Radiol. 2017, 40, 1185–1195. [Google Scholar] [CrossRef]
- Tao, Y.; Wang, M.; Chen, E.; Tang, H. Liver regeneration: Analysis of the main relevant signaling molecules. Mediat. Inflamm. 2017, 2017, 4256352. [Google Scholar] [CrossRef]
- Uebi, T.; Umeda, M.; Imai, T. Estrogen induces estrogen receptor alpha expression and hepatocyte proliferation in the livers of male mice. Genes Cells 2015, 20, 217–223. [Google Scholar] [CrossRef]
- Chiu, E.J.; Lin, H.L.; Chi, C.W.; Liu, T.Y.; Lui, W.Y. Estrogen therapy for hepatectomy patients with poor liver function? Med. Hypotheses 2002, 58, 516–518. [Google Scholar] [CrossRef]
- Schwabe, R.F.; Brenner, D.A. Mechanisms of liver injury. I. TNF-alpha-induced liver injury: Role of IKK, JNK, and ROS pathways. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G583–G589. [Google Scholar] [CrossRef]
- Han, D.; Ybanez, M.D.; Ahmadi, S.; Yeh, K.; Kaplowitz, N. Redox regulation of tumor necrosis factor signaling. Antioxid. Redox Signal. 2009, 11, 2245–2263. [Google Scholar] [CrossRef]
- Sedger, L.M.; McDermott, M.F. TNF and TNF-receptors: From mediators of cell death and inflammation to therapeutic giants - past, present and future. Cytokine Growth Factor Rev. 2014, 25, 453–472. [Google Scholar] [CrossRef]
- Kishimoto, T. IL-6: From its discovery to clinical applications. Int. Immunol. 2010, 22, 347–352. [Google Scholar] [CrossRef]
- Gabay, C.; Kushner, I. Acute-phase proteins and other systemic responses to inflammation. N. Engl. J. Med. 1999, 340, 448–454. [Google Scholar] [CrossRef]
- Schmidt-Arras, D.; Rose-John, S. IL-6 pathway in the liver: From physiopathology to therapy. J. Hepatol. 2016, 64, 1403–1415. [Google Scholar] [CrossRef] [Green Version]
- Heinrich, P.C.; Castell, J.V.; Andus, T. Interleukin-6 and the acute phase response. Biochem. J. 1990, 265, 621–636. [Google Scholar] [CrossRef]
- Kavanaugh, A. Combination cytokine therapy: The next generation of rheumatoid arthritis therapy? Arthritis Rheum. 2002, 47, 87–92. [Google Scholar] [CrossRef]
- Loyer, P.; Ilyin, G.; Abdel Razzak, Z.; Banchereau, J.; Dezier, J.F.; Campion, J.P.; Guguen-Guillouzo, C.; Guillouzo, A. Interleukin 4 inhibits the production of some acute-phase proteins by human hepatocytes in primary culture. FEBS Lett. 1993, 336, 215–220. [Google Scholar] [CrossRef] [Green Version]
- Petes, C.; Mariani, M.K.; Yang, Y.; Grandvaux, N.; Gee, K. Interleukin (IL)-6 inhibits IL-27-and IL-30-mediated inflammatory responses in human monocytes. Front. Immunol. 2018, 9, 256. [Google Scholar] [CrossRef]
- Jayatilaka, H.; Tyle, P.; Chen, J.J.; Kwak, M.; Ju, J.; Kim, H.J.; Lee, J.S.H.; Wu, P.H.; Gilkes, D.M.; Fan, R.; et al. Synergistic IL-6 and IL-8 paracrine signalling pathway infers a strategy to inhibit tumour cell migration. Nat. Commun. 2017, 8, 15584. [Google Scholar] [CrossRef] [Green Version]
- Kastner, L.; Dwyer, D.; Qin, F.X. Synergistic effect of IL-6 and IL-4 in driving fate revision of natural Foxp3+ regulatory T cells. J. Immunol. 2010, 185, 5778–5786. [Google Scholar] [CrossRef]
- Turner, M.D.; Nedjai, B.; Hurst, T.; Pennington, D.J. Cytokines and chemokines: At the crossroads of cell signalling and inflammatory disease. Biochim. Biophys. Acta 2014, 1843, 2563–2582. [Google Scholar] [CrossRef] [Green Version]
- Streetz, K.L.; Luedde, T.; Manns, M.P.; Trautwein, C. Interleukin 6 and liver regeneration. Gut 2000, 47, 309–312. [Google Scholar] [CrossRef] [Green Version]
- Kwon, Y.J.; Lee, K.G.; Choi, D. Clinical implications of advances in liver regeneration. Clin. Mol. Hepatol. 2015, 21, 7–13. [Google Scholar] [CrossRef]
- Merlen, G.; Ursic-Bedoya, J.; Jourdainne, V.; Kahale, N.; Glenisson, M.; Doignon, I.; Rainteau, D.; Tordjmann, T. Bile acids and their receptors during liver regeneration: “Dangerous protectors”. Mol. Asp. Med. 2017, 56, 25–33. [Google Scholar] [CrossRef]
- Palstra, A.P.; Schnabel, D.; Nieveen, M.C.; Spaink, H.P.; van den Thillart, G.E. Temporal expression of hepatic estrogen receptor 1, vitellogenin1 and vitellogenin2 in european silver eels. Gen. Comp. Endocrinol. 2010, 166, 1–11. [Google Scholar] [CrossRef]
- Nagler, J.J.; Cavileer, T.D.; Verducci, J.S.; Schultz, I.R.; Hook, S.E.; Hayton, W.L. Estrogen receptor mRNA expression patterns in the liver and ovary of female rainbow trout over a complete reproductive cycle. Gen. Comp. Endocrinol. 2012, 178, 556–561. [Google Scholar] [CrossRef] [Green Version]
- Colantoni, A.; Emanuele, M.A.; Kovacs, E.J.; Villa, E.; Van Thiel, D.H. Hepatic estrogen receptors and alcohol intake. Mol. Cell Endocrinol. 2002, 193, 101–104. [Google Scholar] [CrossRef]
- Kovats, S. Estrogen receptors regulate innate immune cells and signaling pathways. Cell. Immunol. 2015, 294, 63–69. [Google Scholar] [CrossRef] [Green Version]
- Iyer, J.K.; Kalra, M.; Kaul, A.; Payton, M.E.; Kaul, R. Estrogen receptor expression in chronic hepatitis C and hepatocellular carcinoma pathogenesis. World J. Gastroenterol. 2017, 23, 6802–6816. [Google Scholar] [CrossRef]
- Porter, L.E.; Elm, M.S.; Van Thiel, D.H.; Eagon, P.K. Hepatic estrogen receptor in human liver disease. Gastroenterology 1987, 92, 735–745. [Google Scholar] [CrossRef]
- Iavarone, M.; Lampertico, P.; Seletti, C.; Francesca Donato, M.; Ronchi, G.; del Ninno, E.; Colombo, M. The clinical and pathogenetic significance of estrogen receptor-beta expression in chronic liver diseases and liver carcinoma. Cancer 2003, 98, 529–534. [Google Scholar] [CrossRef]
- Miceli, V.; Cocciadiferro, L.; Fregapane, M.; Zarcone, M.; Montalto, G.; Polito, L.M.; Agostara, B.; Granata, O.M.; Carruba, G. Expression of wild-type and variant estrogen receptor alpha in liver carcinogenesis and tumor progression. OMICS 2011, 15, 313–317. [Google Scholar] [CrossRef]
- Cui, J.; Shen, Y.; Li, R. Estrogen synthesis and signaling pathways during aging: From periphery to brain. Trends Mol. Med. 2013, 19, 197–209. [Google Scholar] [CrossRef]
- Lax, E.R.; Tamulevicius, P.; Muller, A.; Schriefers, H. Hepatic nuclear estrogen receptor concentrations in the rat--influence of age, sex, gestation, lactation and estrous cycle. J. Steroid Biochem. 1983, 19, 1083–1088. [Google Scholar] [CrossRef]
- Eisenfeld, A.J.; Aten, R.F. Estrogen receptors and androgen receptors in the mammalian liver. J. Steroid Biochem. 1987, 27, 1109–1118. [Google Scholar] [CrossRef]
- Shen, M.; Shi, H. Sex hormones and their receptors regulate liver energy homeostasis. Int. J. Endocrinol. 2015, 2015, 294278. [Google Scholar] [CrossRef]
- Nilsson, B.O.; Olde, B.; Leeb-Lundberg, L.M. G protein-coupled oestrogen receptor 1 (GPER1)/GPR30: A new player in cardiovascular and metabolic oestrogenic signalling. Br. J. Pharmacol. 2011, 163, 1131–1139. [Google Scholar] [CrossRef]
- Weigel, N.L.; Moore, N.L. Steroid receptor phosphorylation: A key modulator of multiple receptor functions. Mol. Endocrinol. 2007, 21, 2311–2319. [Google Scholar] [CrossRef]
- Stanisic, V.; Lonard, D.M.; O’Malley, B.W. Modulation of steroid hormone receptor activity. Prog. Brain Res. 2010, 181, 153–176. [Google Scholar]
- Yeh, Y.T.; Chang, C.W.; Wei, R.J.; Wang, S.N. Progesterone and related compounds in hepatocellular carcinoma: Basic and clinical aspects. Biomed. Res. Int. 2013, 2013, 290575. [Google Scholar] [CrossRef]
- Li, Y.; Xu, A.; Jia, S.; Huang, J. Recent advances in the molecular mechanism of sex disparity in hepatocellular carcinoma. Oncol. Lett. 2019, 17, 4222–4228. [Google Scholar] [CrossRef]
- Liu, W.C.; Liu, Q.Y. Molecular mechanisms of gender disparity in hepatitis B virus-associated hepatocellular carcinoma. World J. Gastroenterol. 2014, 20, 6252–6261. [Google Scholar] [CrossRef]
- Bereshchenko, O.; Bruscoli, S.; Riccardi, C. Glucocorticoids, sex hormones, and immunity. Front. Immunol. 2018, 9, 1332. [Google Scholar] [CrossRef]
- Suzuki, T.; Yu, H.P.; Hsieh, Y.C.; Choudhry, M.A.; Bland, K.I.; Chaudry, I.H. Estrogen-mediated activation of non-genomic pathway improves macrophages cytokine production following trauma-hemorrhage. J. Cell. Physiol. 2008, 214, 662–672. [Google Scholar] [CrossRef]
- Fernandez-Perez, L.; Guerra, B.; Diaz-Chico, J.C.; Flores-Morales, A. Estrogens regulate the hepatic effects of growth hormone, a hormonal interplay with multiple fates. Front. Endocrinol. 2013, 4, 66. [Google Scholar] [CrossRef]
- Lang, T.J. Estrogen as an immunomodulator. Clin. Immunol. 2004, 113, 224–230. [Google Scholar] [CrossRef]
- Kyurkchiev, D.; Ivanova-Todorova, E.; Kyurkchiev, S.D. New target cells of the immunomodulatory effects of progesterone. Reprod. Biomed. Online 2010, 21, 304–311. [Google Scholar] [CrossRef] [Green Version]
- Allen, T.K.; Nazzal, M.N.; Feng, L.; Buhimschi, I.A.; Murtha, A.P. Progestins inhibit tumor necrosis factor alpha-induced matrix metalloproteinase 9 activity via the glucocorticoid receptor in primary amnion epithelial cells. Reprod. Sci. 2019, 26, 1193–1202. [Google Scholar] [CrossRef]
- Ucan, H.B.; Kaplan, M.; Salman, B.; Yilmaz, U.; Mentes, B.B.; Aybay, C. Effect of oophorectomy and exogenous estrogen replacement on liver injury in experimental obstructive jaundice. World J. Gastroenterol. 2008, 14, 2818–2824. [Google Scholar] [CrossRef]
- Yuan, Y.; Shimizu, I.; Shen, M.; Aoyagi, E.; Takenaka, H.; Itagaki, T.; Urata, M.; Sannomiya, K.; Kohno, N.; Tamaki, K.; et al. Effects of estradiol and progesterone on the proinflammatory cytokine production by mononuclear cells from patients with chronic hepatitis C. World J. Gastroenterol. 2008, 14, 2200–2207. [Google Scholar] [CrossRef]
- Bouman, A.; Schipper, M.; Heineman, M.J.; Faas, M. 17beta-estradiol and progesterone do not influence the production of cytokines from lipopolysaccharide-stimulated monocytes in humans. Fertil. Steril. 2004, 82 (Suppl. 3), 1212–1219. [Google Scholar] [CrossRef]
- Hsieh, H.G.; Huang, H.C.; Lee, F.Y.; Chan, C.Y.; Lee, J.Y.; Lee, S.D. Kinetics of cytokine expression in cirrhotic rats. J. Chin. Med. Assoc. 2011, 74, 385–393. [Google Scholar] [CrossRef] [Green Version]
- Lickteig, A.J.; Slitt, A.L.; Arkan, M.C.; Karin, M.; Cherrington, N.J. Differential regulation of hepatic transporters in the absence of tumor necrosis factor-alpha, interleukin-1beta, interleukin-6, and nuclear factor-kappaB in two models of cholestasis. Drug Metab. Dispos. 2007, 35, 402–409. [Google Scholar] [CrossRef]
- Plebani, M.; Panozzo, M.P.; Basso, D.; De Paoli, M.; Biasin, R.; Infantolino, D. Cytokines and the progression of liver damage in experimental bile duct ligation. Clin. Exp. Pharmacol. Physiol. 1999, 26, 358–563. [Google Scholar] [CrossRef]
- Tilg, H. Cytokines and liver diseases. Can. J. Gastroenterol. 2001, 15, 661–668. [Google Scholar] [CrossRef]
- Tu, B.; Gong, J.P.; Feng, H.Y.; Wu, C.X.; Shi, Y.J.; Li, X.H.; Peng, Y.; Liu, C.A.; Li, S.W. Role of NF-kB in multiple organ dysfunction during acute obstructive cholangitis. World. J. Gastroenterol. 2003, 9, 179–183. [Google Scholar] [CrossRef]
- Chen, G.; Shi, J.; Ding, Y.; Yin, H.; Hang, C. Progesterone prevents traumatic brain injury-induced intestinal nuclear factor kappa B activation and proinflammatory cytokines expression in male rats. Mediat. Inflamm. 2007, 2007, 93431. [Google Scholar] [CrossRef]
- Louis, H.; Le Moine, O.; Goldman, M.; Deviere, J. Modulation of liver injury by interleukin-10. Acta Gastroenterol. Belg. 2003, 66, 7–14. [Google Scholar]
- Rodriguez-Garay, E.A. Cholestasis: Human disease and experimental animal models. Ann. Hepatol. 2003, 2, 150–158. [Google Scholar] [CrossRef]
- Li, M.; Cai, S.Y.; Boyer, J.L. Mechanisms of bile acid mediated inflammation in the liver. Mol. Asp. Med. 2017, 56, 45–53. [Google Scholar] [CrossRef]
- Vickers, A.E.; Nelson, K.; McCoy, Z.; Lucier, G.W. Changes in estrogen receptor, DNA ploidy, and estrogen metabolism in rat hepatocytes during a two-stage model for hepatocarcinogenesis using 17 alpha-ethinylestradiol as the promoting agent. Cancer Res. 1989, 49, 6512–6520. [Google Scholar]
- Yager, J.D., Jr. Oral contraceptive steroids as promoters or complete carcinogens for liver in female Sprague-Dawley rats. Environ. Health Perspect. 1983, 50, 109–112. [Google Scholar] [CrossRef]
- Arrese, M.; Pizarro, M.; Solis, N.; Koenig, C.; Accatino, L. Enhanced biliary excretion of canalicular membrane enzymes in ethynylestradiol-induced cholestasis. Effects of ursodeoxycholic acid administration. Biochem. Pharmacol. 1995, 50, 1223–1232. [Google Scholar] [CrossRef]
- Rahner, C.; Stieger, B.; Landmann, L. Structure-function correlation of tight junctional impairment after intrahepatic and extrahepatic cholestasis in rat liver. Gastroenterology 1996, 110, 1564–1578. [Google Scholar] [CrossRef]
- Meier, Y.; Zodan, T.; Lang, C.; Zimmermann, R.; Kullak-Ublick, G.A.; Meier, P.J.; Stieger, B.; Pauli-Magnus, C. Increased susceptibility for intrahepatic cholestasis of pregnancy and contraceptive-induced cholestasis in carriers of the 1331T > C polymorphism in the bile salt export pump. World J. Gastroenterol. 2008, 14, 38–45. [Google Scholar] [CrossRef]
- Down, R.H.; Whiting, M.J.; Watts, J.M.; Jones, W. Effect of synthetic oestrogens and progestagens in oral contraceptives on bile lipid composition. Gut 1983, 24, 253–259. [Google Scholar] [CrossRef]
- Ikejima, K.; Enomoto, N.; Iimuro, Y.; Ikejima, A.; Fang, D.; Xu, J.; Forman, D.T.; Brenner, D.A.; Thurman, R.G. Estrogen increases sensitivity of hepatic Kupffer cells to endotoxin. Am. J. Physiol. 1998, 274, G669–G676. [Google Scholar] [CrossRef]
- Konno, A.; Enomoto, N.; Takei, Y.; Hirose, M.; Ikejima, K.; Sato, N. Oral contraceptives worsen endotoxin-induced liver injury in rats. Alcohol Clin. Exp. Res. 2002, 26, 70S–74S. [Google Scholar] [CrossRef]
- Green, R.M.; Beier, D.; Gollan, J.L. Regulation of hepatocyte bile salt transporters by endotoxin and inflammatory cytokines in rodents. Gastroenterology 1996, 111, 193–198. [Google Scholar] [CrossRef]
- Moseley, R.H.; Wang, W.; Takeda, H.; Lown, K.; Shick, L.; Ananthanarayanan, M.; Suchy, F.J. Effect of endotoxin on bile acid transport in rat liver: A potential model for sepsis-associated cholestasis. Am. J. Physiol. 1996, 271, G137–G146. [Google Scholar] [CrossRef]
- Larson, S.P.; Kovilam, O.; Agrawal, D.K. Immunological basis in the pathogenesis of intrahepatic cholestasis of pregnancy. Expert Rev. Clin. Immunol. 2016, 12, 39–48. [Google Scholar] [CrossRef]
- Roma, M.G.; Crocenzi, F.A.; Sanchez Pozzi, E.A. Hepatocellular transport in acquired cholestasis: New insights into functional, regulatory and therapeutic aspects. Clin. Sci. 2008, 114, 567–588. [Google Scholar] [CrossRef]
- Perez, M.J.; Briz, O. Bile-acid-induced cell injury and protection. World J. Gastroenterol. 2009, 15, 1677–1689. [Google Scholar] [CrossRef]
- Roma, M.G.; Crocenzi, F.A.; Mottino, A.D. Dynamic localization of hepatocellular transporters in health and disease. World J. Gastroenterol. 2008, 14, 6786–6801. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, Y.; Moore, R.; Hess, H.A.; Guo, G.L.; Gonzalez, F.J.; Korach, K.S.; Maronpot, R.R.; Negishi, M. Estrogen receptor alpha mediates 17alpha-ethynylestradiol causing hepatotoxicity. J. Biol. Chem. 2006, 281, 16625–16631. [Google Scholar] [CrossRef]
- Mancinelli, R.; Onori, P.; Demorrow, S.; Francis, H.; Glaser, S.; Franchitto, A.; Carpino, G.; Alpini, G.; Gaudio, E. Role of sex hormones in the modulation of cholangiocyte function. World J. Gastrointest. Pathophysiol. 2010, 1, 50–62. [Google Scholar] [CrossRef]
- Sato, K.; Kennedy, L.; Liangpunsakul, S.; Kusumanchi, P.; Yang, Z.; Meng, F.; Glaser, S.; Francis, H.; Alpini, G. Intercellular communication between hepatic cells in liver diseases. Int. J. Mol. Sci. 2019, 20, 2180. [Google Scholar] [CrossRef]
- Hussein, M.A. Prophylactic effect of resveratrol against ethinylestradiol-induced liver cholestasis. J. Med. Food 2013, 16, 246–254. [Google Scholar] [CrossRef]
- Xu, Y.J.; Yu, Z.Q.; Zhang, C.L.; Li, X.P.; Feng, C.Y.; Lei, K.; He, W.X.; Liu, D. Protective effects of ginsenosides on 17alpha-ethynyelstradiol induced intrahepatic cholestasis via anti-oxidative and anti-inflammatory mechanisms in rats. Am. J. Chin. Med. 2017, 45, 1613–1629. [Google Scholar] [CrossRef]
- Udomsuk, L.; Juengwatanatrakul, T.; Putalun, W.; Jarukamjorn, K. Suppression of BSEP and MRP2 in mouse liver by miroestrol and deoxymiroestrol isolated from Pueraria candollei. Phytomedicine Int. J. Phytother. Phytopharm. 2012, 19, 1332–1335. [Google Scholar] [CrossRef]
- Vallejo, M.; Briz, O.; Serrano, M.A.; Monte, M.J.; Marin, J.J. Potential role of trans-inhibition of the bile salt export pump by progesterone metabolites in the etiopathogenesis of intrahepatic cholestasis of pregnancy. J. Hepatol. 2006, 44, 1150–1157. [Google Scholar] [CrossRef]
- Abu-Hayyeh, S.; Martinez-Becerra, P.; Sheikh Abdul Kadir, S.H.; Selden, C.; Romero, M.R.; Rees, M.; Marschall, H.U.; Marin, J.J.; Williamson, C. Inhibition of Na+-taurocholate co-transporting polypeptide-mediated bile acid transport by cholestatic sulfated progesterone metabolites. J. Biol. Chem. 2010, 285, 16504–16512. [Google Scholar] [CrossRef]
- Pincemail, J.; Vanbelle, S.; Gaspard, U.; Collette, G.; Haleng, J.; Cheramy-Bien, J.P.; Charlier, C.; Chapelle, J.P.; Giet, D.; Albert, A.; et al. Effect of different contraceptive methods on the oxidative stress status in women aged 40 48 years from the ELAN study in the province of Liege, Belgium. Hum. Reprod. 2007, 22, 2335–2343. [Google Scholar] [CrossRef]
- Zhu, R.; Wang, Y.; Zhang, L.; Guo, Q. Oxidative stress and liver disease. Hepatol. Res. 2012, 42, 741–749. [Google Scholar] [CrossRef]
- De Groote, D.; Perrier d’Hauterive, S.; Pintiaux, A.; Balteau, B.; Gerday, C.; Claesen, J.; Foidart, J.M. Effects of oral contraception with ethinylestradiol and drospirenone on oxidative stress in women 18–35 years old. Contraception 2009, 80, 187–193. [Google Scholar] [CrossRef]
- Wan, L.; O’Brien, P. Molecular mechanism of 17alpha-ethinylestradiol cytotoxicity in isolated rat hepatocytes. Can. J. Physiol. Pharmacol. 2014, 92, 21–26. [Google Scholar] [CrossRef]
- Cheng, X.; Shimizu, I.; Yuan, Y.; Wei, M.; Shen, M.; Huang, H.; Urata, M.; Sannomiya, K.; Fukuno, H.; Hashimoto-Tamaoki, T.; et al. Effects of estradiol and progesterone on tumor necrosis factor alpha-induced apoptosis in human hepatoma HuH-7 cells. Life Sci. 2006, 79, 1988–1994. [Google Scholar] [CrossRef]
- Kireev, R.A.; Tresguerres, A.C.; Garcia, C.; Borras, C.; Ariznavarreta, C.; Vara, E.; Vina, J.; Tresguerres, J.A. Hormonal regulation of pro-inflammatory and lipid peroxidation processes in liver of old ovariectomized female rats. Biogerontology 2010, 11, 229–243. [Google Scholar] [CrossRef]
- Shimizu, I.; Ito, S. Protection of estrogens against the progression of chronic liver disease. Hepatol. Res. 2007, 37, 239–247. [Google Scholar] [CrossRef]
- Huang, H.; He, J.; Yuan, Y.; Aoyagi, E.; Takenaka, H.; Itagaki, T.; Sannomiya, K.; Tamaki, K.; Harada, N.; Shono, M.; et al. Opposing effects of estradiol and progesterone on the oxidative stress-induced production of chemokine and proinflammatory cytokines in murine peritoneal macrophages. J. Med. Investig. 2008, 55, 133–141. [Google Scholar] [CrossRef] [Green Version]
- Itagaki, T.; Shimizu, I.; Cheng, X.; Yuan, Y.; Oshio, A.; Tamaki, K.; Fukuno, H.; Honda, H.; Okamura, Y.; Ito, S. Opposing effects of oestradiol and progesterone on intracellular pathways and activation processes in the oxidative stress induced activation of cultured rat hepatic stellate cells. Gut 2005, 54, 1782–1789. [Google Scholar] [CrossRef]
- Köse, K.; Yazici, C. The effect of levonorgestrel and melatonin treatments on plasma oxidant-antioxidant system, and lipid/lipoprotein levels in female rats. Turk. J. Med. Sci. 2000, 30, 523–528. [Google Scholar]
- Sissan, M.A.; Menon, V.P.; Leelamma, S. Effects of low-dose oral contraceptive oestrogen and progestin on lipid peroxidation in rats. J. Int. Med. Res. 1995, 23, 272–278. [Google Scholar] [CrossRef]
- Charni-Natan, M.; Aloni-Grinstein, R.; Osher, E.; Rotter, V. Liver and steroid hormones-can a touch of p53 make a difference? Front. Endocrinol. 2019, 10, 374. [Google Scholar] [CrossRef]
- Czekaj, P.; Nowaczyk-Dura, G.; Ciszkowa, V. Effects of ethinylestradiol-norethisterone acetate combinations on the ultrastructure of liver, kidney, ovary and endometrium cells. Folia Biol. 1998, 44, 207–211. [Google Scholar]
- Fernández-Martínez, E.; Wens-Flores, I.; Moreno, M.G.; Ortiz, M.I.; Muriel, P.; Pérez-Álvarez, V. Short-term effects of thalidomide analogs on hepatic glycogen and nitric oxide in endotoxin-challenged rats. Gen. Physiol. Biophys. 2008, 27, 203–210. [Google Scholar]
- Muriel, P.; Deheza, R. Fibrosis and glycogen stores depletion induced by prolonged biliary obstruction in the rat are ameliorated by metadoxine. Liver Int. 2003, 23, 262–268. [Google Scholar] [CrossRef] [Green Version]
- Carrington, L.J.; Bailey, C.J. Effects of natural and synthetic estrogens and progestins on glycogen deposition in female mice. Horm. Res. 1985, 21, 199–203. [Google Scholar] [CrossRef]
- Mandal, B.; Dey, C.D.; Maity, C.R. Effect of combined ethinyl estradiol and norgestrel oral contraceptives on hepatic functions of albino rats. Acta Physiol. Pharmacol. Bulg. 1985, 11, 68–74. [Google Scholar]
- Marczell, E.; Csak, S. Investigation of the liver glycogen level in rats treated with anticonceptives. Arzneimittel Forschung 1977, 27, 1185–1188. [Google Scholar]
- Hamden, K.; Carreau, S.; Boujbiha, M.A.; Lajmi, S.; Aloulou, D.; Kchaou, D.; Elfeki, A. Hyperglycaemia, stress oxidant, liver dysfunction and histological changes in diabetic male rat pancreas and liver: Protective effect of 17 beta-estradiol. Steroids 2008, 73, 495–501. [Google Scholar] [CrossRef]
- Kulcsar, A.; Kulcsar-Gergely, J.; Weisz, G.; Udvardy, M. Hepatic actions of progestogenic steroids. Arzneimittel Forschung 1984, 34, 1301–1305. [Google Scholar]
- Grossmann, M.; Wierman, M.E.; Angus, P.; Handelsman, D.J. Reproductive endocrinology of non-alcoholic fatty liver disease. Endocr. Rev. 2019, 40, 417–446. [Google Scholar] [CrossRef]
- Breikaa, R.M.; Algandaby, M.M.; El-Demerdash, E.; Abdel-Naim, A.B. Biochanin A protects against acute carbon tetrachloride-induced hepatotoxicity in rats. Biosci. Biotechnol. Biochem. 2013, 77, 909–916. [Google Scholar] [CrossRef]
- Breikaa, R.M.; Algandaby, M.M.; El-Demerdash, E.; Abdel-Naim, A.B. Multimechanistic antifibrotic effect of biochanin a in rats: Implications of proinflammatory and profibrogenic mediators. PLoS ONE 2013, 8, e69276. [Google Scholar] [CrossRef]
- Chaturvedi, S.; Malik, M.Y.; Azmi, L.; Shukla, I.; Naseem, Z.; Rao, C.; Agarwal, N.K. Formononetin and biochanin A protects against ritonavir induced hepatotoxicity via modulation of NfkappaB/pAkt signaling molecules. Life Sci. 2018, 213, 174–182. [Google Scholar]
- Liu, X.; Wang, T.; Liu, X.; Cai, L.; Qi, J.; Zhang, P.; Li, Y. Biochanin A protects lipopolysaccharide/D-galactosamine-induced acute liver injury in mice by activating the Nrf2 pathway and inhibiting NLRP3 inflammasome activation. Int. Immunopharmacol. 2016, 38, 324–331. [Google Scholar] [CrossRef]
- Park, H.S.; Hur, H.J.; Kim, S.H.; Park, S.J.; Hong, M.J.; Sung, M.J.; Kwon, D.Y.; Kim, M.S. Biochanin A improves hepatic steatosis and insulin resistance by regulating the hepatic lipid and glucose metabolic pathways in diet-induced obese mice. Mol. Nutr. Food. Res. 2016, 60, 1944–1955. [Google Scholar] [CrossRef]
- Jing, Y.; Hu, T.; Lin, C.; Xiong, Q.; Liu, F.; Yuan, J.; Zhao, X.; Wang, R. Resveratrol downregulates PCSK9 expression and attenuates steatosis through estrogen receptor alpha-mediated pathway in L02cells. Eur. J. Pharmacol. 2019, 855, 216–226. [Google Scholar] [CrossRef]
- Desquiret-Dumas, V.; Gueguen, N.; Leman, G.; Baron, S.; Nivet-Antoine, V.; Chupin, S.; Chevrollier, A.; Vessieres, E.; Ayer, A.; Ferre, M.; et al. Resveratrol induces a mitochondrial complex I-dependent increase in NADH oxidation responsible for sirtuin activation in liver cells. J. Biol. Chem. 2013, 288, 36662–36675. [Google Scholar] [CrossRef]
- Yin, Y.; Liu, H.; Zheng, Z.; Lu, R.; Jiang, Z. Genistein can ameliorate hepatic inflammatory reaction in nonalcoholic steatohepatitis rats. Biomed. Pharmacother. 2019, 111, 1290–1296. [Google Scholar] [CrossRef]
- Chodon, D.; Ramamurty, N.; Sakthisekaran, D. Preliminary studies on induction of apoptosis by genistein on HepG2 cell line. Toxicol. Vitr. Int. J. Publ. Assoc. BIBRA 2007, 21, 887–891. [Google Scholar] [CrossRef]
- Yeh, T.C.; Chiang, P.C.; Li, T.K.; Hsu, J.L.; Lin, C.J.; Wang, S.W.; Peng, C.Y.; Guh, J.H. Genistein induces apoptosis in human hepatocellular carcinomas via interaction of endoplasmic reticulum stress and mitochondrial insult. Biochem. Pharmacol. 2007, 73, 782–792. [Google Scholar] [CrossRef]
- Lee, S.R.; Kwon, S.W.; Lee, Y.H.; Kaya, P.; Kim, J.M.; Ahn, C.; Jung, E.M.; Lee, G.S.; An, B.S.; Jeung, E.B.; et al. Dietary intake of genistein suppresses hepatocellular carcinoma through AMPK-mediated apoptosis and anti-inflammation. BMC Cancer 2019, 19, 1–12. [Google Scholar] [CrossRef]
- Wang, W.; Chen, J.; Mao, J.; Li, H.; Wang, M.; Zhang, H.; Li, H.; Chen, W. Genistein Ameliorates Non-alcoholic Fatty Liver Disease by Targeting the Thromboxane A2 Pathway. J. Agric. Food. Chem. 2018, 66, 5853–5859. [Google Scholar] [CrossRef]
- Liu, H.H.; Zhong, H.J.; Yin, Y.M.; Jiang, Z.Q. Genistein has beneficial effects on hepatic steatosis in high fat-high sucrose diet-treated rats. Biomed. Pharmacother. 2017, 91, 964–969. [Google Scholar] [CrossRef]
- Ganai, A.A.; Khan, A.A.; Malik, Z.A.; Farooqi, H. Genistein modulates the expression of NF-kappaB and MAPK (p-38 and ERK1/2), thereby attenuating d-Galactosamine induced fulminant hepatic failure in Wistar rats. Toxicol. Appl. Pharmacol. 2015, 283, 139–146. [Google Scholar] [CrossRef]
- Lin, X.; Zhang, S.; Huang, R.; Wei, L.; Liang, C.; Chen, Y.; Lv, S.; Liang, S.; Wu, X.; Huang, Q. Protective effect of genistein on lipopolysaccharide/D-galactosamine- induced hepatic failure in mice. Biol. Pharm. Bull. 2014, 37, 625–632. [Google Scholar] [CrossRef]
- Zhao, L.; Wang, Y.; Liu, J.; Wang, K.; Guo, X.; Ji, B.; Wu, W.; Zhou, F. Protective Effects of Genistein and Puerarin against Chronic Alcohol-Induced Liver Injury in Mice via Antioxidant, Anti-inflammatory, and Anti-apoptotic Mechanisms. J. Agric. Food. Chem. 2016, 64, 7291–7297. [Google Scholar] [CrossRef]
- Demiroren, K.; Dogan, Y.; Kocamaz, H.; Ozercan, I.H.; Ilhan, S.; Ustundag, B.; Bahcecioglu, I.H. Protective effects of L-carnitine, N-acetylcysteine and genistein in an experimental model of liver fibrosis. Clin. Res. Hepatol. Gastroenterol. 2014, 38, 63–72. [Google Scholar] [CrossRef]
- Wang, Q.; Wurtz, P.; Auro, K.; Morin-Papunen, L.; Kangas, A.J.; Soininen, P.; Tiainen, M.; Tynkkynen, T.; Joensuu, A.; Havulinna, A.S.; et al. Effects of hormonal contraception on systemic metabolism: Cross-sectional and longitudinal evidence. Int. J. Epidemiol. 2016, 45, 1445–1457. [Google Scholar] [CrossRef]
- Ae Park, S.; Choi, M.S.; Cho, S.Y.; Seo, J.S.; Jung, U.J.; Kim, M.J.; Sung, M.K.; Park, Y.B.; Lee, M.K. Genistein and daidzein modulate hepatic glucose and lipid regulating enzyme activities in C57BL/KsJ-db/db mice. Life Sci. 2006, 79, 1207–1213. [Google Scholar] [CrossRef]
- Park, H.J.; Jeon, Y.K.; You, D.H.; Nam, M.J. Daidzein causes cytochrome c-mediated apoptosis via the Bcl-2 family in human hepatic cancer cells. Food Chem. Toxicol. 2013, 60, 542–549. [Google Scholar] [CrossRef]
- Berk, K.; Drygalski, K.; Harasim-Symbor, E.; Charytoniuk, T.; Ilowska, N.; Lukaszuk, B.; Chabowski, A.; Konstantynowicz-Nowicka, K. The effect of enterolactone on liver lipid precursors of inflammation. Life Sci. 2019, 221, 341–347. [Google Scholar] [CrossRef]
- Charytoniuk, T.; Ilowska, N.; Berk, K.; Drygalski, K.; Chabowski, A.; Konstantynowicz-Nowicka, K. The effect of enterolactone on sphingolipid pathway and hepatic insulin resistance development in HepG2 cells. Life Sci. 2019, 217, 1–7. [Google Scholar] [CrossRef]
- Konstantynowicz-Nowicka, K.; Harasim, E.; Baranowski, M.; Chabowski, A. New evidence for the role of ceramide in the development of hepatic insulin resistance. PLoS ONE 2015, 10, e0116858. [Google Scholar] [CrossRef]
- Costa, R.; Rodrigues, I.; Guardao, L.; Rocha-Rodrigues, S.; Silva, C.; Magalhaes, J.; Ferreira-de-Almeida, M.; Negrao, R.; Soares, R. Xanthohumol and 8-prenylnaringenin ameliorate diabetic-related metabolic dysfunctions in mice. J. Nutr. Biochem. 2017, 45, 39–47. [Google Scholar] [CrossRef]
- Doddapattar, P.; Radovic, B.; Patankar, J.V.; Obrowsky, S.; Jandl, K.; Nusshold, C.; Kolb, D.; Vujic, N.; Doshi, L.; Chandak, P.G.; et al. Xanthohumol ameliorates atherosclerotic plaque formation, hypercholesterolemia, and hepatic steatosis in ApoE-deficient mice. Mol. Nutr. Food Res. 2013, 57, 1718–1728. [Google Scholar]
- Dorn, C.; Kraus, B.; Motyl, M.; Weiss, T.S.; Gehrig, M.; Scholmerich, J.; Heilmann, J.; Hellerbrand, C. Xanthohumol, a chalcon derived from hops, inhibits hepatic inflammation and fibrosis. Mol. Nutr. Food Res. 2010, 54, S205–S213. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Li, N.; Li, F.; Zhu, Q.; Liu, X.; Han, Q.; Wang, Y.; Chen, Y.; Zeng, X.; Lv, Y.; et al. Xanthohumol, a main prenylated chalcone from hops, reduces liver damage and modulates oxidative reaction and apoptosis in hepatitis C virus infected Tupaia belangeri. Int. Immunopharmacol. 2013, 16, 466–474. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L. Molecular regulation of hepatic fibrosis, an integrated cellular response to tissue injury. J. Biol. Chem. 2000, 275, 2247–2250. [Google Scholar] [CrossRef] [PubMed]
- Prosser, C.C.; Yen, R.D.; Wu, J. Molecular therapy for hepatic injury and fibrosis: Where are we? World J. Gastroenterol. 2006, 12, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.C.; Zhang, Q.B.; Qiao, L. Pathogenesis of liver cirrhosis. World J. Gastroenterol. 2014, 20, 7312–7324. [Google Scholar] [CrossRef] [PubMed]
- Bataller, R.; Brenner, D.A. Liver fibrosis. J. Clin. Investig. 2005, 115, 209–218. [Google Scholar] [CrossRef]
- Hemmann, S.; Graf, J.; Roderfeld, M.; Roeb, E. Expression of MMPs and TIMPs in liver fibrosis—A systematic review with special emphasis on anti-fibrotic strategies. J. Hepatol. 2007, 46, 955–975. [Google Scholar] [CrossRef]
- Iimuro, Y.; Brenner, D.A. Matrix metalloproteinase gene delivery for liver fibrosis. Pharm. Res. 2008, 25, 249–258. [Google Scholar] [CrossRef]
- Zhou, Y.; Shimizu, I.; Lu, G.; Itonaga, M.; Okamura, Y.; Shono, M.; Honda, H.; Inoue, S.; Muramatsu, M.; Ito, S. Hepatic stellate cells contain the functional estrogen receptor beta but not the estrogen receptor alpha in male and female rats. Biochem. Biophys. Res. Commun. 2001, 286, 1059–1065. [Google Scholar] [CrossRef]
- Kang, J.S.; Wanibuchi, H.; Morimura, K.; Puatanachokchai, R.; Salim, E.I.; Hagihara, A.; Seki, S.; Fukushima, S. Enhancement by estradiol 3-benzoate in thioacetamide-induced liver cirrhosis of rats. Toxicol. Sci. 2005, 85, 720–726. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.H.; Li, D.G.; Huang, X.; Zong, C.H.; Xu, Q.F.; Lu, H.M. Suppressive effects of 17beta-estradiol on hepatic fibrosis in CCl4-induced rat model. World J. Gastroenterol. 2004, 10, 1315–1320. [Google Scholar] [CrossRef] [PubMed]
- Lu, G.; Shimizu, I.; Cui, X.; Itonaga, M.; Tamaki, K.; Fukuno, H.; Inoue, H.; Honda, H.; Ito, S. Antioxidant and antiapoptotic activities of idoxifene and estradiol in hepatic fibrosis in rats. Life Sci. 2004, 74, 897–907. [Google Scholar] [CrossRef] [PubMed]
- Sener, G.; Arbak, S.; Kurtaran, P.; Gedik, N.; Yegen, B.C. Estrogen protects the liver and intestines against sepsis-induced injury in rats. J. Surg. Res. 2005, 128, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.W.; Gong, J.; Chang, X.M.; Luo, J.Y.; Dong, L.; Hao, Z.M.; Jia, A.; Xu, G.P. Estrogen reduces CCl4-induced liver fibrosis in rats. World J. Gastroenterol. 2002, 8, 883–887. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, I.; Mizobuchi, Y.; Yasuda, M.; Shiba, M.; Ma, Y.R.; Horie, T.; Liu, F.; Ito, S. Inhibitory effect of oestradiol on activation of rat hepatic stellate cells in vivo and in vitro. Gut 1999, 44, 127–136. [Google Scholar] [CrossRef] [Green Version]
- Codes, L.; Matos, L.; Parana, R. Chronic hepatitis C and fibrosis: Evidences for possible estrogen benefits. Braz. J. Infect. Dis. 2007, 11, 371–374. [Google Scholar] [CrossRef]
- Di Martino, V.; Lebray, P.; Myers, R.P.; Pannier, E.; Paradis, V.; Charlotte, F.; Moussalli, J.; Thabut, D.; Buffet, C.; Poynard, T. Progression of liver fibrosis in women infected with hepatitis C: Long-term benefit of estrogen exposure. Hepatology 2004, 40, 1426–1433. [Google Scholar] [CrossRef]
- Shimizu, I.; Kohno, N.; Tamaki, K.; Shono, M.; Huang, H.W.; He, J.H.; Yao, D.F. Female hepatology: Favorable role of estrogen in chronic liver disease with hepatitis B virus infection. World J. Gastroenterol. 2007, 13, 4295–4305. [Google Scholar] [CrossRef]
- Gantus, M.A.; Alves, L.M.; Stipursky, J.; Souza, E.C.; Teodoro, A.J.; Alves, T.R.; Carvalho, D.P.; Martinez, A.M.; Gomes, F.C.; Nasciutti, L.E. Estradiol modulates TGF-beta1 expression and its signaling pathway in thyroid stromal cells. Mol. Cell. Endocrinol. 2011, 337, 71–79. [Google Scholar] [CrossRef]
- Goto, N.; Hiyoshi, H.; Ito, I.; Tsuchiya, M.; Nakajima, Y.; Yanagisawa, J. Estrogen and antiestrogens alter breast cancer invasiveness by modulating the transforming growth factor-beta signaling pathway. Cancer Sci. 2011, 102, 1501–1508. [Google Scholar] [CrossRef]
- Duarte, S.; Baber, J.; Fujii, T.; Coito, A.J. Matrix metalloproteinases in liver injury, repair and fibrosis. Matrix Biol. 2015, 44–46, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Nunez, O.; Fernandez-Martinez, A.; Majano, P.L.; Apolinario, A.; Gomez-Gonzalo, M.; Benedicto, I.; Lopez-Cabrera, M.; Bosca, L.; Clemente, G.; Garcia-Monzon, C.; et al. Increased intrahepatic cyclooxygenase 2, matrix metalloproteinase 2, and matrix metalloproteinase 9 expression is associated with progressive liver disease in chronic hepatitis C virus infection: Role of viral core and NS5A proteins. Gut 2004, 53, 1665–1672. [Google Scholar] [CrossRef] [PubMed]
- Deng, T.; Liu, J.; Zhang, M.; Wang, Y.; Zhu, G.; Wang, J. Inhibition effect of phytoestrogen calycosin on TGF-beta1-induced hepatic stellate cell activation, proliferation, and migration via estrogen receptor beta. Can. J. Physiol. Pharmacol. 2018, 96, 1268–1275. [Google Scholar] [CrossRef] [PubMed]
- Ganai, A.A.; Husain, M. Genistein attenuates D-GalN induced liver fibrosis/chronic liver damage in rats by blocking the TGF-beta/Smad signaling pathways. Chem. Biol. Interact. 2017, 261, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Liu, C.; Zhou, D.; Zhang, L. TGF-beta/SMAD pathway and its regulation in hepatic fibrosis. J. Histochem. Cytochem. 2016, 64, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Geier, A.; Fickert, P.; Trauner, M. Mechanisms of disease: Mechanisms and clinical implications of cholestasis in sepsis. Nat. Clin. Pract. Gastroenterol. Hepatol. 2006, 3, 574–585. [Google Scholar] [CrossRef]
- Kulcsar-Gergely, J.; Kulcsar, A. Different mechanisms in the hepatic actions of estrogens. Exp. Pathol. 1988, 34, 119–123. [Google Scholar] [CrossRef]
- Fischer, G.M.; Cherian, K.; Swain, M.L. Increased synthesis of aortic collagen and elastin in experimental atherosclerosis. Inhibition by contraceptive steroids. Atherosclerosis 1981, 39, 463–467. [Google Scholar] [CrossRef]
- Hansen, M.; Koskinen, S.O.; Petersen, S.G.; Doessing, S.; Frystyk, J.; Flyvbjerg, A.; Westh, E.; Magnusson, S.P.; Kjaer, M.; Langberg, H. Ethinyl oestradiol administration in women suppresses synthesis of collagen in tendon in response to exercise. J. Physiol. 2008, 586, 3005–3016. [Google Scholar] [CrossRef]
- Wreje, U.; Brynhildsen, J.; Aberg, H.; Bystrom, B.; Hammar, M.; von Schoultz, B. Collagen metabolism markers as a reflection of bone and soft tissue turnover during the menstrual cycle and oral contraceptive use. Contraception 2000, 61, 265–270. [Google Scholar] [CrossRef]
- Oza, M.J.; Kulkarni, Y.A. Biochanin A improves insulin sensitivity and controls hyperglycemia in type 2 diabetes. Biomed. Pharmacother. 2018, 107, 1119–1127. [Google Scholar] [CrossRef]
- Ganai, A.A.; Farooqi, H. Bioactivity of genistein: A review of in vitro and in vivo studies. Biomed. Pharmacother. 2015, 76, 30–38. [Google Scholar] [CrossRef]
- Seo, D.B.; Jeong, H.W.; Lee, S.J.; Lee, S.J. Coumestrol Induces Mitochondrial Biogenesis by Activating Sirt1 in Cultured Skeletal Muscle Cells. J. Agric. Food Chem. 2014, 62, 4298–4305. [Google Scholar] [CrossRef]
- Canto, C.; Gerhart-Hines, Z.; Feige, J.N.; Lagouge, M.; Noriega, L.; Milne, J.C.; Elliott, P.J.; Puigserver, P.; Auwerx, J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 2009, 458, 1056–1060. [Google Scholar] [CrossRef]
- Yasuda, M.; Shimizu, I.; Shiba, M.; Ito, S. Suppressive effects of estradiol on dimethylnitrosamine-induced fibrosis of the liver in rats. Hepatology 1999, 29, 719–727. [Google Scholar] [CrossRef]
- Codes, L.; Asselah, T.; Cazals-Hatem, D.; Tubach, F.; Vidaud, D.; Parana, R.; Bedossa, P.; Valla, D.; Marcellin, P. Liver fibrosis in women with chronic hepatitis C: Evidence for the negative role of the menopause and steatosis and the potential benefit of hormone replacement therapy. Gut 2007, 56, 390–395. [Google Scholar] [CrossRef]
- Villa, E.; Vukotic, R.; Camma, C.; Petta, S.; Di Leo, A.; Gitto, S.; Turola, E.; Karampatou, A.; Losi, L.; Bernabucci, V.; et al. Reproductive status is associated with the severity of fibrosis in women with hepatitis C. PLoS ONE 2012, 7, e44624. [Google Scholar] [CrossRef]
- Sarkar, M.; Dodge, J.L.; Greenblatt, R.M.; Kuniholm, M.H.; DeHovitz, J.; Plankey, M.; Kovacs, A.; French, A.L.; Seaberg, E.C.; Ofotokun, I.; et al. Reproductive aging and hepatic fibrosis progression in human immunodeficiency virus/hepatitis C virus-coinfected women. Clin. Infect. Dis. 2017, 65, 1695–1702. [Google Scholar] [CrossRef]
- Leite, A.G.; Duarte, M.I.; Mendes-Correa, M.C. Fibrosis progression in paired liver biopsies from HIV/HCV-coinfected patients without prior treatment of hepatitis C. J. Int. Assoc. Provid. Aids Care 2015, 14, 463–468. [Google Scholar] [CrossRef]
- Yang, J.D.; Abdelmalek, M.F.; Pang, H.; Guy, C.D.; Smith, A.D.; Diehl, A.M.; Suzuki, A. Gender and menopause impact severity of fibrosis among patients with nonalcoholic steatohepatitis. Hepatology 2014, 59, 1406–1414. [Google Scholar] [CrossRef]
- Klair, J.S.; Yang, J.D.; Abdelmalek, M.F.; Guy, C.D.; Gill, R.M.; Yates, K.; Unalp-Arida, A.; Lavine, J.E.; Clark, J.M.; Diehl, A.M.; et al. Nonalcoholic Steatohepatitis Clinical Research, N. A longer duration of estrogen deficiency increases fibrosis risk among postmenopausal women with nonalcoholic fatty liver disease. Hepatology 2016, 64, 85–91. [Google Scholar] [CrossRef]
- Mizobuchi, Y.; Shimizu, I.; Yasuda, M.; Hori, H.; Shono, M.; Ito, S. Retinyl palmitate reduces hepatic fibrosis in rats induced by dimethylnitrosamine or pig serum. J. Hepatol. 1998, 29, 933–943. [Google Scholar] [CrossRef]
- Seifert, W.F.; Bosma, A.; Brouwer, A.; Hendriks, H.F.; Roholl, P.J.; van Leeuwen, R.E.; van Thiel-de Ruiter, G.C.; Seifert-Bock, I.; Knook, D.L. Vitamin A deficiency potentiates carbon tetrachloride-induced liver fibrosis in rats. Hepatology 1994, 19, 193–201. [Google Scholar] [CrossRef]
- Okuyama, R.; Abo, T.; Seki, S.; Ohteki, T.; Sugiura, K.; Kusumi, A.; Kumagai, K. Estrogen administration activates extrathymic T cell differentiation in the liver. J. Exp. Med. 1992, 175, 661–669. [Google Scholar] [CrossRef]
- Fox, H.S.; Bond, B.L.; Parslow, T.G. Estrogen regulates the IFN-gamma promoter. J. Immunol. 1991, 146, 4362–4367. [Google Scholar]
- Escobar, E.; Shimizu, I.; Yasuda, M. Inhibition of hepatofibrogenesis by estradiol. Gastroenterology 1997, 112, A1261. [Google Scholar]
- Omoya, T.; Shimizu, I.; Zhou, Y.; Okamura, Y.; Inoue, H.; Lu, G.; Itonaga, M.; Honda, H.; Nomura, M.; Ito, S. Effects of idoxifene and estradiol on NF-kappaB activation in cultured rat hepatocytes undergoing oxidative stress. Liver 2001, 21, 183–191. [Google Scholar] [CrossRef]
- Desmet, V.J.; van Eyken, P.; Roskams, T. Histopathology of vanishing bile duct diseases. Adv. Clin. Path. 1998, 2, 87–99. [Google Scholar]
- Boyer, J.L. Vanishing Bile Duct Syndrome—From Bench to Bed Side; Kluwer Academic Publishers: Dordrecht, Germany, 1997; pp. 240–246. [Google Scholar]
- Alvaro, D.; Alpini, G.; Onori, P.; Perego, L.; Svegliata Baroni, G.; Franchitto, A.; Baiocchi, L.; Glaser, S.S.; Le Sage, G.; Folli, F.; et al. Estrogens stimulate proliferation of intrahepatic biliary epithelium in rats. Gastroenterology 2000, 119, 1681–1691. [Google Scholar] [CrossRef]
- Kilbourne, E.J.; Scicchitano, M.S. The activation of plasminogen activator inhibitor-1 expression by IL-1beta is attenuated by estrogen in hepatoblastoma HepG2 cells expressing estrogen receptor alpha. Thromb. Haemost. 1999, 81, 423–427. [Google Scholar]
- Rachon, D.; Mysliwska, J.; Suchecka-Rachon, K.; Wieckiewicz, J.; Mysliwski, A. Effects of oestrogen deprivation on interleukin-6 production by peripheral blood mononuclear cells of postmenopausal women. J. Endocrinol. 2002, 172, 387–395. [Google Scholar] [CrossRef] [Green Version]
- Marchesini, G.; Bugianesi, E.; Forlani, G.; Cerrelli, F.; Lenzi, M.; Manini, R.; Natale, S.; Vanni, E.; Villanova, N.; Melchionda, N.; et al. Nonalcoholic fatty liver, steatohepatitis, and the metabolic syndrome. Hepatology 2003, 37, 917–923. [Google Scholar] [CrossRef]
- Loria, P.; Carulli, L.; Bertolotti, M.; Lonardo, A. Endocrine and liver interaction: The role of endocrine pathways in NASH. Nat. Rev. Gastroenterol. Hepatol. 2009, 6, 236–247. [Google Scholar] [CrossRef]
- Lonardo, A.; Carani, C.; Carulli, N.; Loria, P. ‘Endocrine NAFLD’ a hormonocentric perspective of nonalcoholic fatty liver disease pathogenesis. J. Hepatol. 2006, 44, 1196–1207. [Google Scholar] [CrossRef]
- Bubnov, R.V.; Drahulian, M.V.; Buchek, P.V.; Gulko, T.P. High regenerative capacity of the liver and irreversible injury of male reproductive system in carbon tetrachloride-induced liver fibrosis rat model. EPMA J. 2018, 9, 59–75. [Google Scholar] [CrossRef]
- Shimizu, I.; Inoue, H.; Yano, M.; Shinomiya, H.; Wada, S.; Tsuji, Y.; Tsutsui, A.; Okamura, S.; Shibata, H.; Ito, S. Estrogen receptor levels and lipid peroxidation in hepatocellular carcinoma with hepatitis C virus infection. Liver 2001, 21, 342–349. [Google Scholar] [CrossRef]
- Wanless, I.R.; Medline, A. Role of estrogens as promoters of hepatic neoplasia. Lab. Investig. 1982, 46, 313–320. [Google Scholar]
- Fisher, B.; Gunduz, N.; Saffer, E.A.; Zheng, S. Relation of estrogen and its receptor to rat liver growth and regeneration. Cancer Res. 1984, 44, 2410–2415. [Google Scholar]
- Francavilla, A.; Di Leo, A.; Eagon, P.K.; Wu, S.Q.; Ove, P.; Van Thiel, D.H.; Starzl, T.E. Regenerating rat liver: Correlations between estrogen receptor localization and deoxyribonucleic acid synthesis. Gastroenterology 1984, 86, 552–557. [Google Scholar]
- Francavilla, A.; Ove, P.; Polimeno, L.; Sciascia, C.; Coetzee, M.L.; Starzl, T.E. Epidermal growth factor and proliferation in rat hepatocytes in primary culture isolated at different times after partial hepatectomy. Cancer Res. 1986, 46, 1318–1323. [Google Scholar]
- Lingham, R.B.; Stancel, G.M.; Loose-Mitchell, D.S. Estrogen regulation of epidermal growth factor receptor messenger ribonucleic acid. Mol. Endocrinol. 1988, 2, 230–235. [Google Scholar] [CrossRef]
- Lee, C.H.; Edwards, A.M. Stimulation of DNA synthesis and c-fos mRNA expression in primary rat hepatocytes by estrogens. Carcinogenesis 2001, 22, 1473–1481. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, I.; Yasuda, M.; Mizobuchi, Y.; Ma, Y.R.; Liu, F.; Shiba, M.; Horie, T.; Ito, S. Suppressive effect of oestradiol on chemical hepatocarcinogenesis in rats. Gut 1998, 42, 112–119. [Google Scholar] [CrossRef] [Green Version]
- McMahon, B.J.; Alberts, S.R.; Wainwright, R.B.; Bulkow, L.; Lanier, A.P. Hepatitis B-related sequelae. prospective study in 1400 hepatitis B surface antigen-positive Alaska native carriers. Arch. Intern. Med. 1990, 150, 1051–1054. [Google Scholar] [CrossRef]
- Poynard, T.; Ratziu, V.; Charlotte, F.; Goodman, Z.; McHutchison, J.; Albrecht, J. Rates and risk factors of liver fibrosis progression in patients with chronic hepatitis C. J. Hepatol. 2001, 34, 730–739. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Compound | Effect | Immunomodulation and Biochemical Mechanisms | Doses/Concentration | Ref. |
|---|---|---|---|---|
| Estrogens and combined OC (a) Deleterious effects: | ||||
| OC | Cholestasis. Oxidative stress (mitochondrial, and endoplasmic reticulum) in endotoxin-induced liver injury in rodents | Activation of Kupffer cells and production of TNF-α, IL-6, and IL-1β | Female Sprague-Dawley rats: 20 mg/kg Female Wistar rats: 17 alpha-Ethinylestradiol (EE) 35 ng/kg and Norethindrone 2 μ/kg | [111,112,113,114,198] |
| E2 | Enhances cirrhosis induced by thioacetamide in rats | Accumulation of hepatic collagen, LP level, collagen amount, and cirrhosis degree | F344 rats: 100 µg/kg | [24,98,182] |
| (b) Beneficial effects: | ||||
| E2 | Reduces fibrosis in various liver injury models in ovariectomized rats | Decreases TGF-β production, collagen synthesis, and oxidative stress, as well as the MAPK pathways | Female Sprague Dawley: 20 μg/kg/day | [183,186] |
| OC | Reduces liver fibrosis in vitro, female rats or in women | Inhibits collagen synthesis, despite elevated plasma and hepatic TGF-β levels. Inactivates the downstream transcription cascade of TGF-β1 expression and HSC activation. | Wistar rats: 200 μ/kg | [12,135,142,143,165,187,199,200,201,202] |
| High cardiometabolic risk during intake (reversible) | Increase of hepatic glycogen stores in normal rats | Norgestrel and 20 µ/kg EE | ||
| 5841 women (age range 24–49 years) from three population-based cohorts. Women using OC or POCs. Metabolomic profiles were reassessed for 869 women after six years | Increases triglycerides, HDL, Apo C and A-I, insulin, PCR, and SHBG; decreases testosterone; changes fatty acids (decreased ω-6 and increased monounsaturated and saturated) and amino acids (increased Phe and decreased Tyr levels); reduces albumin levels but increases creatinine, glycoprotein acetyls, growth factors (SCGFβ, βNGF, SCF, VEGF, FGF, PDGF-BB), and IL-2rα, IL-12p70, and IL-17. Cytokines IL1β, IL-6, and TNF-α displayed weak and non-significant associations | 30–40 μg EE | ||
| OC, E2 and EE | Hepatoprotective in normal and in diabetic models in mice | Increase of the hepatic glycogen stores in normal rats, lowering the plasma glucose | Female albino mice: 5 µg/kg of 17 beta βE2, 5 µ/kg of EE, 1 mg/kg of P4, 1 mg/kg of norethisterone acetate | [142,144,199] |
| EE and DRSP | Continuous administration of EE and DRSP leads to hyperinsulinemia in female mice | Augmentation of glucose-induced insulin secretion, decreased insulin clearance, and reduced IRβ protein expression in the liver | / | / |
| (c) Bidirectional effects: | ||||
| OC (EE and norgestrel) | Chronic cholestasis in Wistar rats | High LP levels, cytokine imbalance, increased plasma TNF-α and IL-10 in the liver, as well as TGF-β | Wistar rats: 100, 200 μg/kg norgestrel, 10, 20 μg/kg EE/14 and 28 days | [12,127,129,133] |
| 209 Women aged 40–48 years who used OC, compared to non-users and intrauterine users of hormonal and copper devices | Augmentation of LP, antioxidants in plasma were diminished, LDLs elevated while β-carotene and zinc were lowered | Combined OC: EE (0.020–0.035 mg + Progestine (0.075–3.0 mg). IUD (LNG 0.02 mg) | ||
| 32 women 18–35 years old who consumed the OC combination of EE and drospirenone | Combinations of OC inhibit liver PON activity, whilst serum PON activity is augmented; CAT activity in erythrocytes was reduced by all OCs tried. Estrogens (primarily E2) possess antioxidant effects | 0.03 mg of EE and 3 mg drospirenone | ||
| OC in mice | Diminishing ROS, activation of NF-κB and AP-1, pro-inflammatory cytokines. Up-regulates antioxidant enzymes and expression of the Bcl-2 family proteins. | 0.15 mg Desogestrel + 0.03 mg EE; 0.15 mg LNG + 0.03 mg EE and 0.15 mg desogestrel+0.02 mg EE / 21 days each drug | ||
| Diminished LP and TNF-α-induced apoptosis. | ||||
| Progestins (a) Deleterious effects: | ||||
| P4 and its metabolites | Intrahepatic cholestasis of pregnancy in isolated perfused rat liver | High quantities of PM4, PM4-Sul, and epiallopregnanolone-sulfate (PM5-Sul) in the urine of IPC patients has been correlated with failure of biliary canalicular transporters (BSEP, MRP-2, and MRP-3) | 3 µmol to the recirculation media used to in situ perfuse the rat liver | [125,126] |
| P4 and norgestrel | Prooxidant and pro-inflammatory effects in murine | Oxidative stress and LP, production of TNF-α, IL-1β, MIP-2, and MCP-1 | / | [134,135] |
| peritoneal macrophages and cultured rat hepatic stellate cells | Generation of ROS, TGF-β in activated HSC | |||
| P4 | Mononuclear cells from patients with chronic hepatitis C | Production of TNF-α, IL-1β, IL-8, and macrophage chemotactic protein (MCP)-1 | 10−7 mol/L | [94] |
| (b) Beneficial effects: | ||||
| MPA | Anti-inflammatory effects in epithelial cells. | Inhibits the TNF-α-induced matrix metalloproteinase (MMP)-9 via the glucocorticoid receptor | 10−6 M/ 72 h. | [92] |
| Phytoestrogens (a) Deleterious effects: | ||||
| Miroestrol and deoxymiroestrol | Intrahepatic cholestasis in C57BL/6 mice. | Regulates the transporter expression (BSEP and MRP2 mRNA in both male and female mice), though the immunomodulatory effects is not known. | 0.5 mg/kg/day once a day for 7 days | [124] |
| (b) Beneficial effects: | ||||
| Resveratrol and ginsenosides | Prophylactic against EE-induced liver cholestasis | Reduced marker levels of cholestasis, oxidative stress, as well as of the pro-inflammatory cytokines TNF-α, IL-6, and IL-1β | 25 mg/kg/15 days of resveratrol and 30–300 mg/kg of gingenosides, i.g. /5 days | [123] |
| Resveratrol | Protects against atherosclerosis and hepatic steatosis in vitro and in vivo | Down-regulation of SREBP-1c expression through the ERα-mediated pathway in L02cells. Induces a mitochondrial complex I-dependent increase in NADH oxidation resulting in sirtuin activation in HepG2 cells and in mice | In LO2 cells: 10–20 µM HepG2 cells: 1–5 µM. C57BL6/N: 50 mg/kg/day | [153,154] |
| Biochanin A | Protects against acute CCl4-induced hepatotoxicity in Wistar rats | Protects from oxidative stress measured by LPO, GSH, SOD, total antioxidant total, catalase activities, and inhibition of iNOS, COX2, and CD45 expression | Wistar: 25–1600 mg/kg; and 50 mg/kg | [149,151,152,203] |
| Antifibrotic effect in rats. | Decreased the expressions of NF-κB, TGF-β1, MMP9, TNFα | Male Sprague-Dawley with diabetes: 10–40 mg/kg/28 days | ||
| Improves insulin sensitivity, hepatic steatosis, and controls hyperglycemia in type 2 diabetes and obesity models | Antifibrotic effects by decreasing the expressions of NF-κB, TGF-β1, MMP9, and TNFα. | |||
| BCA protected against LPS/GalN-induced acute liver injury in mice | Activating the Nrf2 pathway and inhibiting NLRP3 inflammasome activation | |||
| Improve type 2 diabetes induced in rats | BCA improves insulin sensitivity and increases the expression of SIRT1 histone deacetylase in pancreatic tissue in induced type 2 diabetes | In C57BL/6 mice with obesity: 0.05%/12 weeks (wk) | ||
| Beneficial effects on obesity-mediated hepatic steatosis and insulin resistance of obese mice | BCA increases the expression of PPAR-α and its regulatory proteins in the liver. | |||
| Formononetin and biochanin A | Protection against ritonavir induced hepatotoxicity in adult male Sprague–Dawley rats | Hepatoprotection via modulation of oxidative stress, inflammation, and apoptosis: NFkB/pAkt signaling molecules, caspase-3, NFκB, and eNOS activation | 100 mg/kg, p.o. | [150] |
| Genistein | Inhibition of HCC in mice (C57BL/6 N) treated with DEN at 2 weeks of age and fed with supplemental of genistein | Increase of phospho-AMPK in the liver, Hep3B, and Raw 264.7 cells. Inhibition of NF-κB levels, and down-regulation of TNF and IL-6 | C57BL/6 N: 80 mg/kg/day, for 5 months, from 40 to 62 wk of age | [158,159,160,162,163,196,197,204] |
| Ameliorate NAFLD in C57BL/6 mice, Hep-G2 cells | Inhibition of COX-1 activity as well as its downstream TXA2 biosynthesis | C57BL/6 mice: 1.5–64 mg/kg for 22 wk Hep-G2: 100 μM | ||
| Improved NAFDL in high-fat/high-sucrose diet-treated Sprague–Dawley rats. | In steatosis hepatic via AMPK, thus promoting fatty acid oxidation and inhibiting hepatic lipid synthesis (mRNA levels of FAS and GPAT were lower, but PPARα, CPT-1, and ACO were higher in rats treated with genistein) | Sprague-Dawley rats: 4–8 mg/kg body weight | ||
| Hepatoprotective and anti-fibrotic effects in D-galactosamine (D-GalN)-induced fulminant hepatic failure in Wistar rats | Decreased AST and ALT and increased iNOS, COX-2, NO, and PGE. Suppression of TNF-α, IL-1β, NF-κB, IKKα/β, and MAPK phosphorylation | 5 mg/kg BW/day/30 days, i.g. | ||
| Alleviates hepatic damage induced by chronic alcohol in mice | Decreasing levels of MDA, TNF-α, IL-6, ALT, and LDL. | / | ||
| Inhibition of iNOS, TNF-α, NF-ĸB, and caspases-3 | 0.3 mmol/kg with 50% alcohol once per day for 5 weeks. | |||
| Attenuates DGalN-induced liver fibrosis/chronic liver damage in rats | Hepatoprotection by modulating the NF-κB/MAPK pathways and chronic damage by the Smad7-induced inhibition of TGF-β/Smad2/3 | 5 mg/kg BW i.g./12 wk | ||
| Daidzein | Modulate hepatic glucose and lipid-regulating enzyme activities in C57BL/KsJ-db/db mice | Decrease in blood glucose and HbA(1c) levels, increased the insulin/glucagon ratio in the type 2 diabetic animals | C57BL/KsJ-db/db mice: 0.02% w/w | [166,167] |
| Potent inducer of apoptosis in hepatic cancer cells (SK-HEP-1) | Apoptosis associated with the up-regulation of Bak and down-regulation of Bcl-2 and Bcl-xL proteins, caspases 3 and 9 in SK-HEP-1 cells | 200 μM, 400 μM, or 600 μM | ||
| Coumestrol | Increase in mitochondria number and function in cultured skeletal muscle cells (C2C12). | Activation of SIRT1, ATP levels, glucose uptake, and the protein expression of respiratory chain components. Stimulation of mitochondrial biogenesis. | 5–10 μM | [205,206] |
| Xanthohumol (XN) and 8-prenylnaringenin (8PN) | Ameliorated diabetic-related metabolic dysfunctions in C57Bl/6 mice during 20 weeks. | Promote hepatic and skeletal muscle AMP-activated protein kinase (AMPK), diminishing the expression of target lipogenic enzymes: SREBP-1c, FAS, and acetyl-CoA carboxylase activity. Moreover, both XN and 8PN treatments decreased the VEGFR-1/VEGFB pathway. | C57Bl/6 mice: 10 mg/L of XN and 8PN. | [171,172,173,174] |
| In apolipoprotein-E-deficient (ApoE−/−) mice fed a Western-type diet reducing hepatic lipogenesis. | Induction of SREBP-1c mRNA and CPT-1a or increased fatty acid beta-oxidation. | Female ApoE−/− mice: 300 mg/kg body weight/day for 8 wk | ||
| In vitro: platelet aggregation. | Inhibition of NF-κB, TGF-β1 and IL8 | 1.5 And 3 μM in platelet assay | ||
| In female BALB/c mice with induced NASH | Prevent body weight gain; decreased glycemia, triglycerides, cholesterol, and alkaline phosphatase levels; improved insulin sensitivity in mice, thus suppressing lipogenesis | Female BALB/c: Diet with 1% XN w/w for 3 wk | ||
| Primary human hepatocytes (PHHs) | XN attenuates atherosclerosis | PHH: 25 and 50 μM | ||
| Calycosin (O-methylated isoflavone) | Anti-fibrotic activity in activated HSCs | Inhibition effect on expression of migration, proliferation, activation, and migration of HSC induced by TGF-β1 | 182–780 μM | [195] |
| (b) Bidirectional effects: | ||||
| Enterolactone | Development of hepatic insulin resistance and enhanced apoptosis in HepG2 cells | Increase in COX-2 and TNFα protein expressions, resistance of insulin and apoptosis (caspase 3), and stimulation of the de novo ceramide synthesis pathway | 50 μM of enteronolactone and with palmitic acid 0.5 mM | [169] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soria-Jasso, L.E.; Cariño-Cortés, R.; Muñoz-Pérez, V.M.; Pérez-Hernández, E.; Pérez-Hernández, N.; Fernández-Martínez, E. Beneficial and Deleterious Effects of Female Sex Hormones, Oral Contraceptives, and Phytoestrogens by Immunomodulation on the Liver. Int. J. Mol. Sci. 2019, 20, 4694. https://doi.org/10.3390/ijms20194694
Soria-Jasso LE, Cariño-Cortés R, Muñoz-Pérez VM, Pérez-Hernández E, Pérez-Hernández N, Fernández-Martínez E. Beneficial and Deleterious Effects of Female Sex Hormones, Oral Contraceptives, and Phytoestrogens by Immunomodulation on the Liver. International Journal of Molecular Sciences. 2019; 20(19):4694. https://doi.org/10.3390/ijms20194694
Chicago/Turabian StyleSoria-Jasso, Luis E., Raquel Cariño-Cortés, Víctor Manuel Muñoz-Pérez, Elizabeth Pérez-Hernández, Nury Pérez-Hernández, and Eduardo Fernández-Martínez. 2019. "Beneficial and Deleterious Effects of Female Sex Hormones, Oral Contraceptives, and Phytoestrogens by Immunomodulation on the Liver" International Journal of Molecular Sciences 20, no. 19: 4694. https://doi.org/10.3390/ijms20194694
APA StyleSoria-Jasso, L. E., Cariño-Cortés, R., Muñoz-Pérez, V. M., Pérez-Hernández, E., Pérez-Hernández, N., & Fernández-Martínez, E. (2019). Beneficial and Deleterious Effects of Female Sex Hormones, Oral Contraceptives, and Phytoestrogens by Immunomodulation on the Liver. International Journal of Molecular Sciences, 20(19), 4694. https://doi.org/10.3390/ijms20194694