CircRNA Expression Pattern and ceRNA and miRNA–mRNA Networks Involved in Anther Development in the CMS Line of Brassica campestris

 , ,
, ,
Abstract
:1. Introduction
2. Results
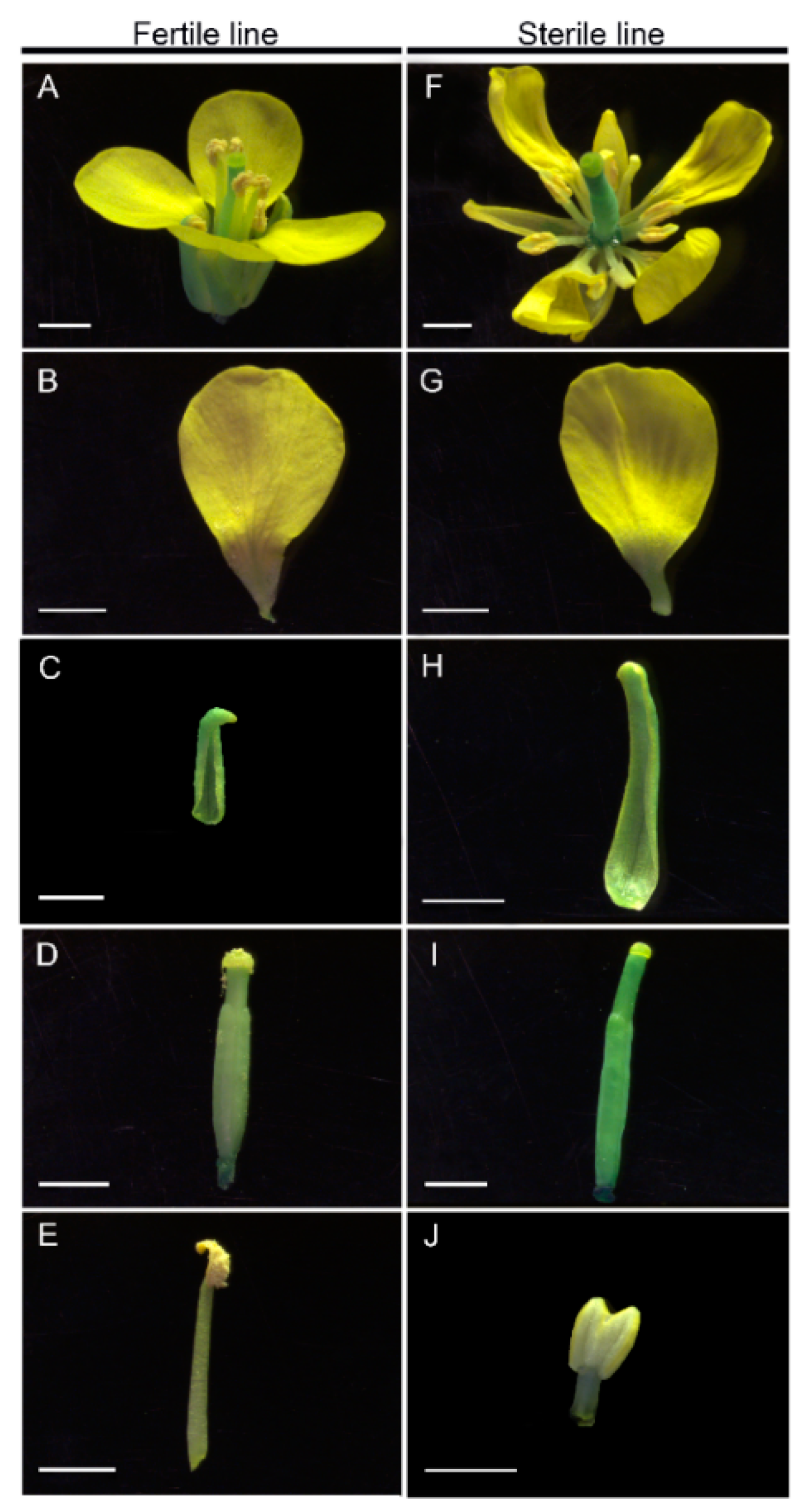
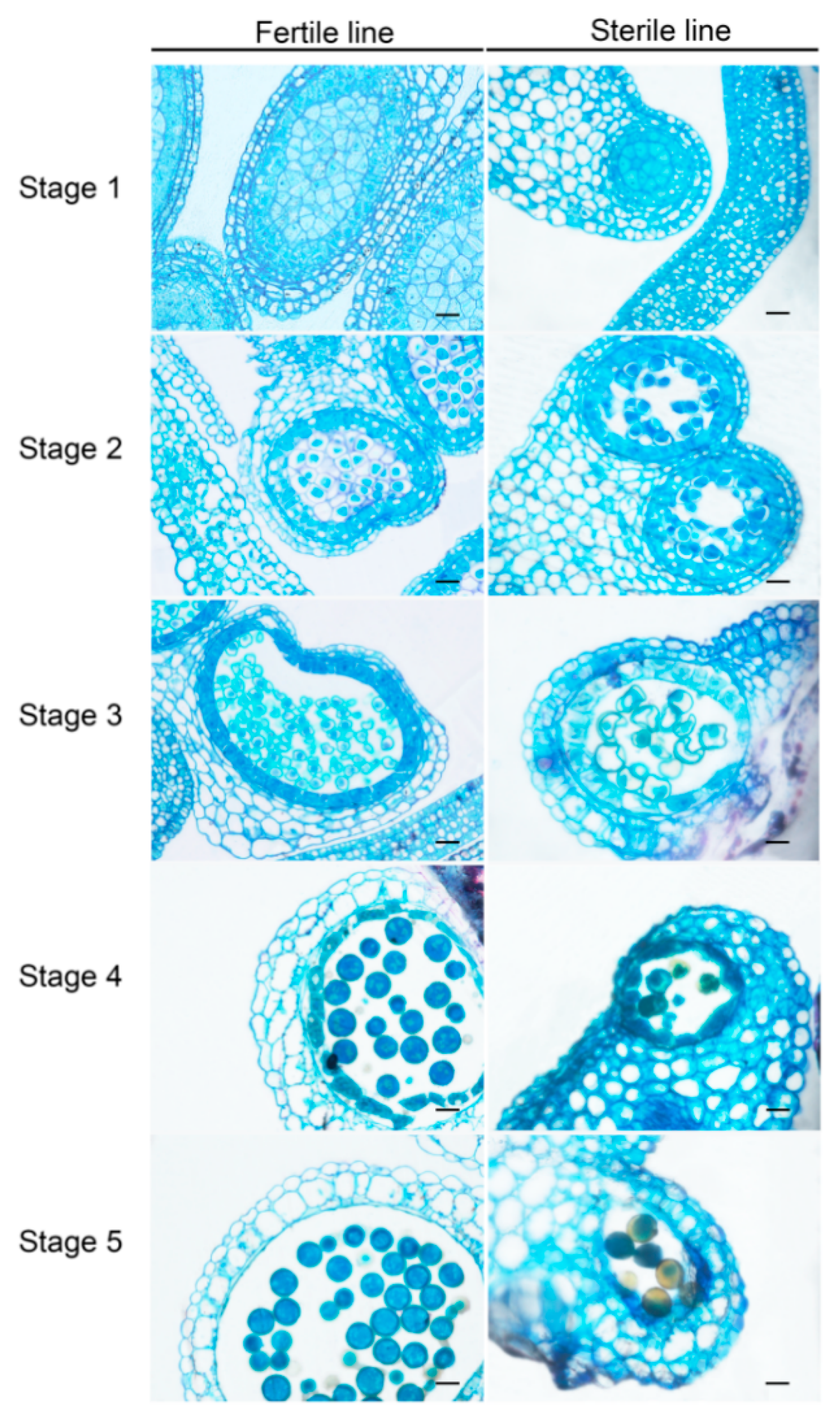
2.1. Establishment of the CMS Line in Brassica Campestris
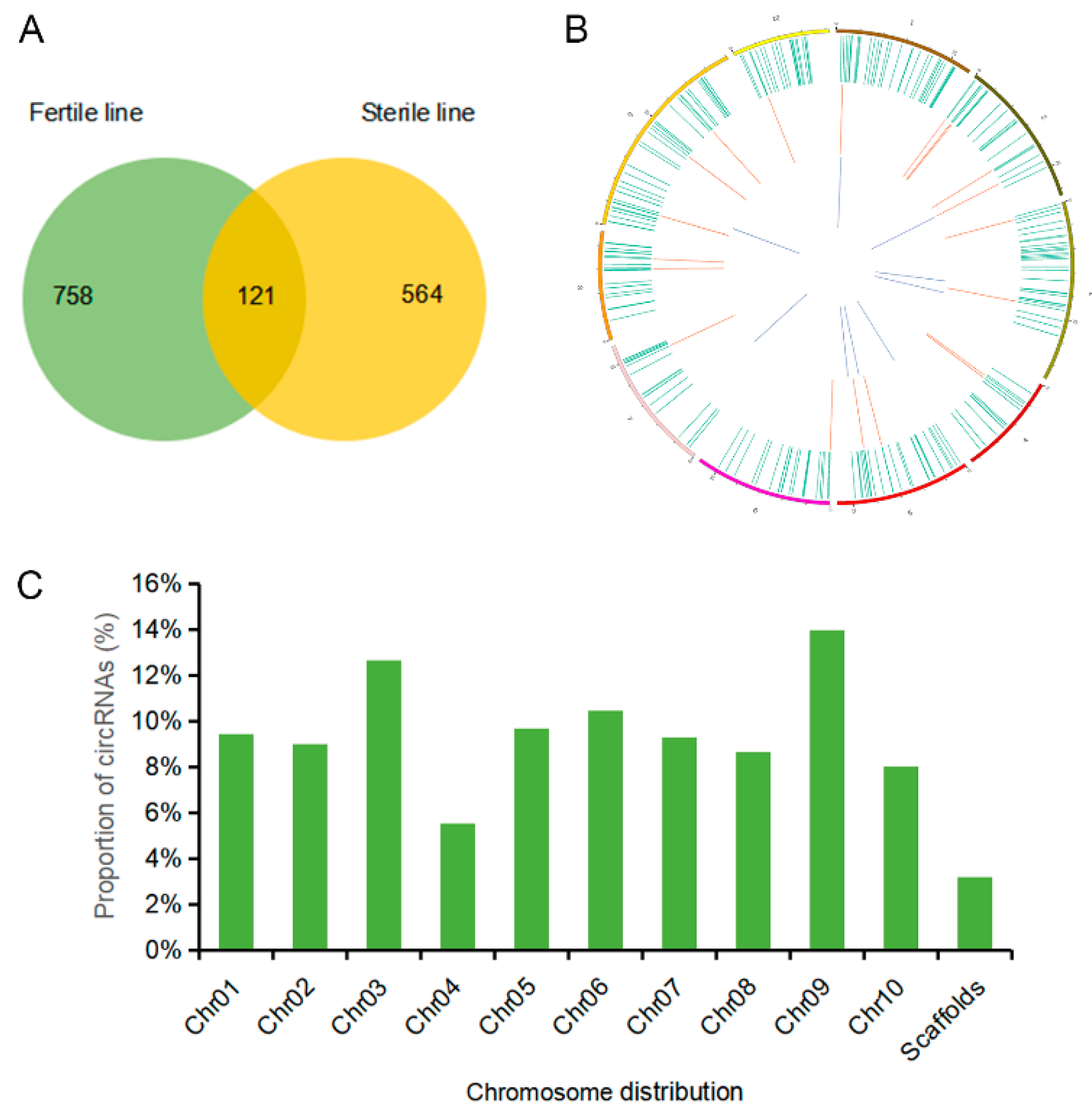
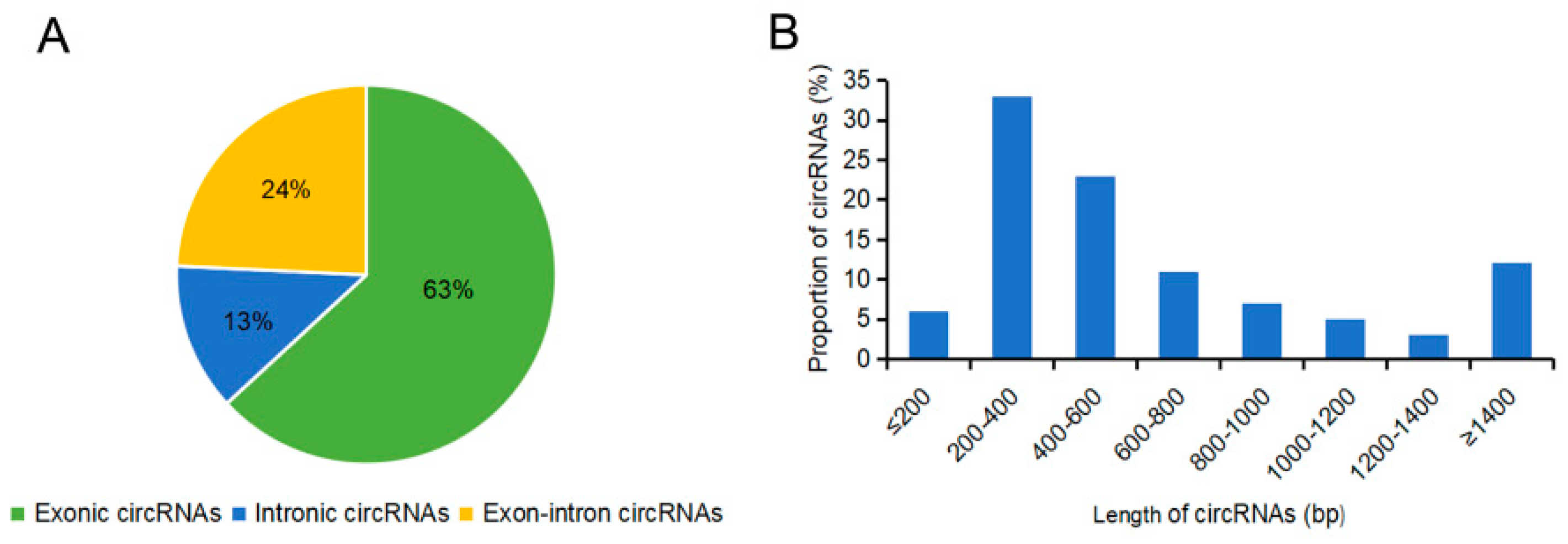
2.2. Identification of circRNAs in the Sterile and Fertile Lines
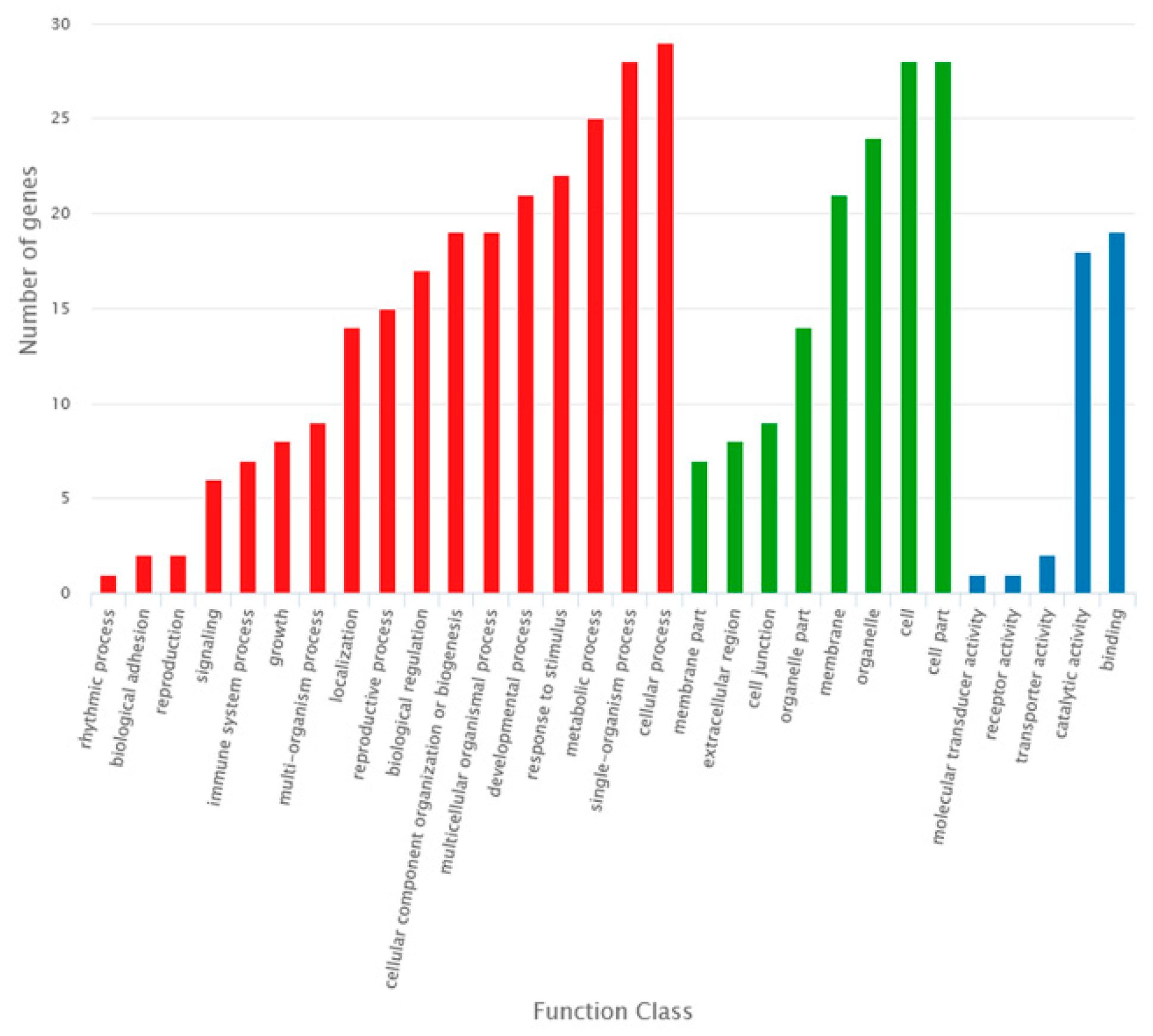
2.3. Identification of DE RNAs in the Sterile Line Compared to the Fertile Line
2.4. Construction of the DEmiRNA–DEmRNA Network
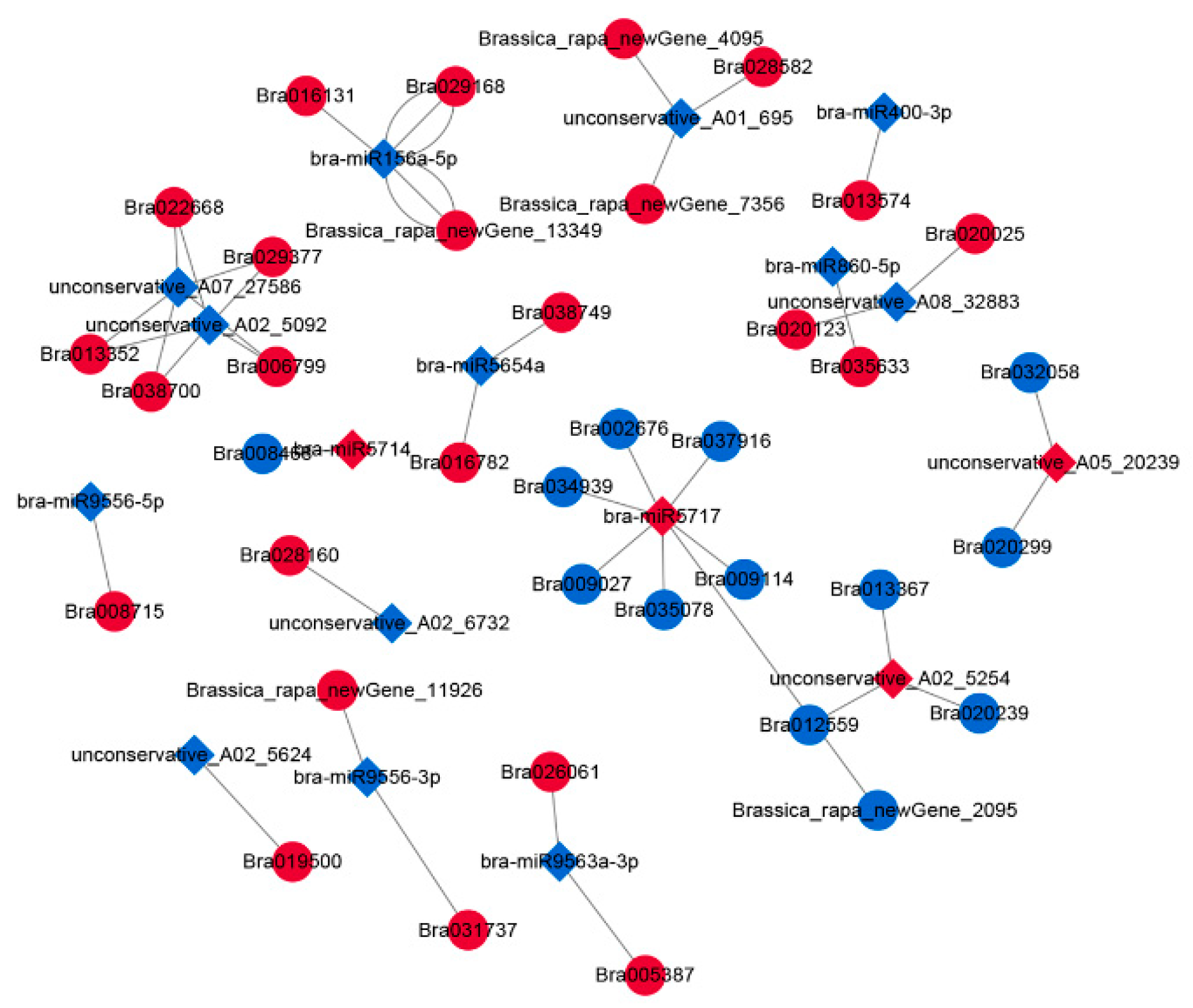
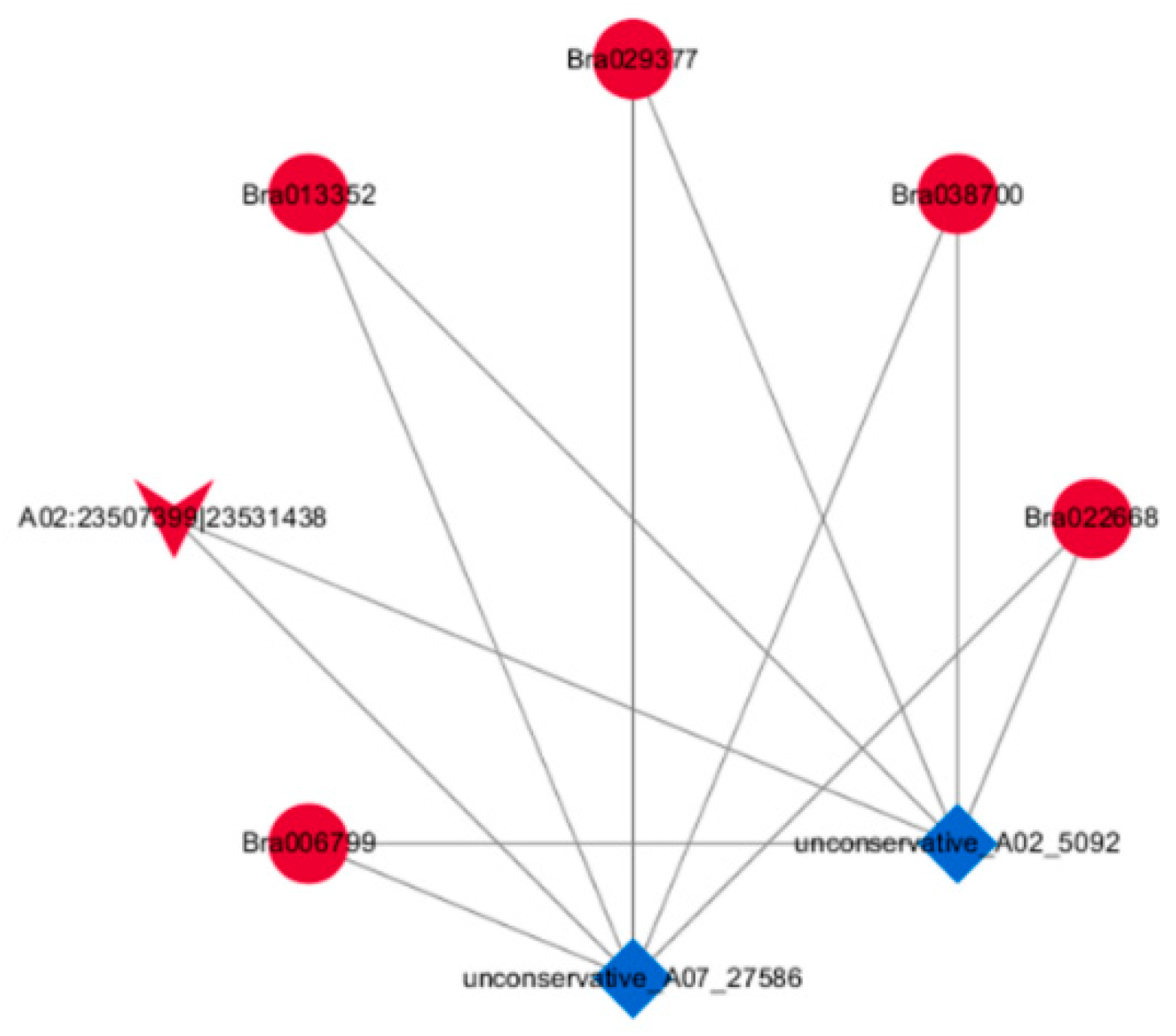
2.5. Construction of the DEcircRNA–DEmiRNA–DEmRNA Network
3. Discussion
3.1. Whole-Transcriptome Sequencing Enriches Understanding of Mechanisms of Anther Development and Male Sterility
3.2. miRNAs Contribute to Pollen Wall Development
3.3. CeRNA Networks Could Provide New Sights into the Regulatory Roles of ncRNAs during Anther Development
4. Materials and Methods
4.1. Plant Materials and Sample Collection
4.2. Morphological and Cytological Observation
4.3. Total RNA Extraction and Detection
4.4. RNA Library Construction and Sequencing
4.5. Sequencing Quality Control and Biological Analysis
4.6. Identifcation of RNAs
4.7. Identification of DE RNAs
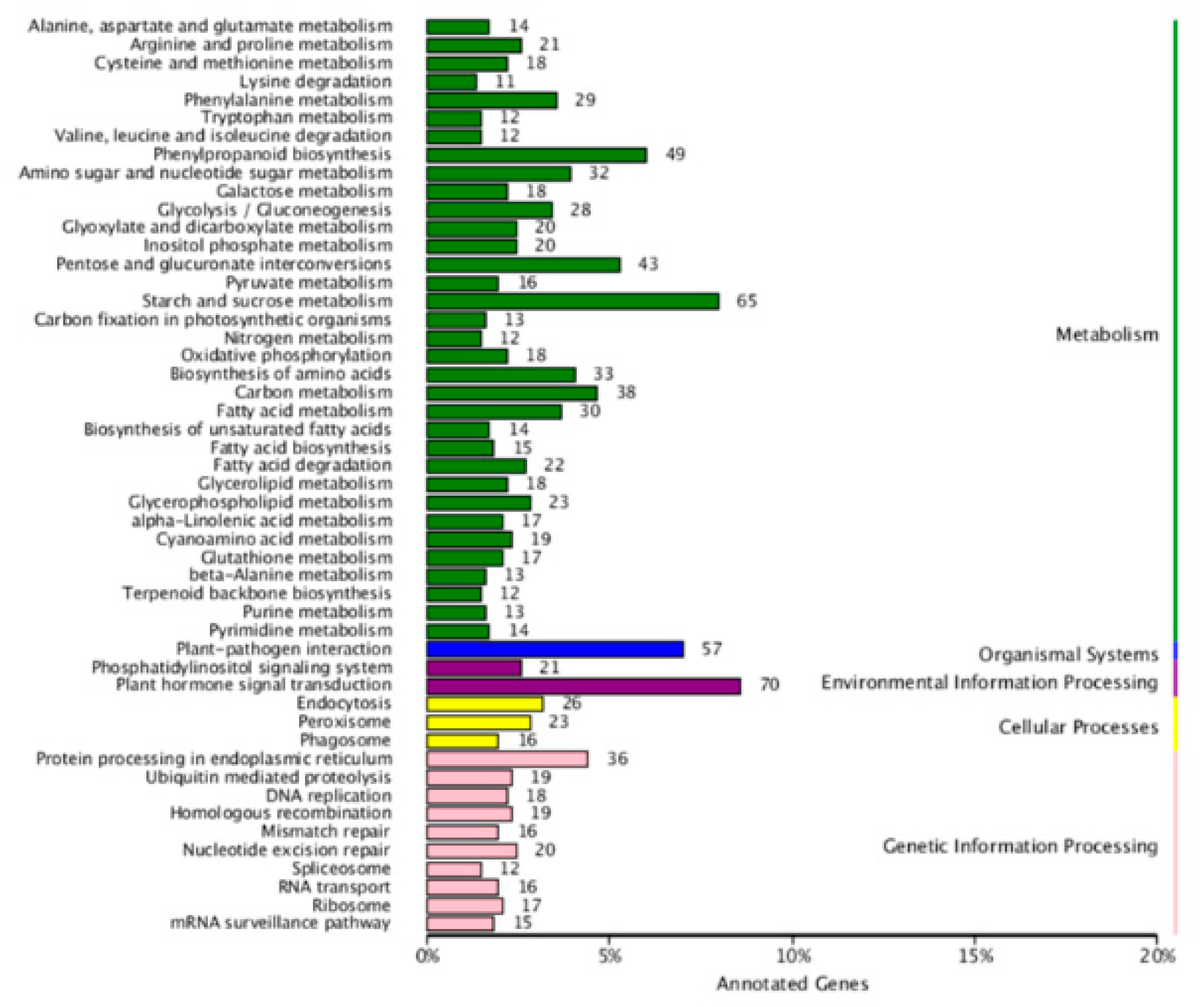
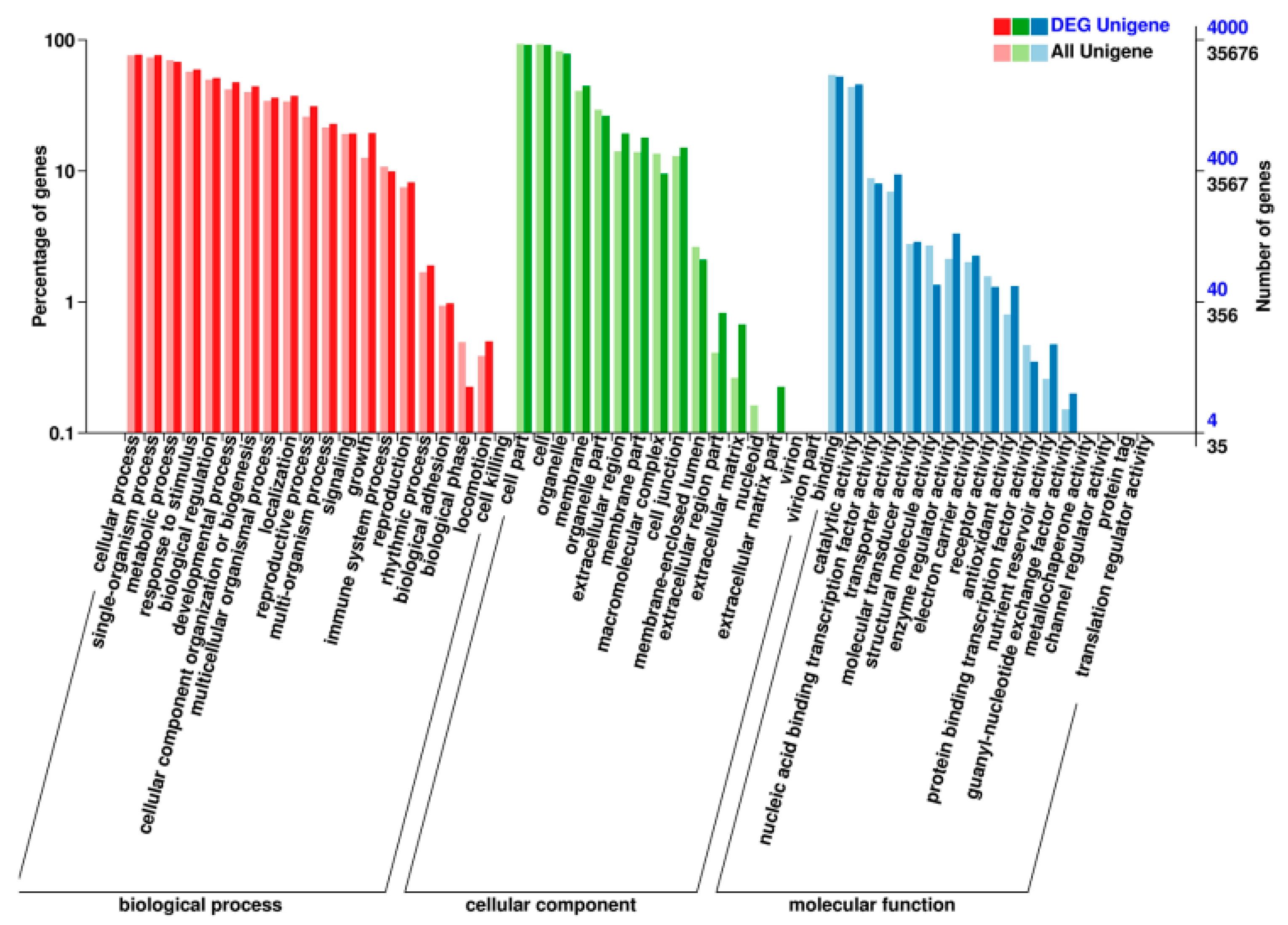
4.8. Target Gene Prediction, GO Analysis, and KEGG Analysis of the Target Genes
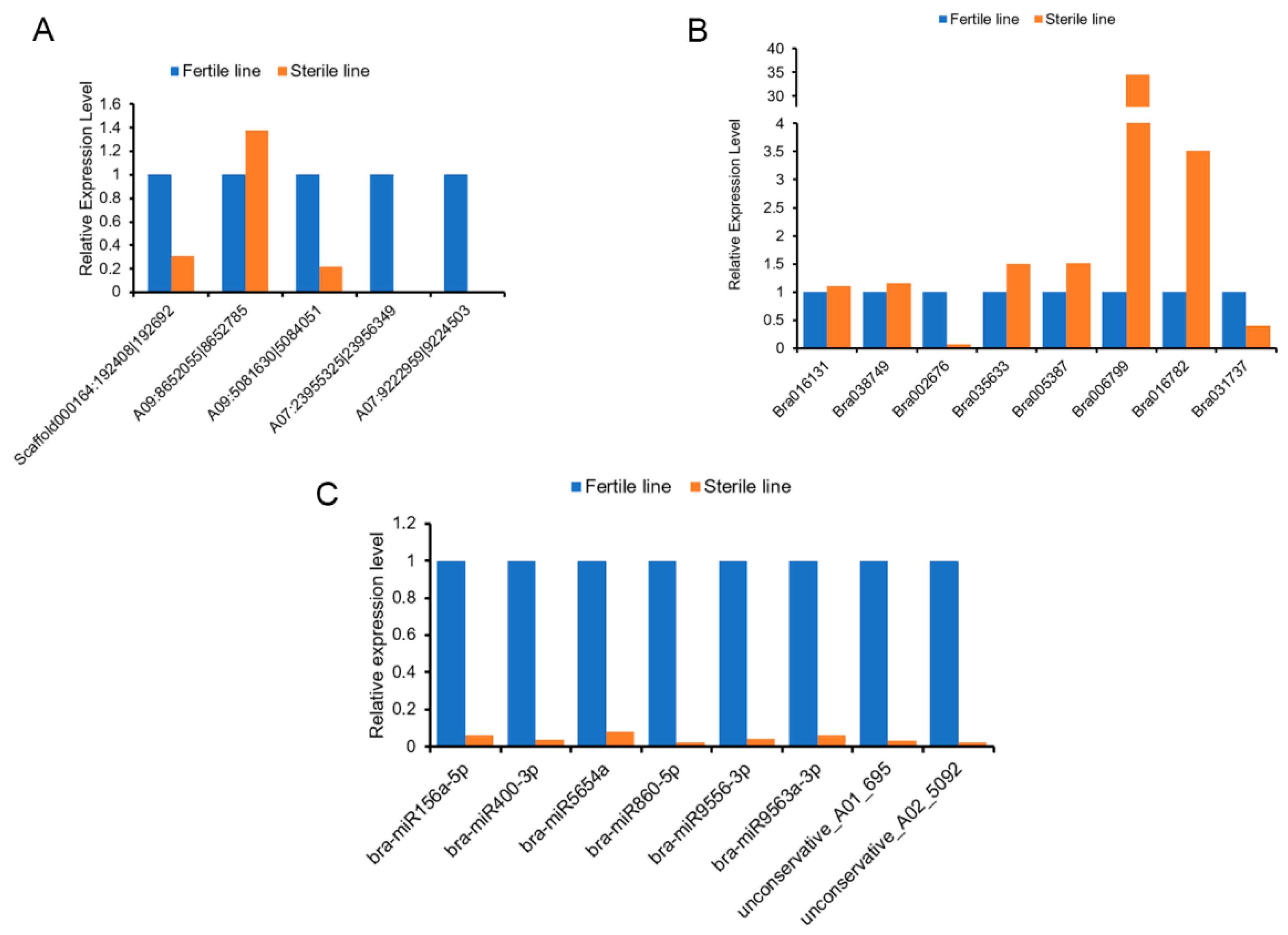
4.9. qRT-PCR Analysis of RNAs
4.10. Construction of miRNA–mRNA and circRNA–miRNA–mRNA Networks
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| circRNAs | circular RNAs |
| ncRNAs | non-coding RNAs |
| miRNA | microRNA |
| ceRNAs | competing endogenous RNAs |
| CMS | cytoplasmic male sterility |
| DE | differentially expressed |
| GO | gene ontology |
| PCD | programmed cell death |
| GMS | genic male sterility |
| pol | Polima |
| TFs | transcription factors |
| DAPI | 4’,6-diamidino-2-phenylindole |
| FDR | false discovery rate |
| BP | biological process |
| CC | cell component |
| MF | molecular function |
| qRT-PCR | quantitative Real-Time PCR |
| TEM | transmission electron microscopy |
References
- Zhang, W.; Xie, Y.; Xu, L.; Wang, Y.; Zhu, X.; Wang, R.; Zhang, Y.; Muleke, E.M.; Liu, L. Identification of microRNAs and their target genes explores miRNA-mediated regulatory network of cytoplasmic male sterility occurrence during anther development in radish (Raphanus sativus L.). Front Plant Sci. 2016, 7, 1054. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Dan, Y.; Zhu, Y. Characterization and use of male sterility in hybrid rice breeding. J. Integr. Plant Biol. 2007, 49, 791–804. [Google Scholar] [CrossRef]
- L’Homme, Y.; Brown, G.G. Organizational differences between cytoplasmic male sterile and male fertile Brassica mitochondrial genomes are confined to a single transposed locus. Nucleic Acids Res. 1993, 21, 1903–1909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, M.; Brown, G.G. Characterization of expression of a mitochondrial gene region associated with the Brassica “Polima” CMS: developmental influences. Curr. Genet. 1993, 24, 316. [Google Scholar] [CrossRef] [PubMed]
- An, H.; Yang, Z.; Yi, B.; Wen, J.; Shen, J.; Tu, J.; Ma, C.; Fu, T. Comparative transcript profiling of the fertile and sterile flower buds of pol CMS in B. napus. BMC Genom. 2014, 15, 258. [Google Scholar] [CrossRef] [PubMed]
- Ding, B.; Hao, M.; Mei, D.; Zaman, Q.U.; Sang, S.; Wang, H.; Wang, W.; Fu, L.; Cheng, H.; Hu, Q. Transcriptome and hormone comparison of three cytoplasmic male sterile systems in Brassica napus. Int. J. Mol. Sci. 2018, 19, 4022. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Zhang, H.; Ruan, H.; Li, Y.; Chen, L.; Wang, T.; Jin, L.; Li, X.; Yang, S.; Gai, J. Exploration of miRNA-mediated fertility regulation network of cytoplasmic male sterility during flower bud development in soybean. 3 Biotech 2019, 9, 22. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Zhang, Z.; Lin, H.; Liu, H.; Chen, J.; Peng, H.; Cao, M.; Rong, T.; Pan, G. Cytoplasmic male sterility-regulated novel microRNAs from maize. Funct. Integr. Genom. 2011, 11, 179–191. [Google Scholar] [CrossRef] [PubMed]
- Song, J.H.; Yang, J.; Pan, F.; Jin, B. Differential expression of microRNAs may regulate pollen development in Brassica oleracea. Genet. Mol. Res. 2015, 14, 15024–15034. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Zhang, H.; Zheng, Y.; Ding, Y. Comparative expression profiling of miRNAs between the cytoplasmic male sterile line MeixiangA and its maintainer line MeixiangB during rice anther development. Planta 2015, 241, 109–123. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Liu, X.; Xu, B.; Zhao, N.; Yang, X.; Zhang, M. Identification of miRNAs and their targets using high-throughput sequencing and degradome analysis in cytoplasmic male-sterile and its maintainer fertile lines of Brassica juncea. BMC Genom. 2013, 14, 9. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.C.; Zhang, X.H.; Yao, Q.J.; Yuan, Y.X.; Li, X.X.; Wei, F.; Zhao, Y.Y.; Zhang, Q.; Wang, Z.Y.; Jiang, W.S. The miRNAs and their regulatory networks responsible for pollen abortion in Ogura-CMS Chinese cabbage revealed by high-throughput sequencing of miRNAs, degradomes, and transcriptomes. Front. Plant Sci. 2015, 6, 894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karreth, F.A.; Pandolfi, P.P. CeRNA cross-talk in cancer: when ce-bling rivalries go awry. Cancer Discov. 2013, 3, 1113–1121. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.L.; Zhang, D.H.; Wu, N.; Xiao, J.H.; Wang, X.; Ma, W. CeRNA in cancer: possible functions and clinical implications. J. Med Genet. 2015, 52, 710–718. [Google Scholar] [CrossRef]
- Lin, X.; Chen, Y. Identification of potentially functional circRNA-miRNA-mRNA regulatory network in hepatocellular carcinoma by integrated microarray analysis. Med Sci Monit Basic Res. 2018, 24, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zhang, W.W.; Peng, F.; Sun, J.Y.; He, Z.Y.; Wu, S.G. Downregulation of hsa_circ_0011946 suppresses the migration and invasion of the breast cancer cell line MCF-7 by targeting RFC3. Cancer Manag. Res. 2018, 10, 535–544. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Feng, C.Y.; Xiang, Z.; Chen, Y.P.; Li, Y.M. CircRNA expression pattern and circRNA-miRNA-mRNA network in the pathogenesis of nonalcoholic steatohepatitis. Oncotarget 2016, 7, 66455–66467. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Ding, X.; Zhang, H.; He, T.; Li, Y.; Wang, T.; Li, X.; Jin, L.; Song, Q.; Yang, S. Comparative analysis of circular RNAs between soybean cytoplasmic male-sterile line NJCMS1A and its maintainer NJCMS1B by high-throughput sequencing. BMC Genom. 2018, 19, 663. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Cao, J.; Zhang, M.; Yao, X.; Xun, X. Production of the Ogura cytoplasmic male sterile (CMS) lines of Chinese Cabbage-pak-choi (Brassica campestris L. ssp. chinensis var. communis) and turnip (B. campestris L. ssp. rapifera) and cytological observation of their sterile organs. Acta Hortic. Sin. 2004, 31, 467–471. [Google Scholar]
- Bernard, A.; Domergue, F.; Pascal, S.; Jetter, R.; Renne, C.; Faure, J.D.; Haslam, R.P.; Napier, J.A.; Lessire, R.; Joubes, J. Reconstitution of plant alkane biosynthesis in yeast demonstrates that Arabidopsis ECERIFERUM1 and ECERIFERUM3 are core components of a very-long-chain alkane synthesis complex. Plant Cell 2012, 24, 3106–3118. [Google Scholar] [CrossRef]
- Rowland, O.; Lee, R.; Franke, R.; Schreiber, L.; Kunst, L. The CER3 wax biosynthetic gene from Arabidopsis thaliana is allelic to WAX2/YRE/FLP1. Febs Lett. 2007, 581, 3538–3544. [Google Scholar] [CrossRef] [PubMed]
- Ariizumi, T.; Hatakeyama, K.; Hinata, K.; Sato, S.; Kato, T.; Tabata, S.; Toriyama, K. A novel male-sterile mutant of Arabidopsis thaliana, faceless pollen-1, produces pollen with a smooth surface and an acetolysis-sensitive exine. Plant Mol. Biol. 2003, 53, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhang, Y.; Deng, C.; Deng, S.; Chen, S. The Arabidopsis Ca2+-Dependent protein kinase CPK12 is involved in plant response to salt stress. Int. J. Mol. Sci. 2018, 19, 4062. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Sun, H.L.; Mei, C.; Wang, X.J.; Yan, L.; Liu, R.; Zhang, X.F.; Wang, X.F.; Zhang, D.P. The Arabidopsis Ca (2+)—dependent protein kinase CPK12 negatively regulates abscisic acid signaling in seed germination and post-germination growth. New Phytol. 2011, 192, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.; Takano, M.; Liu, C.M.; Gasch, A.; Chye, M.L.; Chua, N.H. Expression of three members of the calcium-dependent protein kinase gene family in Arabidopsis thaliana. Plant Mol. Biol. 1996, 30, 1259–1275. [Google Scholar] [CrossRef] [PubMed]
- Gamboa, A.; Paéz-Valencia, J.; Acevedo, G.F.; Vázquez-Moreno, L.; Alvarez-Buylla, R.E. Floral transcription factor AGAMOUS interacts in vitro with a leucine-rich repeat and an acid phosphatase protein complex. Biochem. Biophys. Res. Commun. 2001, 288, 1018–1026. [Google Scholar] [CrossRef] [PubMed]
- Wan, L.L.; Zha, W.J.; Cheng, X.Y.; Liu, C.; Lv, L.; Liu, C.X.; Wang, Z.Q.; Du, B.; Chen, R.Z.; Zhu, L.L. A rice β-1,3-glucanase gene Osg1 is required for callose degradation in pollen development. Planta 2011, 233, 309–323. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, M.; Peragine, A.; Park, M.Y.; Poethig, R.S. A pathway for the biogenesis of trans-acting siRNAs in Arabidopsis. Genes Dev. 2005, 19, 2164–2175. [Google Scholar] [CrossRef]
- Li, Q.W.; Long, F.Y.; Tai, W. Deep sequencing on genome-wide scale reveals the unique composition and expression patterns of microRNAs in developing pollen of Oryza sativa. Genome Biol. 2011, 12, R53. [Google Scholar]
- Huang, L.; Dong, H.; Zhou, D.; Li, M.; Cao, J. Systematic identification of long non-coding RNAs during pollen development and fertilization in Brassica rapa. Plant J. 2018, 96, 203–222. [Google Scholar] [CrossRef]
- Helena, S. The role of non-coding RNAs in cytoplasmic male sterility in flowering plants. Int. J. Mol. Sci. 2017, 18, 2429. [Google Scholar]
- Ding, X.; Li, J.; Zhang, H.; He, T.; Han, S.; Li, Y.; Yang, S.; Gai, J. Identification of miRNAs and their targets by high-throughput sequencing and degradome analysis in cytoplasmic male-sterile line NJCMS1A and its maintainer NJCMS1B of soybean. BMC Genom. 2016, 17, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koichiro, A.; Miyako, U.T.; Maki, K.; Kazuki, H.; Kentaro, Y.; Mikio, N.; Makoto, M. Gibberellin modulates anther development in rice via the transcriptional regulation of GAMYB. Plant Cell 2009, 21, 1453–1472. [Google Scholar]
- Takahiro, K.; Tohru, A.; Maki, K.Y.; Hirofumi, U.; Kinya, T. Abolition of the tapetum suicide program ruins microsporogenesis. Plant Cell Physiol. 2006, 47, 784–787. [Google Scholar]
- Rhoades, M.W.; Reinhart, B.J.; Lim, L.P.; Burge, C.B.; Bartel, B.; Bartel, D.P. Prediction of plant microRNA targets. Cell 2002, 110, 513–520. [Google Scholar] [CrossRef]
- Wang, J.W. Regulation of flowering time by the miR156-mediated age pathway. J. Exp. Bot. 2014, 65, 4723–4730. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.H.; Zhao, Y.X.; Qin, Y.T.; Yue, B.; Zheng, Y.L.; Xiao, H.L. Discovery of microRNAs associated with the S Type cytoplasmic male sterility in maize. J. Integr. Agric. 2013, 12, 229–238. [Google Scholar] [CrossRef]
- Jiang, J.J.; Lyu, M.L.; Liang, Y.; Ma, Z.M.; Cao, J.S. Identification of novel and conserved miRNAs involved in pollen development in Brassica campestris ssp. chinensis by high-throughput sequencing and degradome analysis. BMC Genom. 2014, 15, 146. [Google Scholar]
- Wang, Z.; Qiao, Y.; Zhang, J.; Shi, W.; Zhang, J. Genome wide identification of microRNAs involved in fatty acid and lipid metabolism of Brassica napus by small RNA and degradome sequencing. Gene 2017, 619, 61. [Google Scholar] [CrossRef]
- Hansen, T.B.; Jensen, T.I.; Clausen, B.H.; Bramsen, J.B.; Finsen, B.; Damgaard, C.K.; Kjems, J. Natural RNA circles function as efficient microRNA sponges. Nature 2013, 495, 384–388. [Google Scholar] [CrossRef]
- Zheng, Q.; Bao, C.; Guo, W.; Li, S.; Chen, J.; Chen, B.; Luo, Y.; Lyu, D.; Li, Y.; Shi, G. Circular RNA profiling reveals an abundant circHIPK3 that regulates cell growth by sponging multiple miRNAs. Nat. Commun. 2016, 7, 11215. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Liu, X.; Gai, X.; Ren, J.; Liu, X.; Cai, Y.; Wang, Q.; Ren, H. Cucumis sativus L. WAX2 plays a pivotal role in wax biosynthesis, influencing pollen fertility and plant biotic and abiotic stress responses. Plant Cell Physiol. 2015, 56, 1339–1354. [Google Scholar] [PubMed]
- Lyu, M.L.; Yu, Y.J.; Jiang, J.J.; Song, L.; Liang, Y.; Ma, Z.; Xiong, X.; Cao, J. BcMF26a and BcMF26b are duplicated polygalacturonase genes with divergent expression patterns and functions in pollen development and pollen tube formation in Brassica campestris. PloS one. 2015, 10, e0131173. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Dong, H.; Zhang, F.; Qiu, L.; Wang, F.; Cao, J.; Huang, L. BcMF8, a putative arabinogalactan protein-encoding gene, contributes to pollen wall development, aperture formation and pollen tube growth in Brassica campestris. Ann. Bot. 2014, 113, 777. [Google Scholar] [CrossRef] [PubMed]
- Jens, M. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013, 495, 333–338. [Google Scholar]
- Friedlander, M.R.; Mackowiak, S.D.; Li, N.; Chen, W.; Rajewsky, N. Mirdeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2012, 40, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Ashburmer, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Kanehisa, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, 277–280. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| circRNA ID | Host Gene ID | Type | FDR | Log2FC | Status |
|---|---|---|---|---|---|
| A02:2218194|2218504 | – | Intergenic region | 0.003556881 | −5.326588229 | Down |
| Scaffold000212:46573|47221 | – | Intergenic region | 0.017264214 | −3.898467742 | Down |
| A01:1717686|1718114 | Bra011550 | Intron | 0.024218267 | −3.569160763 | Down |
| A08:17623961|17626439 | – | Intergenic region | 0.027029147 | −3.459397447 | Down |
| A02:2598446|2599366 | – | Intergenic region | 0.03231053 | −3.277304232 | Down |
| A03:23207122|23207987 | Bra012618 | Exon | 0.034434505 | −3.211114134 | Down |
| A05:21205001|21205592 | Bra027353 | Exon | 0.036783873 | −3.141740631 | Down |
| A08:14993249|14994216 | Bra010648 | Exon | 0.039392172 | −3.068861972 | Down |
| A03:25432732|25433123 | Bra019257 | Intron | 0.036783873 | 3.141740631 | Up |
| A03:20154550|20158652 | – | Intergenic region | 0.007140366 | 4.712416081 | Up |
| miRNA ID | FDR | Log2FC | Status |
|---|---|---|---|
| unconservative_A07_27582 | 8.05 × 10−9 | −5.710103115 | Down |
| unconservative_A06_21945 | 6.89 × 10−11 | −5.240482741 | Down |
| unconservative_Scaffold000096_42992 | 6.89 × 10−11 | −5.240482741 | Down |
| unconservative_Scaffold000777_48008 | 6.89 × 10−11 | −5.240482741 | Down |
| unconservative_A02_5624 | 7.51 × 10−11 | −4.410730302 | Down |
| unconservative_A10_42699 | 1.57 × 10−7 | −4.101491939 | Down |
| unconservative_A06_21764 | 3.57 × 10−21 | −4.07323988 | Down |
| unconservative_A06_23706 | 3.61 × 10−8 | −4.053517585 | Down |
| unconservative_A08_32883 | 3.23 × 10−18 | −3.88622319 | Down |
| unconservative_A03_12748 | 4.87 × 10−16 | −3.37012631 | Down |
| GO Term ID | GO Term Description | Unigene Numeber | DE Gene Number | Expected Number | KS |
|---|---|---|---|---|---|
| GO:0009827 | plant-type cell wall modification | 1070 | 360 | 121.45 | <1 × 10−30 |
| GO:0009860 | pollen tube growth | 1366 | 407 | 155.05 | <1 × 10−30 |
| GO:0030048 | actin filament-based movement | 271 | 85 | 30.76 | 2.60 × 10−10 |
| GO:0010584 | pollen exine formation | 326 | 103 | 37 | 9.30 × 10−10 |
| GO:0045490 | pectin catabolic process | 193 | 54 | 21.91 | 6.80 × 10−7 |
| GO:0048235 | pollen sperm cell differentiation | 116 | 40 | 13.17 | 2.40 × 10−6 |
| GO:0015770 | sucrose transport | 66 | 25 | 7.49 | 1.70 × 10−5 |
| GO:0009821 | alkaloid biosynthetic process | 42 | 13 | 4.77 | 1.80 × 10−5 |
| GO:1901700 | response to oxygen-containing compound | 11730 | 1436 | 1331.44 | 1.90 × 10−5 |
| GO:0009737 | response to abscisic acid | 4007 | 511 | 454.82 | 3.60 × 10−5 |
| GO Term ID | GO Term Description | Unigene Numeber | DE Gene Number | Expected Number | KS |
|---|---|---|---|---|---|
| GO:0003785 | actin monomer binding | 22 | 13 | 2.52 | 1.20 × 10−5 |
| GO:0030599 | pectinesterase activity | 182 | 51 | 20.87 | 1.20 × 10−5 |
| GO:0045330 | aspartyl esterase activity | 102 | 33 | 11.7 | 1.60 × 10−5 |
| GO:0090353 | polygalacturonase inhibitor activity | 14 | 5 | 1.61 | 3.20 × 10−5 |
| GO:0008705 | methionine synthase activity | 12 | 5 | 1.38 | 5.40 × 10−5 |
| GO:0019863 | IgE binding | 65 | 17 | 7.45 | 6.90 × 10−5 |
| GO:0009044 | xylan 1,4-beta-xylosidase activity | 31 | 15 | 3.55 | 0.0001 |
| GO:0004350 | glutamate-5-semialdehyde dehydrogenase activity | 11 | 7 | 1.26 | 0.00014 |
| GO:0004601 | peroxidase activity | 225 | 52 | 25.8 | 0.00023 |
| GO:0004349 | glutamate 5-kinase activity | 13 | 7 | 1.49 | 0.00023 |
| GO Term ID | GO Term Description | Unigene Numeber | DE Gene Number | Expected Number | KS |
|---|---|---|---|---|---|
| GO:0003785 | actin monomer binding | 22 | 13 | 2.52 | 1.20 × 10−5 |
| GO:0030599 | pectinesterase activity | 182 | 51 | 20.87 | 1.20 × 10−5 |
| GO:0045330 | aspartyl esterase activity | 102 | 33 | 11.7 | 1.60 × 10−5 |
| GO:0090353 | polygalacturonase inhibitor activity | 14 | 5 | 1.61 | 3.20 × 10−5 |
| GO:0008705 | methionine synthase activity | 12 | 5 | 1.38 | 5.40 × 10−5 |
| GO:0019863 | IgE binding | 65 | 17 | 7.45 | 6.90 × 10−5 |
| GO:0009044 | xylan 1,4-beta-xylosidase activity | 31 | 15 | 3.55 | 0.0001 |
| GO:0004350 | glutamate-5-semialdehyde dehydrogenase activity | 11 | 7 | 1.26 | 0.00014 |
| GO:0004601 | peroxidase activity | 225 | 52 | 25.8 | 0.00023 |
| GO:0004349 | glutamate 5-kinase activity | 13 | 7 | 1.49 | 0.00023 |
| miRNA ID | Status | Gene ID | Protein | Biological Process |
|---|---|---|---|---|
| bra-miR9563a-3p | Down | Bra005387 | UDP-glucose 4-epimerase, UDP-arabinose 4-epimerase | Sugar metabolism; pollen tube development; defense response by callose deposition |
| unconservative_A02_5254 | Up | Bra012559 | Xyloglucan:xyloglucosyl transferase | Callose metabolic process |
| Bra013367 | ||||
| Bra020239 | Unknown | Cellulose biosynthetic process; cell redox homeostasis regulation; tapetum programmed cell death | ||
| bra-miR9556-3p | Down | Brassica_rapa_newGene_11926 | Unknown | Flavonoid biosynthesis process |
| Bra031737 | Unknown | Tapetum programmed cell death | ||
| unconservative_A05_20239 | Up | Bra020299 | Unknown | Glucuronoxylan metabolic process; xylan biosynthetic process |
| Bra032058 | Unknown | Tapetum programmed cell death | ||
| bra-miR156a-5p | Down | Brassica_rapa_newGene_13349 | Unknown | Vegetative to reproductive phase transition of meristem; glucuronoxylan metabolism; xylan biosynthetic process |
| Bra016131 | Unknown | Unknown | ||
| bra-miR9556-5p | Down | Bra008715 | Unknown | Lignin metabolic process; glucuronoxylan biosynthetic process; cell wall organization process; tapetum programmed cell death |
| unconservative_A08_32883 | Down | Bra020025 | Unknown | Lipid metabolic process; xylan metabolic process; cell wall biogenesis |
| Bra020123 | Unknown | Flavonoid biosynthetic process | ||
| bra-miR5717 | Up | Bra002676 | Glycerophosphodiester phosphodiesterase | Pollen tube growth process; pollen wall development; starch metabolism; lipid metabolism |
| Bra009027 | Unknown | Pollen tube growth process; pollen wall development | ||
| Bra035078 | Unknown | Pollen tube growth process; pollen wall development | ||
| Bra034939 | N-acylphosphatidylet-hanolamine-specific phospholipase D | Pollen wall development | ||
| Bra037916 | S-adenosylmethionine-dependent methyltransferase | Pollen wall development; pectin metabolic process |
| miRNA ID | Target Gene ID | Phytohormone-Related Biological Process |
|---|---|---|
| unconservative_A05_20239 | Bra032058 | Be involved in salicylic acid biosynthetic process and response to salicylic acid |
| bra-miR9563a-3p | Bra005387 | Be involved in abscisic acid-activated signaling pathway and response to ethylene |
| unconservative_A02_5254 | Bra013367 | Response to auxin |
| bra-miR5717 | Bra034939 | Response to abscisic acid |
| Bra037916 | Response to abscisic acid | |
| bra-miR9556-5p | Bra008715 | Be involved in salicylic acid mediated signaling pathway and brassinosteroid biosynthetic process |
| Gene ID | Homologue to Arabidopsis | Description of the Homologue to Arabidopsis | Role of the Homologue to Arabidopsis | Reference |
|---|---|---|---|---|
| Bra006799 | AT5G57800 | Encodes a transmembrane protein with similarity to the sterol desaturase family at the N-terminus and to the short-chain dehydrogenase/reductase family at the C-terminus. | Be involved in cuticle membrane and wax biosynthesis, influencing pollen fertility as well as plant biotic and abiotic stress responses, etc. | [20,21,22] |
| Bra013352 | AT4G18810 | NAD(P)-binding Rossmann-fold superfamily protein | Unknown | – |
| Bra022668 | AT5G53730 | Late embryogenesis abundant (LEA) hydroxyproline-rich glycoprotein family | Unknown | – |
| Bra029377 | AT5G23580 | A Member of a unique family of enzymes containing a single polypeptide chain with a kinase domain at the amino terminus and a putative calcium-binding EF hands structure at the carboxyl terminus | Be involved in plant response to salt stress, a Ca (2+) -dependent protein kinase balancer in abscisic acid signaling | [23,24,25] |
| Bra038700 | AT3G12145 | A novel leucine-rich repeat protein | Interacts directly with MADS domain transcription factor during Arabidopsis thaliana flower development | [26] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liang, Y.; Zhang, Y.; Xu, L.; Zhou, D.; Jin, Z.; Zhou, H.; Lin, S.; Cao, J.; Huang, L. CircRNA Expression Pattern and ceRNA and miRNA–mRNA Networks Involved in Anther Development in the CMS Line of Brassica campestris. Int. J. Mol. Sci. 2019, 20, 4808. https://doi.org/10.3390/ijms20194808
Liang Y, Zhang Y, Xu L, Zhou D, Jin Z, Zhou H, Lin S, Cao J, Huang L. CircRNA Expression Pattern and ceRNA and miRNA–mRNA Networks Involved in Anther Development in the CMS Line of Brassica campestris. International Journal of Molecular Sciences. 2019; 20(19):4808. https://doi.org/10.3390/ijms20194808
Chicago/Turabian StyleLiang, Yuwei, Yuzhi Zhang, Liai Xu, Dong Zhou, Zongmin Jin, Huiyan Zhou, Sue Lin, Jiashu Cao, and Li Huang. 2019. "CircRNA Expression Pattern and ceRNA and miRNA–mRNA Networks Involved in Anther Development in the CMS Line of Brassica campestris" International Journal of Molecular Sciences 20, no. 19: 4808. https://doi.org/10.3390/ijms20194808
APA StyleLiang, Y., Zhang, Y., Xu, L., Zhou, D., Jin, Z., Zhou, H., Lin, S., Cao, J., & Huang, L. (2019). CircRNA Expression Pattern and ceRNA and miRNA–mRNA Networks Involved in Anther Development in the CMS Line of Brassica campestris. International Journal of Molecular Sciences, 20(19), 4808. https://doi.org/10.3390/ijms20194808