Micro RNA Transcriptome Profile in Canine Oral Melanoma

 , ,
, , 
Abstract
:1. Introduction
2. Results
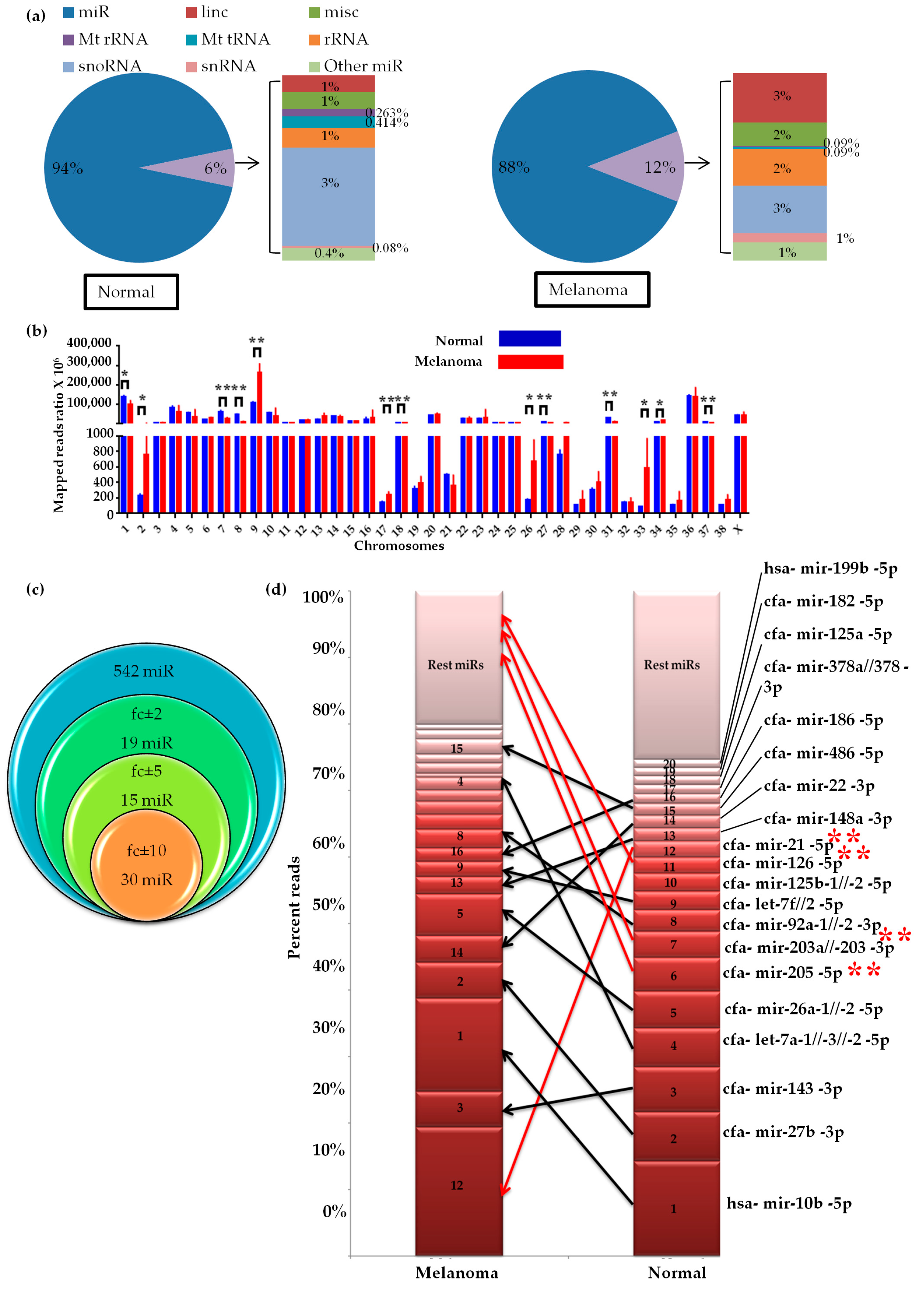
2.1. Small RNA Profile of Canine Oral Melanoma
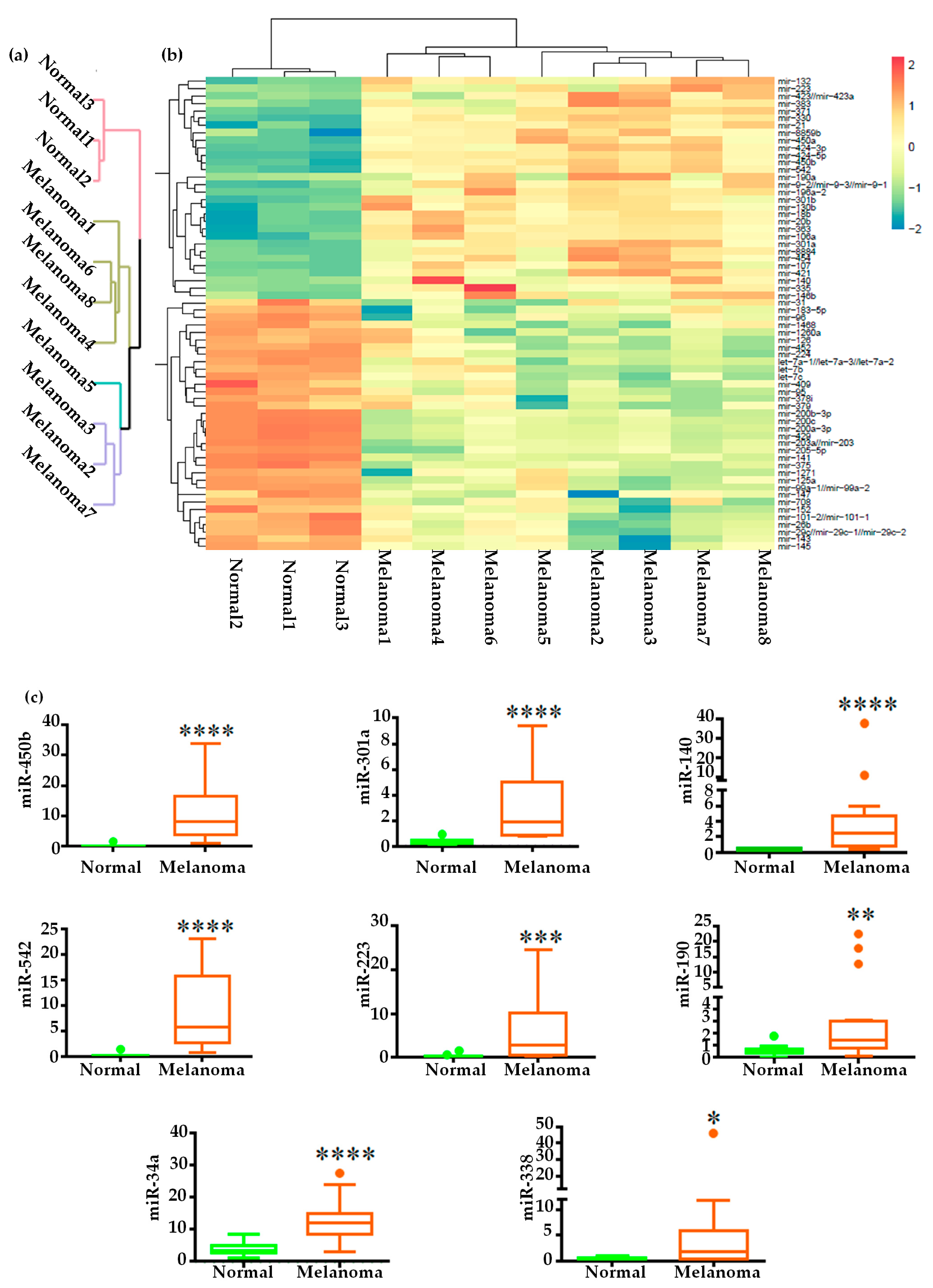
2.2. Global miRNAs Expression in Canine Oral Melanoma
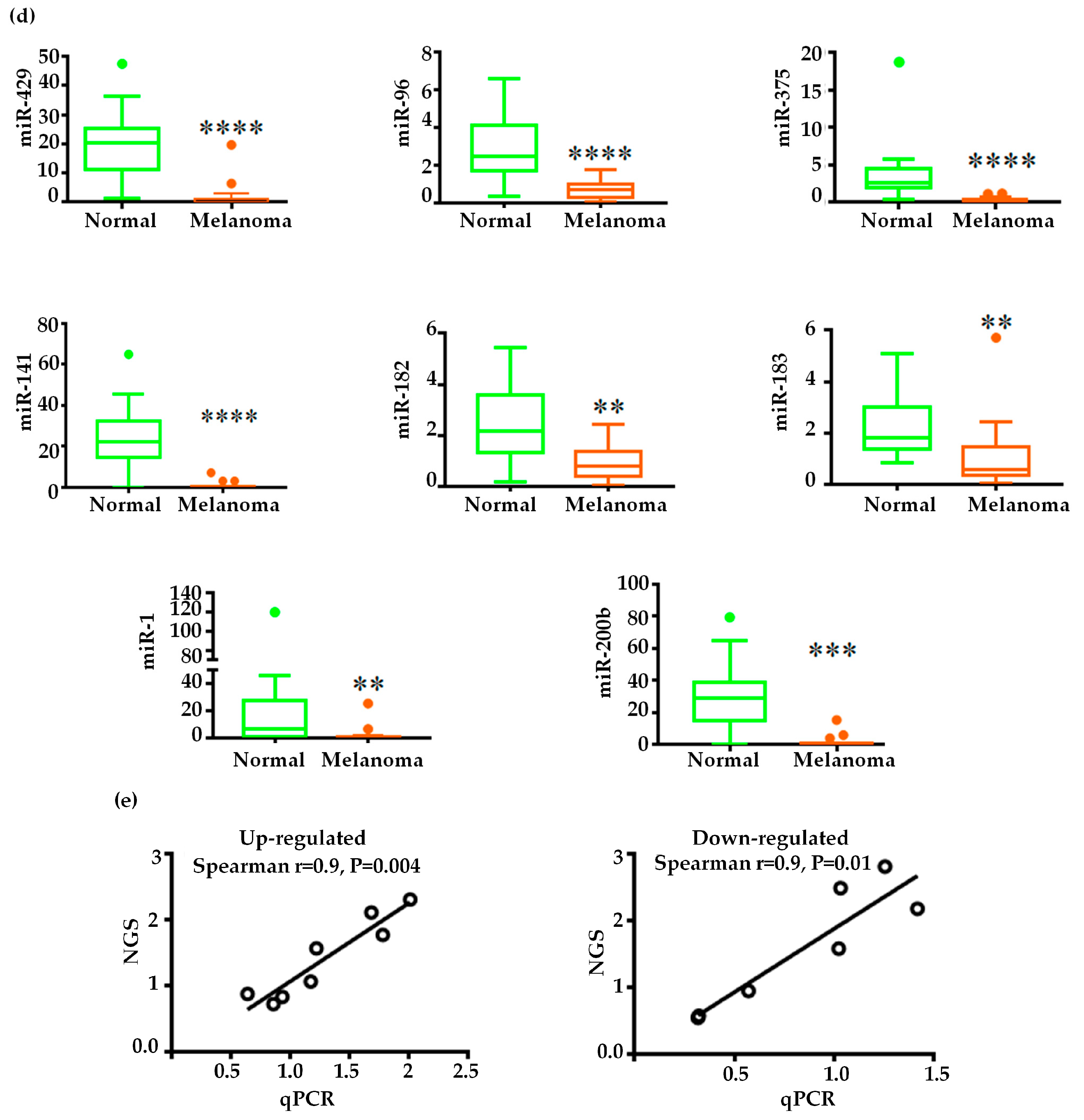
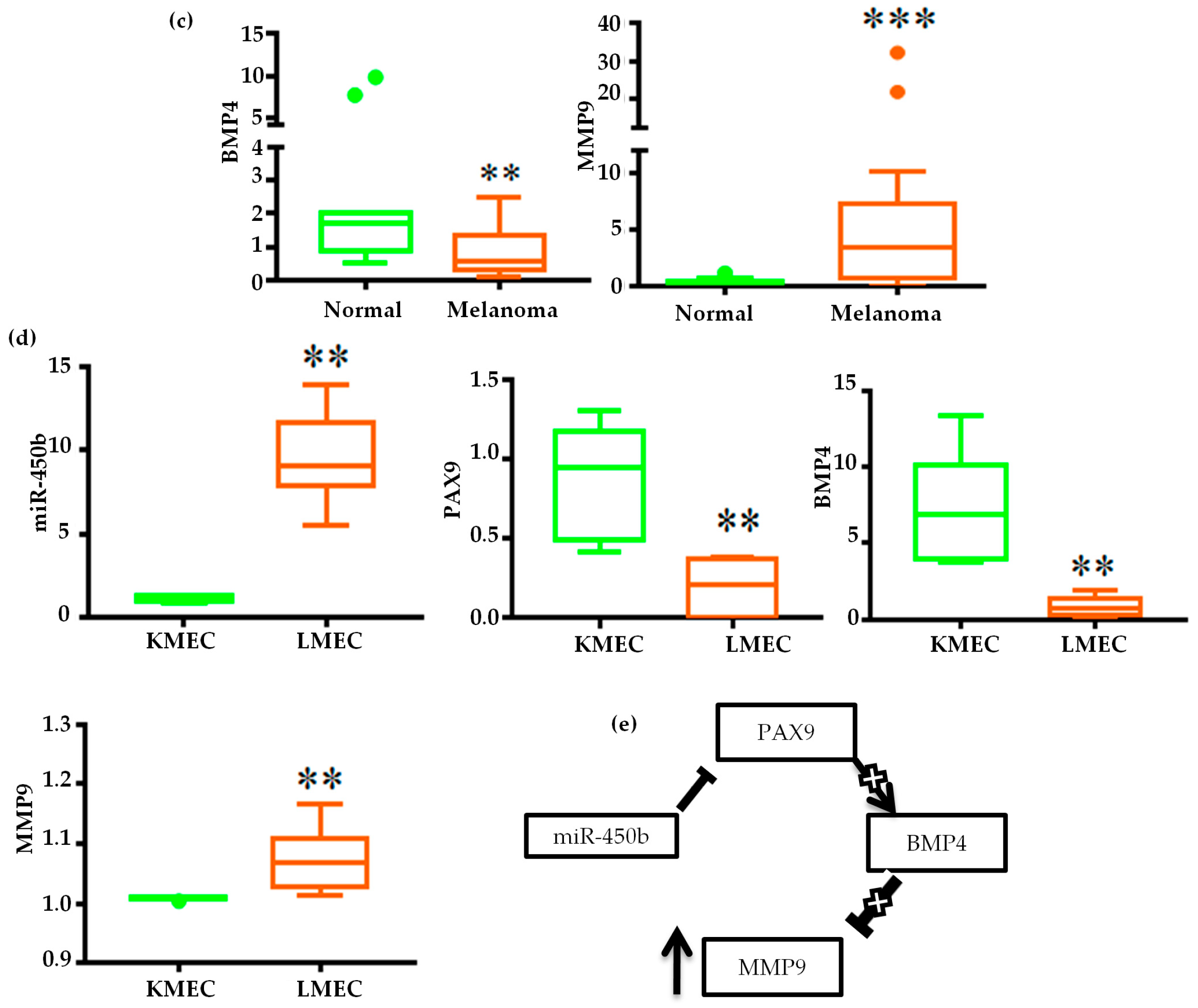
2.3. Validation of miRNA Expression
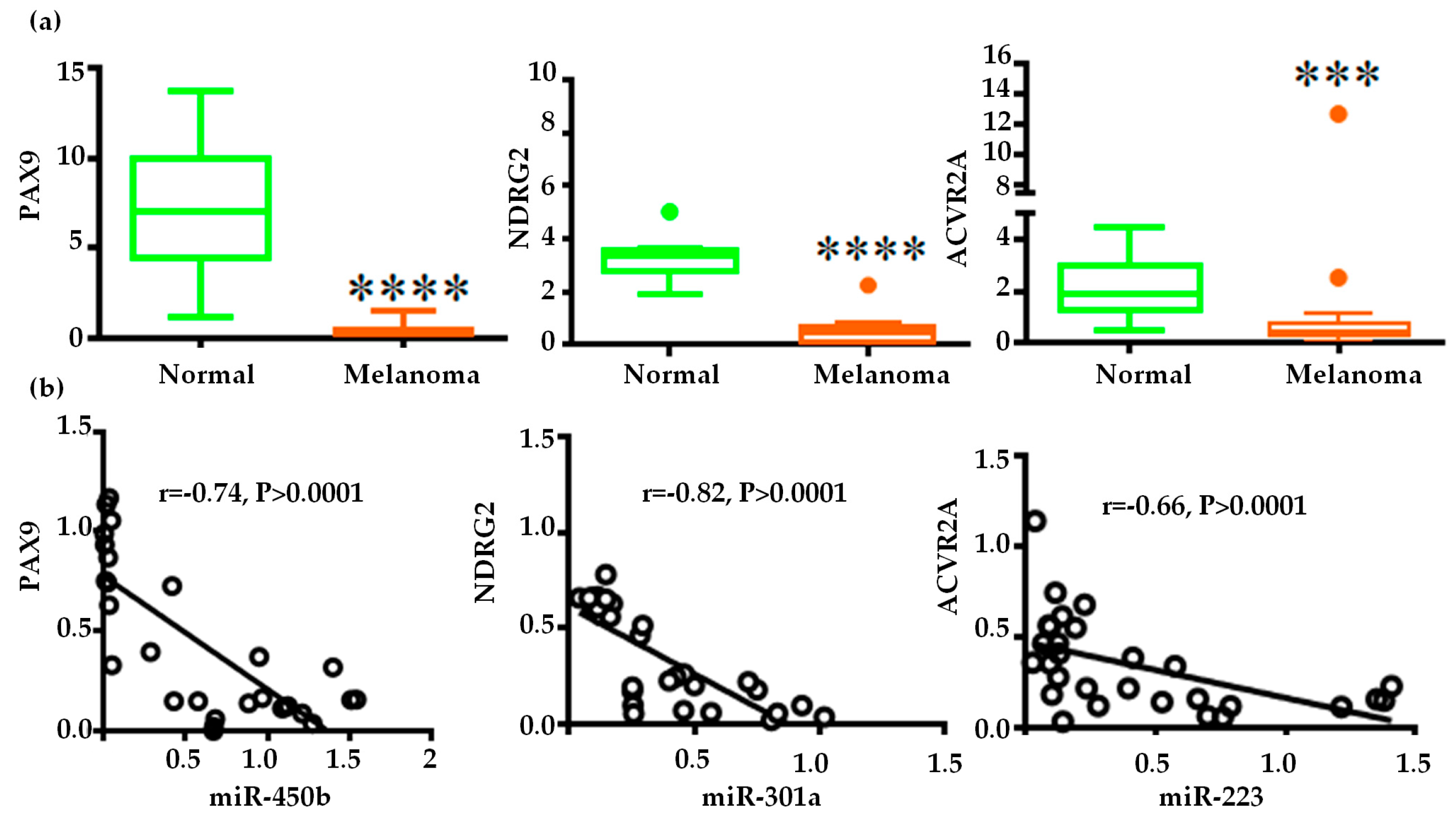
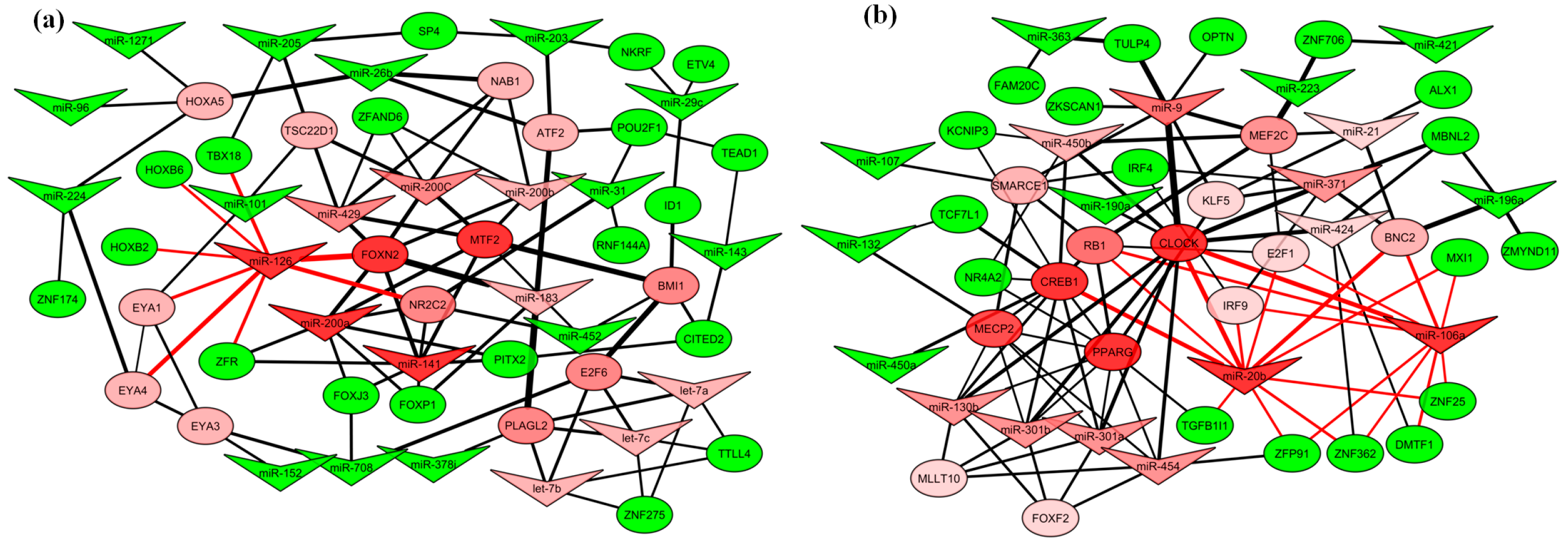
2.4. Gene Regulatory Function of Oncogenic miRNAs
2.5. Gene Ontology and KEGG Pathway Analysis of the Differentially Expressed miRNAs
2.6. miRNA–Transcription Factor Interaction Network between Dog and Human Melanoma
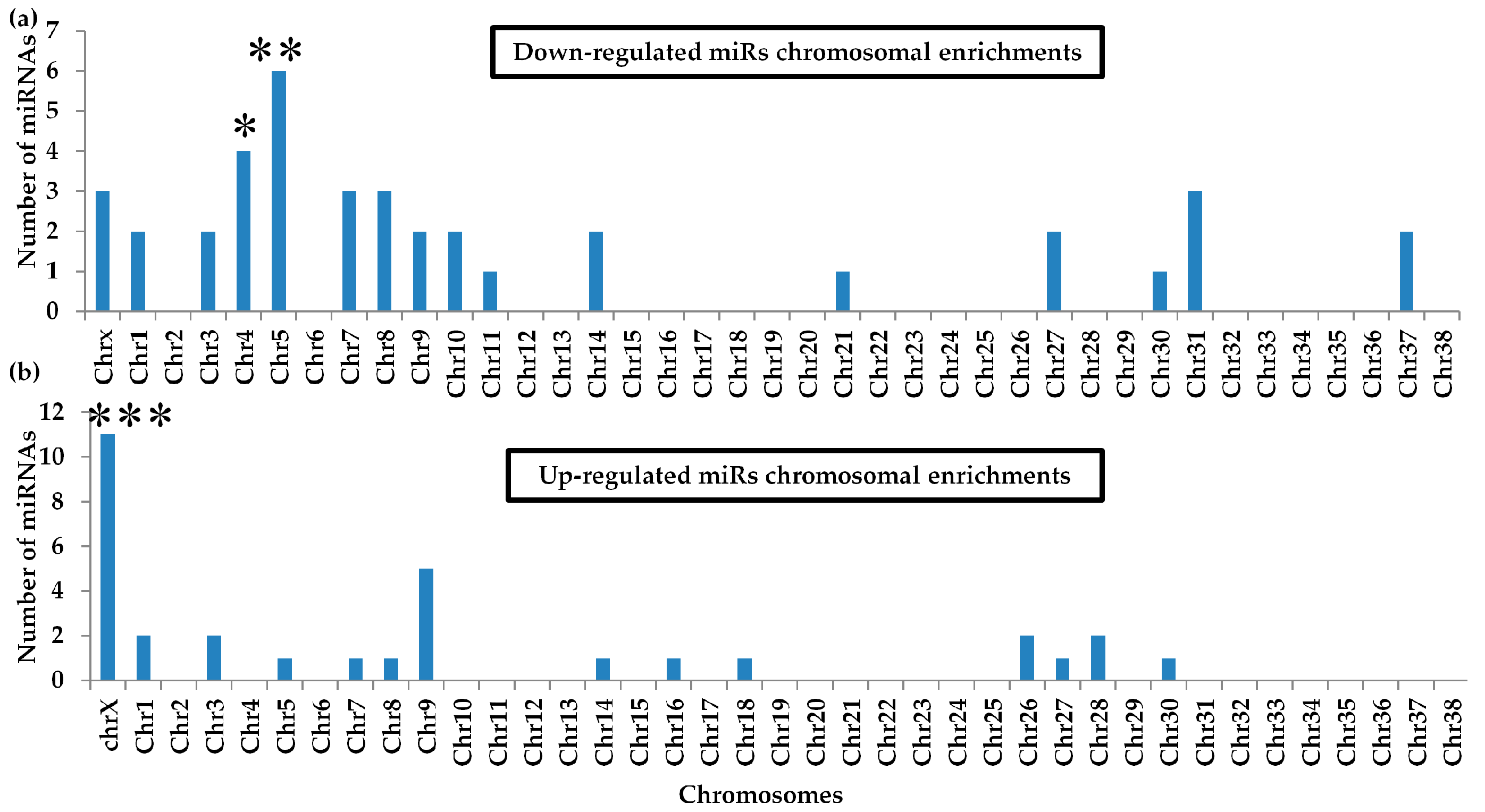
2.7. Differential miRNA Chromosomal Enrichments
3. Discussion
4. Materials and Methods
4.1. Clinical Samples and Canine Melanoma Cell Lines
4.2. RNA Extraction and Sequencing
4.3. Bioinformatics Analysis of Small RNA Reads
4.4. Edge Analysis
4.5. Expression Analysis by qPCR
4.6. Gene Ontology, Pathway Analysis and Network Construction
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgements
Conflicts of Interest
Abbreviations
| MiRNAs | MicroRNAs |
| COM | Canine oral melanoma |
| qPCR | Quantitative real-time PCR |
| NGS | Next generation Sequencing |
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2018. CA Cancer J. Clin. 2018, 67, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Akbani, R.; Akdemir, K.C.; Aksoy, B.A.; Albert, M.; Ally, A.; Amin, S.B.; Arachchi, H.; Arora, A.; Auman, J.T.; Ayala, B.; et al. Genomic classification of cutaneous melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef] [PubMed]
- Hayward, N.K.; Wilmott, J.S.; Waddell, N.; Johansson, P.A.; Field, M.A.; Nones, K.; Patch, A.M.; Kakavand, H.; Alexandrov, L.B.; Burke, H.; et al. Whole-genome landscapes of major melanoma subtypes. Nature 2017, 545, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, B.; Adissu, H.A.; Wei, B.R.; Michael, H.T.; Merlino, G.; Mark Simpson, R. Naturally occurring canine melanoma as a predictive comparative oncology model for human mucosal and other triple wild-type melanomas. Int. J. Mol. Sci. 2018, 19, 394. [Google Scholar] [CrossRef] [PubMed]
- Gillard, M.; Cadieu, E.; De Brito, C.; Abadie, J.; Vergier, B.; Devauchelle, P.; Degorce, F.; Dréano, S.; Primot, A.; Dorso, L.; et al. Naturally occurring melanomas in dogs as models for non-UV pathways of human melanomas. Pigment Cell Melanoma Res. 2014, 27, 90–102. [Google Scholar] [CrossRef] [PubMed]
- Simpson, R.M.; Bastian, B.C.; Michael, H.T.; Webster, J.D.; Prasad, M.L.; Conway, C.M.; Prieto, V.M.; Gary, J.M.; Goldschmidt, M.H.; Esplin, D.G.; et al. Sporadic naturally occurring melanoma in dogs as a preclinical model for human melanoma. Pigment Cell Melanoma Res. 2014, 27, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Mochizuki, H.; Kennedy, K.; Shapiro, S.G.; Breen, M.B. BRAF mutations in canine cancers. PLoS ONE 2015, 10, e0129534. [Google Scholar] [CrossRef] [PubMed]
- Chu, P.Y.; Pan, S.L.; Liu, C.H.; Lee, J.; Yeh, L.S.; Liao, A.T. KIT gene exon 11 mutations in canine malignant melanoma. Vet. J. 2013, 196, 226–230. [Google Scholar] [CrossRef]
- Rupaimoole, R.; Slack, F.J. MicroRNA therapeutics: Towards a new era for the management of cancer and other diseases. Nat. Rev. Drug Discov. 2017, 16, 203–222. [Google Scholar] [CrossRef]
- Noguchi, S.; Mori, T.; Hoshino, Y.; Yamada, N.; Maruo, K.; Akao, Y. MicroRNAs as tumour suppressors in canine and human melanoma cells and as a prognostic factor in canine melanomas. Vet. Comp. Oncol. 2013, 11, 113–123. [Google Scholar] [CrossRef]
- Noguchi, S.; MORI, T.; Hoshino, Y.; Nami, Y.; Nakagawa, T.; Sasaki, N.; Akao, Y.; Maruo, K. Comparative study of anti-oncogenic microRNA-145 in canine and human malignant melanoma. J. Vet. Med. Sci. 2012, 74, 1108090601. [Google Scholar] [CrossRef] [PubMed]
- Islam, F.; Gopalan, V.; Vider, J.; Wahab, R.; Ebrahimi, F.; Lu, C.T.; Kasem, K.; Lam, A.K.Y. MicroRNA-186-5p overexpression modulates colon cancer growth by repressing the expression of the FAM134B tumour inhibitor. Exp. Cell Res. 2017, 357, 260–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedrich, M.; Pracht, K.; Mashreghi, M.F.; Jäck, H.M.; Radbruch, A.; Seliger, B. The role of the miR-148/-152 family in physiology and disease. Eur. J. Immunol. 2017, 47, 2026–2038. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Li, Y.; Ding, M.; Zhang, H.; Xu, X.; Tang, J. Molecular mechanisms and clinical applications of MIR-22 in regulating malignant progression in human cancer (Review). Int. J. Oncol. 2017, 50, 345–355. [Google Scholar] [CrossRef] [PubMed]
- Ushio, N.; Rahman, M.M.; Maemura, T.; Lai, Y.C.; Iwanaga, T.; Kawaguchi, H.; Miyoshi, N.; Momoi, Y.; Miura, N. Identification of dysregulated microRNAs in canine malignant melanoma. Oncol. Lett. 2019, 17, 1080–1088. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.M.; Li, J.M.F.M.; Guo, L.L.; Xiao, H.T.; Dong, L.; Wang, F.; Huang, F.B.; Cao, D.; Qin, T.; Yin, X.H.; et al. TGF-β1 suppression of microRNA-450b-5p expression: A novel mechanism for blocking myogenic differentiation of rhabdomyosarcoma. Oncogene 2013, 33, 2075. [Google Scholar] [CrossRef]
- Guo, Y.J.; Liu, J.X.; Guan, Y.W. Hypoxia induced upregulation of miR-301a/b contributes to increased cell autophagy and viability of prostate cancer cells by targeting NDRG2. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 101–108. [Google Scholar]
- Yang, L.; Li, Y.; Wang, X.; Mu, X.; Qin, D.; Huang, W.; Alshahrani, S.; Nieman, M.; Peng, J.; Essandoh, K.; et al. Overexpression of miR-223 tips the balance of pro- and anti-hypertrophic signaling cascades toward physiologic cardiac hypertrophy. J. Biol. Chem. 2016, 291, 15700–15713. [Google Scholar] [CrossRef]
- Zhou, J.; Gao, Y.; Lan, Y.; Jia, S.; Jiang, R. Pax9 regulates a molecular network involving Bmp4, Fgf10, Shh signaling and the Osr2 transcription factor to control palate morphogenesis. Development 2013, 140, 4709–4718. [Google Scholar] [CrossRef] [Green Version]
- Laulan, N.B.; St-Pierre, Y. Bone morphogenetic protein 4 (BMP-4) and epidermal growth factor (EGF) inhibit metalloproteinase-9 (MMP-9) expression in cancer cells. Oncoscience 2015, 2, 309–316. [Google Scholar] [CrossRef]
- Inoue, K.; Ohashi, E.; Kadosawa, T.; Hong, S.-H.; Matsunaga, S.; Mochizuki, M.; Nishimura, R.; Sasaki, N. Establishment and characterization of four canine melanoma cell Lines. J. Vet. Med. Sci. 2004, 66, 1437–1440. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Li, Y.; Lv, X.; Li, J.; Wang, X.; Lei, Z.; Li, X. Expression of MicroRNA-301a and its functional roles in malignant melanoma. Cell Physiol. Biochem. 2016, 40, 230–244. [Google Scholar] [CrossRef] [PubMed]
- Larsen, A.C.; Mikkelsen, L.H.; Borup, R.; Kiss, K.; Toft, P.B.; Von Buchwald, C.; Coupland, S.E.; Prause, J.U.; Heegaard, S. MicroRNA expression profile in conjunctival melanoma. Investig. Ophthalmol. Vis. Sci. 2016, 57, 4205–4212. [Google Scholar] [CrossRef] [PubMed]
- Mirzaei, H.H.R.H.; Gholamin, S.; Shahidsales, S.; Sahebkar, A.; Jafaari, M.R.; Mirzaei, H.H.R.H.; Hassanian, S.M.; Avan, A. MicroRNAs as potential diagnostic and prognostic biomarkers in melanoma. Eur. J. Cancer 2016, 53, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Brenn, T.; Brown, E.R.S.; Doherty, V.; Melton, D.W. Differential expression of microRNAs during melanoma progression: MiR-200c, miR-205 and miR-211 are downregulated in melanoma and act as tumour suppressors. Br. J. Cancer 2012, 106, 553–561. [Google Scholar] [CrossRef] [PubMed]
- Fattore, L.; Costantini, S.; Malpicci, D.; Ruggiero, C.F.; Ascierto, P.A.; Croce, C.M.; Mancini, R.; Ciliberto, G. MicroRNAs in melanoma development and resistance to target therapy. Oncotarget 2015, 8, 22262–22278. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Fisher, D.E.; Flaherty, K.T. Cell-state dynamics and therapeutic resistance in melanoma from the perspective of MITF and IFNγ pathways. Nat. Rev. Clin. Oncol. 2019, 16, 549–562. [Google Scholar] [CrossRef] [PubMed]
- Garraway, L.A.; Widlund, H.R.; Rubin, M.A.; Getz, G.; Berger, A.J.; Ramaswamy, S.; Beroukhim, R.; Milner, D.A.; Granter, S.R.; Du, J.; et al. Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature 2005, 436, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Laganà, A.; Russo, F.; Sismeiro, C.; Giugno, R.; Pulvirenti, A.; Ferro, A. Variability in the incidence of miRNAs and genes in fragile sites and the role of repeats and CpG islands in the distribution of genetic material. PLoS ONE 2010, 5, e11166. [Google Scholar] [CrossRef] [PubMed]
- Calin, G.A.; Sevignani, C.; Dumitru, C.D.; Hyslop, T.; Noch, E.; Yendamuri, S.; Shimizu, M.; Rattan, S.; Bullrich, F.; Negrini, M.; et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc. Natl. Acad. Sci. USA 2004, 101, 2999–3004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calin, G.A.; Dumitru, C.D.; Shimizu, M.; Bichi, R.; Zupo, S.; Noch, E.; Aldler, H.; Rattan, S.; Keating, M.; Rai, K.; et al. Frequent deletions and down-regulation of micro-RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. USA 2002, 99, 15524–15529. [Google Scholar] [CrossRef] [PubMed]
- Dambal, S.; Shah, M.; Mihelich, B.; Nonn, L. The microRNA-183 cluster: The family that plays together stays together. Nucleic Acids Res. 2015, 43, 7173–7188. [Google Scholar] [CrossRef] [PubMed]
- Lindblad-Toh, K.; Wade, C.M.; Mikkelsen, T.S.; Karlsson, E.K.; Jaffe, D.B.; Kamal, M.; Clamp, M.; Chang, J.L.; Kulbokas, E.J.; Zody, M.C.; et al. Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature 2005, 438, 803–819. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.; Yang, Y.; Lee, M.S.; Yoo, Y.D.; Lee, H.G.; Lim, J.S. NDRG2 gene expression in B16F10 melanoma cells restrains melanogenesis via inhibition of Mitf expression. Pigment Cell Melanoma Res. 2008, 21, 653–664. [Google Scholar] [CrossRef] [PubMed]
- Hata, S.; Hamada, J.-I.; Maeda, K.; Murai, T.; Tada, M.; Furukawa, H.; Tsutsumida, A.; Saito, A.; Yamamoto, Y.; Moriuchi, T. PAX4 has the potential to function as a tumor suppressor in human melanoma. Int. J. Oncol. 2008, 33, 1065–1071. [Google Scholar]
- Sand, M.; Skrygan, M.; Sand, D.; Georgas, D.; Gambichler, T.; Hahn, S.A.; Altmeyer, P.; Bechara, F.G. Comparative microarray analysis of microRNA expression profiles in primary cutaneous malignant melanoma, cutaneous malignant melanoma metastases, and benign melanocytic nevi. Cell Tissue Res. 2013, 351, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Shon, S.K.; Kim, A.; Kim, J.Y.; Kim, K., II; Yang, Y.; Lim, J.S. Bone morphogenetic protein-4 induced by NDRG2 expression inhibits MMP-9 activity in breast cancer cells. Biochem. Biophys. Res. Commun. 2009, 385, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Brachelente, C.; Cappelli, K.; Capomaccio, S.; Porcellato, I.; Silvestri, S.; Bongiovanni, L.; De Maria, R.; Verini Supplizi, A.; Mechelli, L.; Sforna, M. Transcriptome Analysis of Canine Cutaneous Melanoma and Melanocytoma Reveals a Modulation of Genes Regulating Extracellular Matrix Metabolism and Cell Cycle. Sci. Rep. 2017, 7, 6386. [Google Scholar] [CrossRef]
- Dong, Y.; Fu, C.; Guan, H.; Zhang, Z.; Zhou, T.; Li, B.S. Prognostic significance of miR-126 in various cancers: A meta-analysis. Onco Targets Ther. 2016, 9, 2547–2555. [Google Scholar] [CrossRef]
- Liu, F.; Zhang, F.; Li, X.; Liu, Q.; Liu, W.; Song, P.; Qiu, Z.; Dong, Y.; Xiang, H. Prognostic role of miR-17-92 family in human cancers: Evaluation of multiple prognostic outcomes. Oncotarget 2017, 8, 69125–69138. [Google Scholar] [CrossRef]
- Scoggins, C.R.; Ross, M.I.; Reintgen, D.S.; Noyes, R.D.; Goydos, J.S.; Beitsch, P.D.; Urist, M.M.; Ariyan, S.; Sussman, J.J.; Edwards, M.J.; et al. Gender-related differences in outcome for melanoma patients. Ann. Surg. 2006, 243, 693–700. [Google Scholar] [CrossRef] [PubMed]
- Mihajlovic, M.; Vlajkovic, S.; Jovanovic, P.; Stefanovic, V. Primary mucosal melanomas: A comprehensive review. Int. J. Clin. Exp. Pathol. 2012, 5, 739–753. [Google Scholar] [PubMed]
- Muñoz-Rodríguez, J.L.; Vrba, L.; Futscher, B.W.; Hu, C.; Komenaka, I.K.; Meza-Montenegro, M.M.; Gutierrez-Millan, L.E.; Daneri-Navarro, A.; Thompson, P.A.; Martinez, M.E. Differentially expressed MicroRNAs in postpartum breast cancer in Hispanic women. PLoS ONE 2015, 10, e0124340. [Google Scholar] [CrossRef] [PubMed]
- Ghorai, A.; Ghosh, U. miRNA gene counts in chromosomes vary widely in a species and biogenesis of miRNA largely depends on transcription or post-transcriptional processing of coding genes. Front. Genet. 2014, 5, 100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozomara, A.; Griffiths-Jones, S. MiRBase: Annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014, 42, D68–D73. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; Smyth, G.K. Small-sample estimation of negative binomial dispersion, with applications to SAGE data. Biostatistics 2008, 9, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 2008, 3, 1103–1108. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Tan, Q.; Kir, J.; Liu, D.; Bryant, D.; Guo, Y.; Stephens, R.; Baseler, M.W.; Lane, H.C.; et al. DAVID Bioinformatics Resources: Expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res. 2007, 35, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 2005, 120, 15–20. [Google Scholar] [CrossRef]
- Wong, N.; Wang, X. miRDB: An online resource for microRNA target prediction and functional annotations. Nucleic Acids Res. 2015, 43, 146–152. [Google Scholar] [CrossRef]
- Lopes, C.T.; Franz, M.; Kazi, F.; Donaldson, S.L.; Morris, Q.; Bader, G.D.; Dopazo, J. Cytoscape Web: An interactive web-based network browser. Bioinformatics 2011, 27, 2347–2348. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef] [PubMed]
- Scardoni, G.; Petterlini, M.; Laudanna, C. Analyzing biological network parameters with CentiScaPe. Bioinformatics 2009, 25, 2857–2859. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Down-Regulated miRNA’s Target Genes Ontology | ||
|---|---|---|
| Biological Process | ||
| Terms | FE 1 | FDR 2 |
| GO:0018105~peptidyl-serine phosphorylation | 2.323 | 0.001 |
| GO:0045944~positive regulation of transcription from RNA polymerase II promoter | 1.461 | 0.025 |
| Cellular Component | ||
| GO:0005634~nucleus | 1.277 | 1.65 × 10−6 |
| GO:0005654~nucleoplasm | 1.374 | 2.49 × 10−5 |
| GO:0005737~cytoplasm | 1.232 | 1.09 × 10−4 |
| GO:0005911~cell-cell junction | 2.11 | 0.068146 |
| Molecular Function | ||
| GO:0004702~receptor signaling protein serine/threonine kinase activity | 2.932 | 9.98 × 10−4 |
| GO:0005201~extracellular matrix structural constituent | 3.373 | 0.002697 |
| GO:0003682~chromatin binding | 1.709 | 0.002892 |
| GO:0003714~transcription corepressor activity | 2.104 | 0.057312 |
| Up-Regulated miRNA’s Target Genes Ontology | ||
| Biological Process | ||
| Terms | FE 1 | FDR 2 |
| GO:0042787~protein ubiquitination involved in ubiquitin-dependent protein catabolic process | 2.208 | 0.004 |
| Cellular Component | ||
| GO:0005654~nucleoplasm | 1.441 | 1.35 × 10−7 |
| GO:0005737~cytoplasm | 1.228 | 2.87 × 10−4 |
| GO:0005794~Golgi apparatus | 1.590 | 8.03 × 10−4 |
| GO:0005634~nucleus | 1.228 | 0.001041 |
| Molecular Function | ||
| GO:0061630~ubiquitin protein ligase activity | 2.027 | 0.012 |
| GO:0044212~transcription regulatory region DNA binding | 2.127 | 0.019 |
| Down-Regulated miRNAs Target Genes Pathway | ||
|---|---|---|
| Terms | FE 1 | FDR 2 |
| cfa05206:MicroRNAs in cancer | 2.522 | 6.72 × 10−7 |
| cfa04010:MAPK signaling pathway | 1.917 | 1.20 × 10−4 |
| cfa04151:PI3K-Akt signaling pathway | 1.762 | 1.65 × 10−4 |
| cfa04360:Axon guidance | 2.307 | 4.96 × 10−4 |
| cfa05205:Proteoglycans in cancer | 1.981 | 8.64 × 10−4 |
| cfa04910:Insulin signaling pathway | 2.209 | 8.88 × 10−4 |
| cfa04152:AMPK signaling pathway | 2.230 | 0.003 |
| cfa04510:Focal adhesion | 1.901 | 0.003 |
| cfa04722:Neurotrophin signaling pathway | 2.164 | 0.010 |
| cfa04012:ErbB signaling pathway | 2.384 | 0.013 |
| cfa04512:ECM-receptor interaction | 2.384 | 0.013 |
| Up-regulated miRNAs target genes pathway | ||
| Terms | FE 1 | FDR 2 |
| cfa04144:Endocytosis | 2.424 | 1.31 × 10−11 |
| cfa04810:Regulation of actin cytoskeleton | 2.273 | 2.78 × 10−7 |
| cfa05200:Pathways in cancer | 1.604 | 0.010 |
| cfa04710:Circadian rhythm | 3.536 | 0.060 |
| cfa05410:Hypertrophic cardiomyopathy (HCM) | 2.398 | 0.063 |
| cfa05414:Dilated cardiomyopathy | 2.346 | 0.065 |
| cfa04713:Circadian entrainment | 2.210 | 0.098 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rahman, M.M.; Lai, Y.-C.; Husna, A.A.; Chen, H.-w.; Tanaka, Y.; Kawaguchi, H.; Miyoshi, N.; Nakagawa, T.; Fukushima, R.; Miura, N. Micro RNA Transcriptome Profile in Canine Oral Melanoma. Int. J. Mol. Sci. 2019, 20, 4832. https://doi.org/10.3390/ijms20194832
Rahman MM, Lai Y-C, Husna AA, Chen H-w, Tanaka Y, Kawaguchi H, Miyoshi N, Nakagawa T, Fukushima R, Miura N. Micro RNA Transcriptome Profile in Canine Oral Melanoma. International Journal of Molecular Sciences. 2019; 20(19):4832. https://doi.org/10.3390/ijms20194832
Chicago/Turabian StyleRahman, Md. Mahfuzur, Yu-Chang Lai, Al Asmaul Husna, Hui-wen Chen, Yuiko Tanaka, Hiroaki Kawaguchi, Noriaki Miyoshi, Takayuki Nakagawa, Ryuji Fukushima, and Naoki Miura. 2019. "Micro RNA Transcriptome Profile in Canine Oral Melanoma" International Journal of Molecular Sciences 20, no. 19: 4832. https://doi.org/10.3390/ijms20194832
APA StyleRahman, M. M., Lai, Y.-C., Husna, A. A., Chen, H.-w., Tanaka, Y., Kawaguchi, H., Miyoshi, N., Nakagawa, T., Fukushima, R., & Miura, N. (2019). Micro RNA Transcriptome Profile in Canine Oral Melanoma. International Journal of Molecular Sciences, 20(19), 4832. https://doi.org/10.3390/ijms20194832