Functional Genomics of the Retina to Elucidate its Construction and Deconstruction


Abstract
:1. Introduction
2. Retinal Development is Controlled by a Complex Gene Network
3. Inherited Retinal Diseases Caused by Photoreceptor Degeneration
4. Genomics and the Data Explosion
5. Retinal Specific Expression Patterns Revealed by Transcriptomics
6. The Function of Retinal Proteins Assessed Using Proteomics
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AMD | Age-related macular degeneration |
| ARMS2 | Age-Related Maculopathy Susceptibility 2 |
| bHLH | Basic helix-loop-helix |
| CERN | Conseil européen pour la recherche nucléaire - European Organization for Nuclear Research |
| CFH | Complement factor H |
| CNTF | Ciliary neurotrophic factor |
| CpG | Cytosine-phosphate-guanine |
| CRX | Cone rod homeobox |
| GDP/GTP | Guanosine di/tri phosphate |
| GWAS | Genome-wide association study |
| HTRA1 | HtrA Serine Peptidase 1 |
| LHC | Large hadron collider |
| NAD+ | Nicotinamide adenine dinucleotide |
| NRE2E3 | Nuclear receptor subfamily 2, group E, member 3 |
| NRL | Neural retina leucine zipper |
| OTX | Orthodenticle homeobox |
| PAX6 | Paired Box 6 |
| RB1 | Retinoblastoma 1 |
| rd1 | Retinal degeneration-1 |
| RHO | Rhodopsin |
| ROS | Reactive oxygen species |
| RP | Retinitis pigmentosa |
| RPE | Retinal pigmented epithelium |
| RPE65 | Retinal Pigment Epithelium-Specific 65 KDa Protein |
| RPGR | Retinitis pigmentosa GTPase regulator |
| SLC16A8 | Solute Carrier Family 16 Member 8 |
| SOS | Son of sevenless |
References
- Kolb, H. Simple Anatomy of the Retina. In Webvision: The Organization of the Retina and Visual System; Kolb, H., Fernandez, E., Nelson, R., Eds.; University of Utah Health Sciences Center: Salt Lake City, UT, USA, 1995. [Google Scholar]
- Wolff, T. Pattern formation in the Drosophila retina. Dev. Drosoph. Melanogaster 1993, 2, 1277–1325. [Google Scholar]
- Courgeon, M.; Desplan, C. Coordination of neural patterning in the Drosophila visual system. Curr. Opin. Neurobiol. 2019, 56, 153–159. [Google Scholar] [CrossRef]
- Nie, J.; Mahato, S.; Mustill, W.; Tipping, C.; Bhattacharya, S.S.; Zelhof, A.C. Cross species analysis of Prominin reveals a conserved cellular role in invertebrate and vertebrate photoreceptor cells. Dev. Biol. 2012, 371, 312–320. [Google Scholar] [CrossRef] [PubMed]
- Darwin, C. On the Origin of Species by Means of Natural Selection, or Preservation of Favoured Races in the Struggle for Life; John Murray: London, UK, 1859. [Google Scholar]
- Halder, G.; Callaerts, P.; Gehring, W.J. Induction of ectopic eyes by targeted expression of the eyeless gene in Drosophila. Science 1995, 267, 1788–1792. [Google Scholar] [CrossRef] [PubMed]
- Gehring, J.W. The evolution of vision. Wiley Interdiscip. Rev. Dev. Biol. 2014, 3, 1–40. [Google Scholar] [CrossRef] [PubMed]
- Bhaya, D. Light matters: Phototaxis and signal transduction in unicellular cyanobacteria. Mol. Microbiol. 2004, 53, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Dong, G.; Golden, S.S. How a cyanobacterium tells time. Curr. Opin. Microbiol. 2008, 11, 541–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Neill, J.S.; van Ooijen, G.; Dixon, L.E.; Troein, C.; Corellou, F.; Bouget, F.Y.; Reddy, A.B.; Millar, A.J. Circadian rhythms persist without transcription in a eukaryote. Nature 2011, 469, 554–558. [Google Scholar] [CrossRef]
- O’Neill, J.S.; Reddy, A.B. Circadian clocks in human red blood cells. Nature 2011, 469, 498–503. [Google Scholar] [CrossRef] [Green Version]
- Plachetzki, D.C.; Fong, C.R.; Oakley, T.H. Cnidocyte discharge is regulated by light and opsin-mediated phototransduction. BMC Biol. 2012, 10, 17. [Google Scholar] [CrossRef]
- Feinberg, T.E.; Mallatt, J.M. Consciousness Gets a Head Start Vertebrate Brains, Vision, and the Cambrian Birth of the Mental Image. In The Ancient Origins of Consciousness; MIT Press: Cambridge, MA, USA, 2016; pp. 69–100. [Google Scholar]
- Fain, G.L.; Hardie, R.; Laughlin, S.B. Phototransduction and the evolution of photoreceptors. Curr. Biol. 2010, 20, R114–R124. [Google Scholar] [CrossRef] [PubMed]
- Therrien, M.; Morrison, D.K.; Wong, A.M.; Rubin, G.M. A genetic screen for modifiers of a kinase suppressor of Ras-dependent rough eye phenotype in Drosophila. Genetics 2000, 156, 1231–1242. [Google Scholar] [PubMed]
- Rogge, R.D.; Karlovich, C.A.; Banerjee, U. Genetic dissection of a neurodevelopmental pathway: Son of sevenless functions downstream of the sevenless and EGF receptor tyrosine kinases. Cell 1991, 64, 39–48. [Google Scholar] [CrossRef]
- Bonfini, L.; Karlovich, C.A.; Dasgupta, C.; Banerjee, U. The Son of sevenless gene product: A putative activator of Ras. Science 1992, 255, 603–606. [Google Scholar] [CrossRef] [PubMed]
- Price, J.; Turner, D.; Cepko, C. Lineage analysis in the vertebrate nervous system by retrovirus-mediated gene transfer. Proc. Natl. Acad. Sci. USA 1987, 84, 156–160. [Google Scholar] [CrossRef]
- Livesey, F.J.; Cepko, C.L. Vertebrate neural cell-fate determination: Lessons from the retina. Nat. Rev. Neurosci. 2001, 2, 109–118. [Google Scholar] [CrossRef]
- Adler, R.; Hatlee, M. Plasticity and differentiation of embryonic retinal cells after terminal mitosis. Science 1989, 243, 391–393. [Google Scholar] [CrossRef]
- Furukawa, T.; Morrow, E.M.; Cepko, C.L. Crx, a novel otx-like homeobox gene, shows photoreceptor-specific expression and regulates photoreceptor differentiation. Cell 1997, 91, 531–541. [Google Scholar] [CrossRef]
- Chen, S.; Wang, Q.L.; Nie, Z.; Sun, H.; Lennon, G.; Copeland, N.G.; Gilbert, D.J.; Jenkins, N.A.; Zack, D.J. Crx, a novel Otx-like paired-homeodomain protein, binds to and transactivates photoreceptor cell-specific genes. Neuron 1997, 19, 1017–1030. [Google Scholar] [CrossRef]
- Swaroop, A.; Kim, D.; Forrest, D. Transcriptional regulation of photoreceptor development and homeostasis in the mammalian retina. Nat. Rev. Neurosci. 2010, 11, 563–576. [Google Scholar] [CrossRef] [Green Version]
- Swaroop, A.; Xu, J.Z.; Pawar, H.; Jackson, A.; Skolnick, C.; Agarwal, N. A conserved retina-specific gene encodes a basic motif/leucine zipper domain. Proc. Natl. Acad. Sci. USA 1992, 89, 266–270. [Google Scholar] [CrossRef] [PubMed]
- Mears, A.J.; Kondo, M.; Swain, P.K.; Takada, Y.; Bush, R.A.; Saunders, T.L.; Sieving, P.A.; Swaroop, A. Nrl is required for rod photoreceptor, development. Nat. Genet. 2001, 29, 447–452. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Khan, N.W.; Roger, J.E.; Swaroop, A. Excess cones in the retinal degeneration rd7 mouse, caused by the loss of function of orphan nuclear receptor Nr2e3, originate from early-born photoreceptor precursors. Hum. Mol. Genet. 2011, 20, 4102–4115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eldred, K.C.; Hadyniak, S.E.; Hussey, K.A.; Brenerman, B.; Zhang, P.W.; Chamling, X.; Sluch, V.M.; Welsbie, D.S.; Hattar, S.; Taylor, J.; et al. Thyroid hormone signaling specifies cone subtypes in human retinal organoids. Science 2018, 362, eaau6348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reichman, S.; Slembrouck, A.; Gagliardi, G.; Chaffiol, A.; Terray, A.; Nanteau, C.; Potey, A.; Belle, M.; Rabesandratana, O.; Duebel, J.; et al. Generation of Storable Retinal Organoids and Retinal Pigmented Epithelium from Adherent Human iPS Cells in Xeno-Free and Feeder-Free Conditions. Stem Cells 2017, 35, 1176–1188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.L.; Singh, H.P.; Wang, L.; Qi, D.L.; Poulos, B.K.; Abramson, D.H.; Jhanwar, S.C.; Cobrinik, D. Rb suppresses human cone-precursor-derived retinoblastoma tumours. Nature 2014, 514, 385–388. [Google Scholar] [CrossRef] [Green Version]
- Singh, H.P.; Wang, S.; Stachelek, K.; Lee, S.; Reid, M.W.; Thornton, M.E.; Craft, C.M.; Grubbs, B.H.; Cobrinik, D. Developmental stage-specific proliferation and retinoblastoma genesis in RB-deficient human but not mouse cone precursors. Proc. Natl. Acad. Sci. USA 2018, 115, E9391–E9400. [Google Scholar] [CrossRef] [Green Version]
- Leveillard, T. Cancer metabolism of cone photoreceptors. Oncotarget 2015, 6, 32285. [Google Scholar] [CrossRef]
- Friend, S.H.; Bernards, R.; Rogelj, S.; Weinberg, R.A.; Rapaport, J.M.; Albert, D.M.; Dryja, T.P. A human DNA segment with properties of the gene that predisposes to retinoblastoma and osteosarcoma. Nature 1986, 323, 643–646. [Google Scholar] [CrossRef]
- Fung, Y.K.; Murphree, A.L.; T’Ang, A.; Qian, J.; Hinrichs, S.H.; Benedict, W.F. Structural evidence for the authenticity of the human retinoblastoma gene. Science 1987, 236, 1657–1661. [Google Scholar] [CrossRef]
- Lee, W.H.; Bookstein, R.; Hong, F.; Young, L.J.; Shew, J.Y.; Lee, E.Y. Human retinoblastoma susceptibility gene: Cloning, identification, and sequence. Science 1987, 235, 1394–1399. [Google Scholar] [CrossRef] [PubMed]
- Dyson, N.J. RB1: A prototype tumor suppressor and an enigma. Genes Dev. 2016, 30, 1492–1502. [Google Scholar] [CrossRef] [PubMed]
- Elliott, J.; Cayouette, M.; Gravel, C. The CNTF/LIF signaling pathway regulates developmental programmed cell death and differentiation of rod precursor cells in the mouse retina in vivo. Dev. Biol. 2006, 300, 583–598. [Google Scholar] [CrossRef] [Green Version]
- Skowronska-Krawczyk, D.; Ballivet, M.; Dynlacht, B.D.; Matter, J.M. Highly specific interactions between bHLH transcription factors and chromatin during retina development. Development 2004, 131, 4447–4454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elgin, S.C.; Reuter, G. Position-effect variegation, heterochromatin formation, and gene silencing in Drosophila. Cold Spring Harb. Perspect. Biol. 2013, 5, a017780. [Google Scholar] [CrossRef]
- Merbs, S.L.; Khan, M.A.; Hackler, L.; Oliver, V.F.; Wan, J.; Qian, J.; Zack, D.J. Cell-specific DNA methylation patterns of retina-specific genes. PLoS ONE 2012, 7, e32602. [Google Scholar] [CrossRef]
- Mo, A.; Luo, C.; Davis, F.P.; Mukamel, E.A.; Henry, G.L.; Nery, J.R.; Urich, M.A.; Picard, S.; Lister, R.; Eddy, S.R.; et al. Epigenomic landscapes of retinal rods and cones. Elife 2016, 5, e11613. [Google Scholar] [CrossRef]
- Farinelli, P.; Perera, A.; Arango-Gonzalez, B.; Trifunovic, D.; Wagner, M.; Carell, T.; Biel, M.; Zrenner, E.; Michalakis, S.; Paquet-Durand, F.; et al. DNA methylation and differential gene regulation in photoreceptor cell death. Cell Death Dis. 2014, 5, e1558. [Google Scholar] [CrossRef]
- Corso-Diaz, X.; Jaeger, C.; Chaitankar, V.; Swaroop, A. Epigenetic control of gene regulation during development and disease: A view from the retina. Prog. Retin. Eye Res. 2018, 65, 1–27. [Google Scholar] [CrossRef]
- Seritrakul, P.; Gross, J.M. Genetic and epigenetic control of retinal development in zebrafish. Curr. Opin. Neurobiol. 2019, 59, 120–127. [Google Scholar] [CrossRef]
- Naruse, Y.; Oh-hashi, K.; Iijima, N.; Naruse, M.; Yoshioka, H.; Tanaka, M. Circadian and light-induced transcription of clock gene Per1 depends on histone acetylation and deacetylation. Mol. Cell. Biol. 2004, 24, 6278–6287. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Cepko, C.L. HDAC4 regulates neuronal survival in normal and diseased retinas. Science 2009, 323, 256–259. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, R.C.; Popova, E.Y.; James, J.; Briones, M.R.; Zhang, S.S.; Barnstable, C.J. Histone Deacetylase 1 Is Essential for Rod Photoreceptor Differentiation by Regulating Acetylation at Histone H3 Lysine 9 and Histone H4 Lysine 12 in the Mouse Retina. J. Biol. Chem. 2017, 292, 2422–2440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Du, J.; Justus, S.; Hsu, C.W.; Bonet-Ponce, L.; Wu, W.H.; Tsai, Y.T.; Wu, W.P.; Jia, Y.; Duong, J.K.; et al. Reprogramming metabolism by targeting sirtuin 6 attenuates retinal degeneration. J. Clin. Investig. 2016, 126, 4659–4673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albadri, S.; Naso, F.; Thauvin, M.; Gauron, C.; Parolin, C.; Duroure, K.; Vougny, J.; Fiori, J.; Boga, C.; Vriz, S.; et al. Redox Signaling via Lipid Peroxidation Regulates Retinal Progenitor Cell Differentiation. Dev. Cell 2019, 50, 73–89. [Google Scholar] [CrossRef] [PubMed]
- Freund, C.L.; Gregory-Evans, C.Y.; Furukawa, T.; Papaioannou, M.; Looser, J.; Ploder, L.; Bellingham, J.; Ng, D.; Herbrick, J.A.; Duncan, A.; et al. Cone-rod dystrophy due to mutations in a novel photoreceptor-specific homeobox gene (CRX) essential for maintenance of the photoreceptor. Cell 1997, 91, 543–553. [Google Scholar] [CrossRef]
- Bessant, D.A.; Payne, A.M.; Mitton, K.P.; Wang, Q.L.; Swain, P.K.; Plant, C.; Bird, A.C.; Zack, D.J.; Swaroop, A.; Bhattacharya, S.S. A mutation in NRL is associated with autosomal dominant retinitis pigmentosa. Nat. Genet. 1999, 21, 355. [Google Scholar] [CrossRef] [PubMed]
- Haider, N.B.; Jacobson, S.G.; Cideciyan, A.V.; Swiderski, R.; Streb, L.M.; Searby, C.; Beck, G.; Hockey, R.; Hanna, D.B.; Gorman, S.; et al. Mutation of a nuclear receptor gene, NR2E3, causes enhanced S cone syndrome, a disorder of retinal cell fate. Nat. Genet. 2000, 24, 127–131. [Google Scholar] [CrossRef]
- Gregory-Evans, K.; Pennesi, M.E.; Weleber, R.G. Chapter 40—Retinitis Pigmentosa and Allied Disorders. In Retina (Fifth Edition); Ryan, S.J., Sadda, S.R., Hinton, D.R., Schachat, A.P., Sadda, S.R., Wilkinson, C.P., Wiedemann, P., Schachat, A.P., Eds.; W.B. Saunders: London, UK, 2013; pp. 761–835. [Google Scholar]
- Duncan, J.L.; Pierce, E.A.; Laster, A.M.; Daiger, S.P.; Birch, D.G.; Ash, J.D.; Iannaccone, A.; Flannery, J.G.; Sahel, J.A.; Zack, D.J.; et al. Inherited retinal degenerations: Current landscape and knowledge gaps. Transl. Vis. Sci. Technol. 2018, 7, 6. [Google Scholar] [CrossRef]
- Hartong, D.T.; Berson, E.L.; Dryja, T.P. Retinitis pigmentosa. Lancet 2006, 368, 1795–1809. [Google Scholar] [CrossRef]
- Hamel, C. Retinitis pigmentosa. Orphanet J. Rare Dis. 2006, 1, 40. [Google Scholar] [CrossRef]
- Portera-Cailliau, C.; Sung, C.H.; Nathans, J.; Adler, R. Apoptotic photoreceptor cell death in mouse models of retinitis pigmentosa. Proc. Natl. Acad. Sci. USA 1994, 91, 974–978. [Google Scholar] [CrossRef]
- Zeitz, C.; Robson, A.G.; Audo, I. Congenital stationary night blindness: An analysis and update of genotype-phenotype correlations and pathogenic mechanisms. Prog. Retin. Eye Res. 2015, 45, 58–110. [Google Scholar] [CrossRef] [PubMed]
- Hamel, C.P. Cone rod dystrophies. Orphanet J. Rare Dis. 2007, 2, 7. [Google Scholar] [CrossRef] [PubMed]
- Allikmets, R.; Singh, N.; Sun, H.; Shroyer, N.F.; Hutchinson, A.; Chidambaram, A.; Gerrard, B.; Baird, L.; Stauffer, D.; Peiffer, A.; et al. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat. Genet. 1997, 15, 236–246. [Google Scholar] [CrossRef] [PubMed]
- Perrault, I.; Rozet, J.M.; Calvas, P.; Gerber, S.; Camuzat, A.; Dollfus, H.; Chatelin, S.; Souied, E.; Ghazi, I.; Leowski, C.; et al. Retinal-specific guanylate cyclase gene mutations in Leber’s congenital amaurosis. Nat. Genet. 1996, 14, 461–464. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.M.; Thompson, D.A.; Srikumari, C.R.; Lorenz, B.; Finckh, U.; Nicoletti, A.; Murthy, K.R.; Rathmann, M.; Kumaramanickavel, G.; Denton, M.J.; et al. Mutations in RPE65 cause autosomal recessive childhood-onset severe retinal dystrophy. Nat. Genet. 1997, 17, 194–197. [Google Scholar] [CrossRef] [PubMed]
- Kohl, S.; Marx, T.; Giddings, I.; Jagle, H.; Jacobson, S.G.; Apfelstedt-Sylla, E.; Zrenner, E.; Sharpe, L.T.; Wissinger, B. Total colourblindness is caused by mutations in the gene encoding the alpha-subunit of the cone photoreceptor cGMP-gated cation channel. Nat. Genet. 1998, 19, 257–925. [Google Scholar] [CrossRef] [PubMed]
- Kohl, S.; Baumann, B.; Broghammer, M.; Jagle, H.; Sieving, P.; Kellner, U.; Spegal, R.; Anastasi, M.; Zrenner, E.; Sharpe, L.T.; et al. Mutations in the CNGB3 gene encoding the beta-subunit of the cone photoreceptor cGMP-gated channel are responsible for achromatopsia (ACHM3) linked to chromosome 8q21. Hum. Mol. Genet. 2000, 9, 2107–2116. [Google Scholar] [CrossRef]
- Hirji, N.; Aboshiha, J.; Georgiou, M.; Bainbridge, J.; Michaelides, M. Achromatopsia: Clinical features, molecular genetics, animal models and therapeutic options. Ophthalmic Genet. 2018, 39, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Cremers, F.P.M.; Boon, C.J.F.; Bujakowska, K.; Zeitz, C. Special Issue Introduction: Inherited Retinal Disease: Novel Candidate Genes, Genotype-Phenotype Correlations, and Inheritance Models. Genes 2018, 9, 215. [Google Scholar] [CrossRef]
- Weil, D.; Blanchard, S.; Kaplan, J.; Guilford, P.; Gibson, F.; Walsh, J.; Mburu, P.; Varela, A.; Levilliers, J.; Weston, M.D.; et al. Defective myosin VIIA gene responsible for Usher syndrome type 1B. Nature 1995, 374, 60–61. [Google Scholar] [CrossRef] [PubMed]
- Eudy, J.D.; Weston, M.D.; Yao, S.; Hoover, D.M.; Rehm, H.L.; Ma-Edmonds, M.; Yan, D.; Ahmad, I.; Cheng, J.J.; Ayuso, C.; et al. Mutation of a gene encoding a protein with extracellular matrix motifs in Usher syndrome type IIa. Science 1998, 280, 1753–1757. [Google Scholar] [CrossRef] [PubMed]
- Katsanis, N.; Beales, P.L.; Woods, M.O.; Lewis, R.A.; Green, J.S.; Parfrey, P.S.; Ansley, S.J.; Davidson, W.S.; Lupski, J.R. Mutations in MKKS cause obesity, retinal dystrophy and renal malformations associated with Bardet-Biedl syndrome. Nat. Genet. 2000, 26, 67–70. [Google Scholar] [CrossRef]
- Mockel, A.; Perdomo, Y.; Stutzmann, F.; Letsch, J.; Marion, V.; Dollfus, H. Retinal dystrophy in Bardet-Biedl syndrome and related syndromic ciliopathies. Prog. Retin. Eye Res. 2011, 30, 258–274. [Google Scholar] [CrossRef] [PubMed]
- Nachury, M.V.; Loktev, A.V.; Zhang, Q.; Westlake, C.J.; Peranen, J.; Merdes, A.; Slusarski, D.C.; Scheller, R.H.; Bazan, J.F.; Sheffield, V.C.; et al. A core complex of BBS proteins cooperates with the GTPase Rab8 to promote ciliary membrane biogenesis. Cell 2007, 129, 1201–1213. [Google Scholar] [CrossRef]
- Ait-Ali, N.; Fridlich, R.; Millet-Puel, G.; Clerin, E.; Delalande, F.; Jaillard, C.; Blond, F.; Perrocheau, L.; Reichman, S.; Byrne, L.C.; et al. Rod-derived cone viability factor promotes cone survival by stimulating aerobic glycolysis. Cell 2015, 161, 817–832. [Google Scholar] [CrossRef]
- Delyfer, M.N.; Raffelsberger, W.; Mercier, D.; Korobelnik, J.F.; Gaudric, A.; Charteris, D.G.; Tadayoni, R.; Metge, F.; Caputo, G.; Barale, P.O.; et al. Transcriptomic analysis of human retinal detachment reveals both inflammatory response and photoreceptor death. PLoS ONE 2011, 6, e28791. [Google Scholar] [CrossRef]
- Kole, C.; Berdugo, N.; da Silva, C.; Ait-Ali, N.; Millet-Puel, G.; Pagan, D.; Blond, F.; Poidevin, L.; Ripp, R.; Fontaine, V.; et al. Identification of an Alternative Splicing Product of the Otx2 Gene Expressed in the Neural Retina and Retinal Pigmented Epithelial Cells. PLoS ONE 2016, 11, e0150758. [Google Scholar] [CrossRef]
- Humphries, P.; Kenna, P.; Farrar, G.J. On the molecular genetics of retinitis pigmentosa. Science 1992, 256, 804–808. [Google Scholar] [CrossRef]
- Thompson, D.A.; Gal, A. Vitamin A metabolism in the retinal pigment epithelium: Genes, mutations, and diseases. Prog. Retin. Eye Res. 2003, 22, 683–703. [Google Scholar] [CrossRef]
- Acland, G.M.; Aguirre, G.D.; Ray, J.; Zhang, Q.; Aleman, T.S.; Cideciyan, A.V.; Pearce-Kelling, S.E.; Anand, V.; Zeng, Y.; Maguire, A.M.; et al. Gene therapy restores vision in a canine model of childhood blindness. Nat. Genet. 2001, 28, 92–95. [Google Scholar] [CrossRef] [PubMed]
- Maguire, A.M.; Simonelli, F.; Pierce, E.A.; Pugh, E.N.; Mingozzi, F.; Bennicelli, J.; Banfi, S.; Marshall, K.A.; Testa, F.; Surace, E.M.; et al. Safety and efficacy of gene transfer for Leber’s congenital amaurosis. N. Engl. J. Med. 2008, 358, 2240–2248. [Google Scholar] [CrossRef] [PubMed]
- Russell, S.; Bennett, J.; Wellman, J.A.; Chung, D.C.; Yu, Z.F.; Tillman, A.; Wittes, J.; Pappas, J.; Elci, O.; McCague, S.; et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: A randomised, controlled, open-label, phase 3 trial. Lancet 2017, 390, 849–860. [Google Scholar] [CrossRef]
- Bainbridge, J.W.; Smith, A.J.; Barker, S.S.; Robbie, S.; Henderson, R.; Balaggan, K.; Viswanathan, A.; Holder, G.E.; Stockman, A.; Tyler, N.; et al. Effect of gene therapy on visual function in Leber’s congenital amaurosis. N. Engl. J. Med. 2008, 358, 2231–2239. [Google Scholar] [CrossRef] [PubMed]
- Cideciyan, A.V.; Jacobson, S.G.; Beltran, W.A.; Sumaroka, A.; Swider, M.; Iwabe, S.; Roman, A.J.; Olivares, M.B.; Schwartz, S.B.; Komaromy, A.M.; et al. Human retinal gene therapy for Leber congenital amaurosis shows advancing retinal degeneration despite enduring visual improvement. Proc. Natl. Acad. Sci. USA 2013, 110, E517–E525. [Google Scholar] [CrossRef] [Green Version]
- Patel, U.; Boucher, M.; de Leseleuc, L.; Visintini, S. Voretigene Neparvovec: An Emerging Gene Therapy for the Treatment of Inherited Blindness. In CAD TH Issue in Emerging Health Technologies; Canadian Agency for Drugs and Technologies in Health: Ottawa, ON, Canada, 2018; pp. 1–11. [Google Scholar]
- Hamel, C.P.; Tsilou, E.; Pfeffer, B.A.; Hooks, J.J.; Detrick, B.; Redmond, T.M. Molecular cloning and expression of RPE65, a novel retinal pigment epithelium-specific microsomal protein that is post-transcriptionally regulated in vitro. J. Biol. Chem. 1993, 268, 15751–15757. [Google Scholar]
- Strom, S.P.; Clark, M.J.; Martinez, A.; Garcia, S.; Abelazeem, A.A.; Matynia, A.; Parikh, S.; Sullivan, L.S.; Bowne, S.J.; Daiger, S.P.; et al. De Novo Occurrence of a Variant in ARL3 and Apparent Autosomal Dominant Transmission of Retinitis Pigmentosa. PLoS ONE 2016, 11, e0150944. [Google Scholar] [CrossRef]
- Wright, Z.C.; Singh, R.K.; Alpino, R.; Goldberg, A.F.; Sokolov, M.; Ramamurthy, V. ARL3 regulates trafficking of prenylated phototransduction proteins to the rod outer segment. Hum. Mol. Genet. 2016, 25, 2031–2044. [Google Scholar] [CrossRef] [Green Version]
- Xu, M.; Eblimit, A.; Wang, J.; Li, J.; Wang, F.; Zhao, L.; Wang, X.; Xiao, N.; Li, Y.; Wong, L.J.; et al. ADIPOR1 Is Mutated in Syndromic Retinitis Pigmentosa. Hum. Mutat. 2016, 37, 246–249. [Google Scholar] [CrossRef]
- Sluch, V.M.; Banks, A.; Li, H.; Crowley, M.A.; Davis, V.; Xiang, C.; Yang, J.; Demirs, J.T.; Vrouvlianis, J.; Leehy, B.; et al. ADIPOR1 is essential for vision and its RPE expression is lost in the Mfrp(rd6) mouse. Sci. Rep. 2018, 8, 14339. [Google Scholar] [CrossRef] [PubMed]
- Davidson, A.E.; Millar, I.D.; Urquhart, J.E.; Burgess-Mullan, R.; Shweikh, Y.; Parry, N.; O’Sullivan, J.; Maher, G.J.; McKibbin, M.; Downes, S.M.; et al. Missense mutations in a retinal pigment epithelium protein, bestrophin-1, cause retinitis pigmentosa. Am. J. Hum. Genet. 2009, 85, 581–592. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Stanton, J.B.; Wu, J.; Yu, K.; Hartzell, H.C.; Peachey, N.S.; Marmorstein, L.Y.; Marmorstein, A.D. Suppression of Ca2+ signaling in a mouse model of Best disease. Hum. Mol. Genet. 2010, 19, 1108–1118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guziewicz, K.E.; Zangerl, B.; Lindauer, S.J.; Mullins, R.F.; Sandmeyer, L.S.; Grahn, B.H.; Stone, E.M.; Acland, G.M.; Aguirre, G.D. Bestrophin gene mutations cause canine multifocal retinopathy: A novel animal model for best disease. Investig. Ophthalmol. Vis. Sci. 2007, 48, 1959–1967. [Google Scholar] [CrossRef] [PubMed]
- Zangerl, B.; Wickstrom, K.; Slavik, J.; Lindauer, S.J.; Ahonen, S.; Schelling, C.; Lohi, H.; Guziewicz, K.E.; Aguirre, G.D. Assessment of canine BEST1 variations identifies new mutations and establishes an independent bestrophinopathy model (cmr3). Mol. Vis. 2010, 16, 2791–2804. [Google Scholar]
- Datta, R.; Waheed, A.; Bonapace, G.; Shah, G.N.; Sly, W.S. Pathogenesis of retinitis pigmentosa associated with apoptosis-inducing mutations in carbonic anhydrase IV. Proc. Natl. Acad. Sci. USA 2009, 106, 3437–3442. [Google Scholar] [CrossRef] [Green Version]
- Tran, N.M.; Zhang, A.; Zhang, X.; Huecker, J.B.; Hennig, A.K.; Chen, S. Mechanistically distinct mouse models for CRX-associated retinopathy. PLoS Genet. 2014, 10, e1004111. [Google Scholar] [CrossRef]
- Occelli, L.M.; Tran, N.M.; Narfstrom, K.; Chen, S.; Petersen-Jones, S.M. CrxRdy Cat: A large animal model for CRX-associated leber congenital amaurosis. Investig. Ophthalmol. Vis. Sci. 2016, 57, 3780–3792. [Google Scholar] [CrossRef]
- Sullivan, L.S.; Bowne, S.J.; Birch, D.G.; Hughbanks-Wheaton, D.; Heckenlively, J.R.; Lewis, R.A.; Garcia, C.A.; Ruiz, R.S.; Blanton, S.H.; Northrup, H.; et al. Prevalence of disease-causing mutations in families with autosomal dominant retinitis pigmentosa: A screen of known genes in 200 families. Investig. Ophthalmol. Vis. Sci. 2006, 47, 3052–3064. [Google Scholar] [CrossRef]
- Yokokura, S.; Wada, Y.; Nakai, S.; Sato, H.; Yao, R.; Yamanaka, H.; Ito, S.; Sagara, Y.; Takahashi, M.; Nakamura, Y.; et al. Targeted disruption of FSCN2 gene induces retinopathy in mice. Investig. Ophthalmol. Vis. Sci. 2005, 46, 2905–2915. [Google Scholar] [CrossRef]
- Pennesi, M.E.; Howes, K.A.; Baehr, W.; Wu, S.M. Guanylate cyclase-activating protein (GCAP) 1 rescues cone recovery kinetics in GCAP1/GCAP2 knockout mice. Proc. Natl. Acad. Sci. USA 2003, 100, 6783–6788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sullivan, L.S.; Koboldt, D.C.; Bowne, S.J.; Lang, S.; Blanton, S.H.; Cadena, E.; Avery, C.E.; Lewis, R.A.; Webb-Jones, K.; Wheaton, D.H.; et al. A Dominant Mutation in Hexokinase 1 (HK1) Causes Retinitis Pigmentosa. Investig. Ophthalmol. Vis. Sci. 2014, 55, 7147–7158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tam, L.C.; Kiang, A.S.; Kennan, A.; Kenna, P.F.; Chadderton, N.; Ader, M.; Palfi, A.; Aherne, A.; Ayuso, C.; Campbell, M.; et al. Therapeutic benefit derived from RNAi-mediated ablation of IMPDH1 transcripts in a murine model of autosomal dominant retinitis pigmentosa (RP10). Hum. Mol. Genet. 2008, 17, 2084–2100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manes, G.; Meunier, I.; Avila-Fernandez, A.; Banfi, S.; le Meur, G.; Zanlonghi, X.; Corton, M.; Simonelli, F.; Brabet, P.; Labesse, G.; et al. Mutations in IMPG1 cause vitelliform macular dystrophies. Am. J. Hum. Genet. 2013, 93, 571–578. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.S.; Ray, J.W.; Waseem, N.; Johnson, K.; Brooks, M.J.; Hugosson, T.; Breuer, D.; Branham, K.E.; Krauth, D.S.; Bowne, S.J.; et al. Mutations in a BTB-Kelch protein, KLHL7, cause autosomal-dominant retinitis pigmentosa. Am. J. Hum. Genet. 2009, 84, 792–800. [Google Scholar] [CrossRef]
- Blanco-Kelly, F.; Garcia Hoyos, M.; Lopez Martinez, M.A.; Lopez-Molina, M.I.; Riveiro-Alvarez, R.; Fernandez-San Jose, P.; Avila-Fernandez, A.; Corton, M.; Millan, J.M.; Garcia Sandoval, B.; et al. Dominant Retinitis Pigmentosa, p.Gly56Arg Mutation in NR2E3: Phenotype in a Large Cohort of 24 Cases. PLoS ONE 2016, 11, e0149473. [Google Scholar] [CrossRef]
- Akhmedov, N.B.; Piriev, N.I.; Chang, B.; Rapoport, A.L.; Hawes, N.L.; Nishina, P.M.; Nusinowitz, S.; Heckenlively, J.R.; Roderick, T.H.; Kozak, C.A.; et al. A deletion in a photoreceptor-specific nuclear receptor mRNA causes retinal degeneration in the rd7 mouse. Proc. Natl. Acad. Sci. USA 2000, 97, 5551–5556. [Google Scholar] [CrossRef]
- Graziotto, J.J.; Farkas, M.H.; Bujakowska, K.; Deramaudt, B.M.; Zhang, Q.; Nandrot, E.F.; Inglehearn, C.F.; Bhattacharya, S.S.; Pierce, E.A. Three Gene-Targeted Mouse Models of RNA Splicing Factor RP Show Late-Onset RPE and Retinal Degeneration. Investig. Ophthalmol. Vis. Sci. 2011, 52, 190–198. [Google Scholar] [CrossRef] [Green Version]
- Ruzickova, S.; Stanek, D. Mutations in spliceosomal proteins and retina degeneration. RNA Biol. 2017, 14, 544–552. [Google Scholar] [CrossRef]
- Chen, X.; Liu, Y.; Sheng, X.; Tam, P.O.; Zhao, K.; Chen, X.; Rong, W.; Liu, Y.; Liu, X.; Pan, X.; et al. PRPF4 mutations cause autosomal dominant retinitis pigmentosa. Hum. Mol. Genet. 2014, 23, 2926–2939. [Google Scholar] [CrossRef] [Green Version]
- Farkas, M.H.; Lew, D.S.; Sousa, M.E.; Bujakowska, K.; Chatagnon, J.; Bhattacharya, S.S.; Pierce, E.A.; Nandrot, E.F. Mutations in pre-mRNA processing factors 3, 8, and 31 cause dysfunction of the retinal pigment epithelium. Am. J. Pathol. 2014, 184, 2641–2652. [Google Scholar] [CrossRef] [PubMed]
- Kajiwara, K.; Hahn, L.B.; Mukai, S.; Travis, G.H.; Berson, E.L.; Dryja, T.P. Mutations in the human retinal degeneration slow gene in autosomal dominant retinitis pigmentosa. Nature 1991, 354, 480–483. [Google Scholar] [CrossRef] [PubMed]
- Hawes, N.L.; Smith, R.S.; Chang, B.; Davisson, M.; Heckenlively, J.R.; John, S.W. Mouse fundus photography and angiography: A catalogue of normal and mutant phenotypes. Mol. Vis. 1999, 5, 22. [Google Scholar] [PubMed]
- Fingert, J.H.; Oh, K.; Chung, M.; Scheetz, T.E.; Andorf, J.L.; Johnson, R.M.; Sheffield, V.C.; Stone, E.M. Association of a novel mutation in the retinol dehydrogenase 12 (RDH12) gene with autosomal dominant retinitis pigmentosa. Arch. Ophthalmol. 2008, 126, 1301–1307. [Google Scholar] [CrossRef] [PubMed]
- Maeda, A.; Maeda, T.; Sun, W.; Zhang, H.; Baehr, W.; Palczewski, K. Redundant and unique roles of retinol dehydrogenases in the mouse retina. Proc. Natl. Acad. Sci. USA 2007, 104, 19565–19570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olsson, J.E.; Gordon, J.W.; Pawlyk, B.S.; Roof, D.; Hayes, A.; Molday, R.S.; Mukai, S.; Cowley, G.S.; Berson, E.L.; Dryja, T.P. Transgenic mice with a rhodopsin mutation (Pro23His): A mouse model of autosomal dominant retinitis pigmentosa. Neuron 1992, 9, 815–830. [Google Scholar] [CrossRef]
- Kennan, A.; Aherne, A.; Palfi, A.; Humphries, M.; McKee, A.; Stitt, A.; Simpson, D.A.; Demtroder, K.; Orntoft, T.; Ayuso, C.; et al. Identification of an IMPDH1 mutation in autosomal dominant retinitis pigmentosa (RP10) revealed following comparative microarray analysis of transcripts derived from retinas of wild-type and Rho(-/-) mice. Hum. Mol. Genet. 2002, 11, 547–557. [Google Scholar] [CrossRef] [PubMed]
- Orhan, E.; Dalkara, D.; Neuille, M.; Lechauve, C.; Michiels, C.; Picaud, S.; Leveillard, T.; Sahel, J.A.; Naash, M.I.; Lavail, M.M.; et al. Genotypic and phenotypic characterization of P23H line 1 rat model. PLoS ONE 2015, 10, e0127319. [Google Scholar] [CrossRef]
- Wang, W.; Lee, S.J.; Scott, P.A.; Lu, X.; Emery, D.; Liu, Y.; Ezashi, T.; Roberts, M.R.; Ross, J.W.; Kaplan, H.J.; et al. Two-Step Reactivation of Dormant Cones in Retinitis Pigmentosa. Cell. Rep. 2016, 15, 372–385. [Google Scholar] [CrossRef] [Green Version]
- Kostic, C.; Arsenijevic, Y. Animal modelling for inherited central vision loss. J. Pathol. 2016, 238, 300–310. [Google Scholar] [CrossRef]
- Clarke, G.; Goldberg, A.F.X.; Vidgen, D.; Collins, L.; Ploder, L.; Schwarz, L.; Molday, L.L.; Rossant, J.; Szel, A.; Molday, R.S.; et al. Rom-1 is required for rod photoreceptor viability and the regulation of disk morphogenesis. Nat. Genet. 2000, 25, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Zuo, J.; Pierce, E.A. The retinitis pigmentosa 1 protein is a photoreceptor microtubule- associated protein. J. Neurosci. 2004, 24, 6427–6436. [Google Scholar] [CrossRef] [PubMed]
- Song, D.; Grieco, S.; Li, Y.; Hunter, A.; Chu, S.; Zhao, L.; Song, Y.; DeAngelis, R.A.; Shi, L.Y.; Liu, Q.; et al. A murine RP1 missense mutation causes protein mislocalization and slowly progressive photoreceptor degeneration. Am. J. Pathol. 2014, 184, 2721–2729. [Google Scholar] [CrossRef] [PubMed]
- Abid, A.; Ismail, M.; Mehdi, S.Q.; Khaliq, S. Identification of novel mutations in the SEMA4A gene associated with retinal degenerative diseases. J. Med. Genet. 2006, 43, 378–381. [Google Scholar] [CrossRef] [PubMed]
- Rice, D.S.; Huang, W.; Jones, H.A.; Hansen, G.; Ye, G.L.; Xu, N.; Wilson, E.A.; Troughton, K.; Vaddi, K.; Newton, R.C.; et al. Severe retinal degeneration associated with disruption of semaphorin 4A. Investig. Ophthalmol. Vis. Sci. 2004, 45, 2767–2777. [Google Scholar] [CrossRef] [PubMed]
- Coussa, R.G.; Chakarova, C.; Ajlan, R.; Taha, M.; Kavalec, C.; Gomolin, J.; Khan, A.; Lopez, I.; Ren, H.; Waseem, N.; et al. Genotype and Phenotype Studies in Autosomal Dominant Retinitis Pigmentosa (adRP) of the French Canadian Founder Population. Investig. Ophthalmol. Vis. Sci. 2015, 56, 8297–8305. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, X.; Xu, Q.; Gao, X.; Tam, P.O.; Zhao, K.; Zhang, X.; Chen, L.J.; Jia, W.; Zhao, Q.; et al. SPP2 Mutations Cause Autosomal Dominant Retinitis Pigmentosa. Sci. Rep. 2015, 5, 14867. [Google Scholar] [CrossRef] [PubMed]
- Schob, C.; Orth, U.; Gal, A.; Kindler, S.; Chakarova, C.F.; Bhattacharya, S.S.; Ruther, K. Mutations in TOPORS: A rare cause of autosomal dominant retinitis pigmentosa in continental Europe? Ophthalmic Genet. 2009, 30, 96–98. [Google Scholar] [CrossRef]
- Marshall, H.; Bhaumik, M.; Aviv, H.; Moore, D.; Yao, M.; Dutta, J.; Rahim, H.; Gounder, M.; Ganesan, S.; Saleem, A.; et al. Deficiency of the dual ubiquitin/SUMO ligase Topors results in genetic instability and an increased rate of malignancy in mice. BMC Mol. Biol. 2010, 11, 31. [Google Scholar] [CrossRef]
- Weng, J.; Mata, N.L.; Azarian, S.M.; Tzekov, R.T.; Birch, D.G.; Travis, G.H. Insights into the function of Rim protein in photoreceptors and etiology of Stargardt’s disease from the phenotype in abcr knockout mice. Cell 1999, 98, 13–23. [Google Scholar] [CrossRef]
- Makelainen, S.; Godia, M.; Hellsand, M.; Viluma, A.; Hahn, D.; Makdoumi, K.; Zeiss, C.J.; Mellersh, C.; Ricketts, S.L.; Narfstrom, K.; et al. An ABCA4 loss-of-function mutation causes a canine form of Stargardt disease. PLoS Genet. 2019, 15, e1007873. [Google Scholar] [CrossRef] [PubMed]
- Branham, K.; Matsui, H.; Biswas, P.; Guru, A.A.; Hicks, M.; Suk, J.J.; Li, H.; Jakubosky, D.; Long, T.; Telenti, A.; et al. Establishing the involvement of the novel gene AGBL5 in retinitis pigmentosa by whole genome sequencing. Physiol. Genom. 2016, 48, 922–927. [Google Scholar] [CrossRef]
- Zhou, Y.; Li, S.; Huang, L.; Yang, Y.; Zhang, L.; Yang, M.; Liu, W.; Ramasamy, K.; Jiang, Z.; Sundaresan, P.; et al. A splicing mutation in aryl hydrocarbon receptor associated with retinitis pigmentosa. Hum. Mol. Genet. 2018, 27, 2563–2572. [Google Scholar] [CrossRef]
- Kim, S.Y.; Yang, H.J.; Chang, Y.S.; Kim, J.W.; Brooks, M.; Chew, E.Y.; Wong, W.T.; Fariss, R.N.; Rachel, R.A.; Cogliati, T.; et al. Deletion of aryl hydrocarbon receptor AHR in mice leads to subretinal accumulation of microglia and RPE atrophy. Investig. Ophthalmol. Vis. Sci. 2014, 55, 6031–6040. [Google Scholar] [CrossRef] [PubMed]
- Arno, G.; Carss, K.J.; Hull, S.; Zihni, C.; Robson, A.G.; Fiorentino, A.; Consortium, U.K.I.R.D.; Hardcastle, A.J.; Holder, G.E.; Cheetham, M.E.; et al. Biallelic Mutation of ARHGEF18, Involved in the Determination of Epithelial Apicobasal Polarity, Causes Adult-Onset Retinal Degeneration. Am. J. Hum. Genet. 2017, 100, 334–342. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Nishimura, D.; Seo, S.; Vogel, T.; Morgan, D.A.; Searby, C.; Bugge, K.; Stone, E.M.; Rahmouni, K.; Sheffield, V.C. Bardet-Biedl syndrome 3 (Bbs3) knockout mouse model reveals common BBS-associated phenotypes and Bbs3 unique phenotypes. Proc. Natl. Acad. Sci. USA 2011, 108, 20678–20683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davidson, A.E.; Schwarz, N.; Zelinger, L.; Stern-Schneider, G.; Shoemark, A.; Spitzbarth, B.; Gross, M.; Laxer, U.; Sosna, J.; Sergouniotis, P.; et al. Mutations in ARL2BP, encoding ADP-ribosylation-factor-like 2 binding protein, cause autosomal-recessive retinitis pigmentosa. Am. J. Hum. Genet. 2013, 93, 321–329. [Google Scholar] [CrossRef]
- Davis, R.E.; Swiderski, R.E.; Rahmouni, K.; Nishimura, D.Y.; Mullins, R.F.; Agassandian, K.; Philp, A.R.; Searby, C.C.; Andrews, M.P.; Thompson, S.; et al. A knockin mouse model of the Bardet-Biedl syndrome 1 M390R mutation has cilia defects, ventriculomegaly, retinopathy, and obesity. Proc. Natl. Acad. Sci. USA 2007, 104, 19422–19427. [Google Scholar] [CrossRef]
- Nishimura, D.Y.; Fath, M.; Mullins, R.F.; Searby, C.; Andrews, M.; Davis, R.; Andorf, J.L.; Mykytyn, K.; Swiderski, R.E.; Yang, B.L.; et al. Bbs2-null mice have neurosensory deficits, a defect in social dominance, and retinopathy associated with mislocalization of rhodopsin. Proc. Natl. Acad. Sci. USA 2004, 101, 16588–16593. [Google Scholar] [CrossRef]
- Audo, I.; Lancelot, M.E.; Mohand-Said, S.; Antonio, A.; Germain, A.; Sahel, J.A.; Bhattacharya, S.S.; Zeitz, C. Novel C2orf71 Mutations Account for similar to 1% of Cases in a Large French arRP Cohort. Hum. Mutat. 2011, 32, E2091–E2103. [Google Scholar] [CrossRef]
- Kevany, B.M.; Zhang, N.; Jastrzebska, B.; Palczewski, K. Animals deficient in C2Orf71, an autosomal recessive retinitis pigmentosa-associated locus, develop severe early-onset retinal degeneration. Hum. Mol. Genet. 2015, 24, 2627–2640. [Google Scholar] [CrossRef] [PubMed]
- Ravesh, Z.; el Asrag, M.E.; Weisschuh, N.; McKibbin, M.; Reuter, P.; Watson, C.M.; Baumann, B.; Poulter, J.A.; Sajid, S.; Panagiotou, E.S.; et al. Novel C8orf37 mutations cause retinitis pigmentosa in consanguineous families of Pakistani origin. Mol. Vis. 2015, 21, 236–243. [Google Scholar] [PubMed]
- Garanto, A.; Mandal, N.A.; Egido-Gabas, M.; Marfany, G.; Fabrias, G.; Anderson, R.E.; Casas, J.; Gonzalez-Duarte, R. Specific sphingolipid content decrease in Cerkl knockdown mouse retinas. Exp. Eye Res. 2013, 110, 96–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Jiao, X.; D’Atri, I.; Ono, F.; Nelson, R.; Chan, C.C.; Nakaya, N.; Ma, Z.; Ma, Y.; Cai, X.; et al. Mutation in the intracellular chloride channel CLCC1 associated with autosomal recessive retinitis pigmentosa. PLoS Genet. 2018, 14, e1007504. [Google Scholar] [CrossRef]
- Tian, G.; Lee, R.; Ropelewski, P.; Imanishi, Y. Impairment of Vision in a Mouse Model of Usher Syndrome Type III. Investig. Ophthalmol. Vis. Sci. 2016, 57, 866–875. [Google Scholar] [CrossRef] [PubMed]
- Gopal, S.R.; Chen, D.H.C.; Chou, S.W.; Zang, J.J.; Neuhauss, S.C.F.; Stepanyan, R.; McDermott, B.M.; Alagramam, K.N. Zebrafish Models for the Mechanosensory Hair Cell Dysfunction in Usher Syndrome 3 Reveal That Clarin-1 Is an Essential Hair Bundle Protein. J. Neurosci. 2015, 35, 10188–10201. [Google Scholar] [CrossRef]
- Trudeau, M.C.; Zagotta, W.N. An intersubunit interaction regulates trafficking of rod cyclic nucleotide-gated channels and is disrupted in an inherited form of blindness. Neuron 2002, 34, 197–207. [Google Scholar] [CrossRef]
- Huttl, S.; Michalakis, S.; Seeliger, M.; Luo, D.G.; Acar, N.; Geiger, H.; Hudl, K.; Mader, R.; Haverkamp, S.; Moser, M.; et al. Impaired channel targeting and retinal degeneration in mice lacking the cyclic nucleotide-gated channel subunit CNGB1. J. Neurosci. 2005, 25, 130–138. [Google Scholar] [CrossRef]
- Winkler, P.A.; Ekenstedt, K.J.; Occelli, L.M.; Frattaroli, A.V.; Bartoe, J.T.; Venta, P.J.; Petersen-Jones, S.M. A large animal model for CNGB1 autosomal recessive retinitis pigmentosa. PLoS ONE 2013, 8, 72229. [Google Scholar] [CrossRef]
- Mehalow, A.K.; Kameya, S.; Smith, R.S.; Hawes, N.L.; Denegre, J.M.; Young, J.A.; Bechtold, L.; Haider, N.B.; Tepass, U.; Heckenlively, J.R.; et al. CRB1 is essential for external limiting membrane integrity and photoreceptor morphogenesis in the mammalian retina. Hum. Mol. Genet. 2003, 12, 2179–2189. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.; Andrieu-Soler, C.; Kowalczuk, L.; Paz Cortes, M.; Berdugo, M.; Dernigoghossian, M.; Halili, F.; Jeanny, J.C.; Goldenberg, B.; Savoldelli, M.; et al. A new CRB1 rat mutation links Muller glial cells to retinal telangiectasia. J. Neurosci. 2015, 35, 6093–6106. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Guo, L.; Cai, S.P.; Dai, M.; Yang, Q.; Yu, W.; Yan, N.; Zhou, X.; Fu, J.; Guo, X.; et al. Exome sequencing identifies compound heterozygous mutations in CYP4V2 in a pedigree with retinitis pigmentosa. PLoS ONE 2012, 7, e33673. [Google Scholar] [CrossRef] [PubMed]
- Lockhart, C.M.; Nakano, M.; Rettie, A.E.; Kelly, E.J. Generation and characterization of a murine model of Bietti crystalline dystrophy. Investig. Ophthalmol. Vis. Sci. 2014, 55, 5572–5581. [Google Scholar] [CrossRef] [PubMed]
- Venturini, G.; Koskiniemi-Kuendig, H.; Harper, S.; Berson, E.L.; Rivolta, C. Two specific mutations are prevalent causes of recessive retinitis pigmentosa in North American patients of Jewish ancestry. Genet. Med. 2015, 17, 285–290. [Google Scholar] [CrossRef] [Green Version]
- Zuchner, S.; Dallman, J.; Wen, R.; Beecham, G.; Naj, A.; Farooq, A.; Kohli, M.A.; Whitehead, P.L.; Hulme, W.; Konidari, I.; et al. Whole-exome sequencing links a variant in DHDDS to retinitis pigmentosa. Am. J. Hum. Genet. 2011, 88, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Ajmal, M.; Khan, M.I.; Neveling, K.; Khan, Y.M.; Azam, M.; Waheed, N.K.; Hamel, C.P.; Ben-Yosef, T.; de Baere, E.; Koenekoop, R.K.; et al. A missense mutation in the splicing factor gene DHX38 is associated with early-onset retinitis pigmentosa with macular coloboma. J. Med. Genet. 2014, 51, 444–448. [Google Scholar] [CrossRef] [PubMed]
- Abu-Safieh, L.; Alrashed, M.; Anazi, S.; Alkuraya, H.; Khan, A.O.; Al-Owain, M.; Al-Zahrani, J.; Al-Abdi, L.; Hashem, M.; Al-Tarimi, S.; et al. Autozygome-guided exome sequencing in retinal dystrophy patients reveals pathogenetic mutations and novel candidate disease genes. Genome Res. 2013, 23, 236–247. [Google Scholar] [CrossRef] [PubMed]
- Littink, K.W.; van den Born, L.I.; Koenekoop, R.K.; Collin, R.W.J.; Zonneveld, M.N.; Blokland, E.A.W.; Khan, H.; Theelen, T.; Hoyng, C.B.; Cremers, F.P.; et al. Mutations in the EYS gene account for approximately 5% of autosomal recessive retinitis pigmentosa and cause a fairly homogeneous phenotype. Ophthalmology 2010, 117, 2026–2033. [Google Scholar] [CrossRef] [PubMed]
- Arai, Y.; Maeda, A.; Hirami, Y.; Ishigami, C.; Kosugi, S.; Mandai, M.; Kurimoto, Y.; Takahashi, M. Retinitis Pigmentosa with EYS Mutations is the Most Prevalent Inherited Retinal Dystrophy in Japanese Populations. J. Ophthalmol. 2015, 2015. [Google Scholar] [CrossRef]
- Yu, M.; Liu, Y.; Li, J.; Natale, B.N.; Cao, S.Q.; Wang, D.L.; Amack, J.D.; Hu, H.Y. Eyes shut homolog is required for maintaining the ciliary pocket and survival of photoreceptors in zebrafish. Biol. Open 2016, 5, 1662–1673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Schil, K.; Klevering, B.J.; Leroy, B.P.; Pott, J.W.R.; Bandah-Rozenfeld, D.; Zonneveld-Vrieling, M.N.; Sharon, D.; den Hollander, A.I.; Cremers, F.P.M.; de Baere, E.; et al. A Nonsense Mutation in FAM161A Is a Recurrent Founder Allele in Dutch and Belgian Individuals With Autosomal Recessive Retinitis Pigmentosa. Investig. Ophthalmol. Vis. Sci. 2015, 56, 7418–7426. [Google Scholar] [CrossRef] [PubMed]
- Karlstetter, M.; Sorusch, N.; Caramoy, A.; Dannhausen, K.; Aslanidis, A.; Fauser, S.; Boesl, M.R.; Nagel-Wolfrum, K.; Tamm, E.R.; Jagle, H.; et al. Disruption of the retinitis pigmentosa 28 gene Fam161a in mice affects photoreceptor ciliary structure and leads to progressive retinal degeneration. Hum. Mol. Genet. 2014, 23, 5197–5210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Downs, L.M.; Mellersh, C.S. An Intronic SINE Insertion in FAM161A that Causes Exon-Skipping Is Associated with Progressive Retinal Atrophy in Tibetan Spaniels and Tibetan Terriers. PLoS ONE 2014, 9, e93990. [Google Scholar] [CrossRef] [PubMed]
- Haer-Wigman, L.; Newman, H.; Leibu, R.; Bax, N.M.; Baris, H.N.; Rizel, L.; Banin, E.; Massarweh, A.; Roosing, S.; Lefeber, D.J.; et al. Non-syndromic retinitis pigmentosa due to mutations in the mucopolysaccharidosis type IIIC gene, heparan-alpha-glucosaminide N-acetyltransferase (HGSNAT). Hum. Mol. R Genet. 2015, 24, 3742–3751. [Google Scholar] [CrossRef] [PubMed]
- Hartong, D.T.; Dange, M.; Mcgee, T.L.; Berson, E.L.; Dryja, T.P.; Colman, R.F. Insights from retinitis pigmentosa into the roles of isocitrate dehydrogenases in the Krebs cycle. Nat. Genet. 2008, 40, 1230–1234. [Google Scholar] [CrossRef] [Green Version]
- Hull, S.; Owen, N.; Islam, F.; Tracey-White, D.; Plagnol, V.; Holder, G.E.; Michaelides, M.; Carss, K.; Raymond, F.L.; Rozet, J.M.; et al. Nonsyndromic Retinal Dystrophy due to Bi-Allelic Mutations in the Ciliary Transport Gene IFT140. Investig. Ophthalmol. Vis. Sci. 2016, 57, 1053–1062. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.A.; Ah-Cann, C.J.; Welfare, M.F.; Tan, T.Y.; Pope, K.; Caruana, G.; Freckmann, M.L.; Savarirayan, R.; Bertram, J.F.; Dobbie, M.S.; et al. Mouse Strain with an Ift140 Mutation That Results in a Skeletal Ciliopathy Modelling Jeune Syndrome. PLoS Genet. 2013, 9, e1003746. [Google Scholar] [CrossRef]
- Bujakowska, K.M.; Zhang, Q.; Siemiatkowska, A.M.; Liu, Q.; Place, E.; Falk, M.J.; Consugar, M.; Lancelot, M.E.; Antonio, A.; Lonjou, C.; et al. Mutations in IFT172 cause isolated retinal degeneration and Bardet-Biedl syndrome. Hum. Mol. Genet. 2015, 24, 230–242. [Google Scholar] [CrossRef]
- Huangfu, D.W.; Liu, A.M.; Rakeman, A.S.; Murcia, N.S.; Niswander, L.; Anderson, K.V. Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature 2003, 426, 83–87. [Google Scholar] [CrossRef]
- Van Huet, R.A.C.; Collin, R.W.J.; Siemiatkowska, A.M.; Klaver, C.C.W.; Hoyng, C.B.; Simonelli, F.; Khan, M.I.; Qamar, R.; Banin, E.; Cremers, F.P.; et al. IMPG2-Associated Retinitis Pigmentosa Displays Relatively Early Macular Involvement. Investig. Ophthalmol. Vis. Sci. 2014, 55, 3939–3953. [Google Scholar] [CrossRef]
- El Shamieh, S.; Neuille, M.; Terray, A.; Orhan, E.; Condroyer, C.; Demontant, V.; Michiels, C.; Antonio, A.; Boyard, F.; Lancelot, M.E.; et al. Whole-Exome Sequencing Identifies KIZ as a Ciliary Gene Associated with Autosomal-Recessive Rod-Cone Dystrophy. Am. J. Hum. Genet. 2014, 94, 625–633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, D.A.; Li, Y.; McHenry, C.L.; Carlson, T.J.; Ding, X.; Sieving, P.A.; Apfelstedt-Sylla, E.; Gal, A. Mutations in the gene encoding lecithin retinol acyltransferase are associated with early-onset severe retinal dystrophy. Nat. Genet. 2001, 28, 123–124. [Google Scholar] [CrossRef] [PubMed]
- Batten, M.L.; Imanishi, Y.; Maeda, T.; Tu, D.C.; Moise, A.R.; Bronson, D.; Possin, D.; van Gelder, R.N.; Baehr, W.; Palczewski, K. Lecithin-retinol acyltransferase is essential for accumulation of all-trans-retinyl esters in the eye and in the liver. J. Biol. Chem. 2004, 279, 10422–10432. [Google Scholar] [CrossRef] [PubMed]
- Van Huet, R.A.; Siemiatkowska, A.M.; Ozgul, R.K.; Yucel, D.; Hoyng, C.B.; Banin, E.; Blumenfeld, A.; Rotenstreich, Y.; Riemslag, F.C.; den Hollander, A.I.; et al. Retinitis pigmentosa caused by mutations in the ciliary MAK gene is relatively mild and is not associated with apparent extra-ocular features. Acta Ophthalmol. 2015, 93, 83–94. [Google Scholar] [CrossRef]
- Omori, Y.; Chaya, T.; Katoh, K.; Kajimura, N.; Sato, S.; Muraoka, K.; Ueno, S.; Koyasu, T.; Kondo, M.; Furukawa, T. Negative regulation of ciliary length by ciliary male germ cell-associated kinase (Mak) is required for retinal photoreceptor survival. Proc. Natl. Acad. Sci. USA 2010, 107, 22671–22676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, R.S.; McMahon, E.J.; Pop, S.M.; Reap, E.A.; Caricchio, R.; Cohen, P.L.; Earp, H.S.; Matsushima, G.K. Phagocytosis and clearance of apoptotic cells is mediated by MER. Nature 2001, 411, 207–211. [Google Scholar] [CrossRef]
- Everson, R.; Pettitt, L.; Forman, O.P.; Dower-Tylee, O.; McLaughlin, B.; Ahonen, S.; Kaukonen, M.; Komaromy, A.M.; Lohi, H.; Mellersh, C.S.; et al. An intronic LINE-1 insertion in MERTK is strongly associated with retinopathy in Swedish Vallhund dogs. PLoS ONE 2017, 12, e0183021. [Google Scholar] [CrossRef]
- Siemiatkowska, A.M.; van den Born, L.I.; van Hagen, P.M.; Stoffels, M.; Neveling, K.; Henkes, A.; Kipping-Geertsema, M.; Hoefsloot, L.H.; Hoyng, C.B.; Simon, A.; et al. Mutations in the Mevalonate Kinase (MVK) Gene Cause Nonsyndromic Retinitis Pigmentosa. Ophthalmology 2013, 120, 2697–2705. [Google Scholar] [CrossRef]
- Hager, E.J.; Tse, H.M.; Piganelli, J.D.; Gupta, M.; Baetscher, M.; Tse, T.E.; Pappu, A.S.; Steiner, R.D.; Hoffmann, G.F.; Gibson, K.M. Deletion of a single mevalonate kinase (Mvk) allele yields a murine model of hyper-IgD syndrome. J. Inherit. Metab. Dis. 2007, 30, 888–895. [Google Scholar] [CrossRef]
- Nishiguchi, K.M.; Tearle, R.G.; Liu, Y.F.P.; Ohd, E.C.; Miyake, N.; Benaglio, P.; Harper, S.; Koskiniemi-Kuendig, H.; Venturini, G.; Sharon, D.; et al. Whole genome sequencing in patients with retinitis pigmentosa reveals pathogenic DNA structural changes and NEK2 as a new disease gene. Proc. Natl. Acad. Sci. USA 2013, 110, 16139–16144. [Google Scholar] [CrossRef]
- Sonn, S.; Khang, I.; Kim, K.; Rhee, K. Suppression of Nek2A in mouse early embryos confirms its requirement for chromosome segregation. J. Cell Sci. 2004, 117, 5557–5566. [Google Scholar] [CrossRef]
- Wang, F.; Li, H.J.; Xu, M.C.; Li, H.; Zhao, L.; Yang, L.Z.; Zaneveld, J.E.; Wang, K.Q.; Li, Y.M.; Sui, R.F.; et al. A Homozygous Missense Mutation in NEUROD1 Is Associated With Nonsyndromic Autosomal Recessive Retinitis Pigmentosa. Investig. Ophthalmol. Vis. Sci. 2015, 56, 150–155. [Google Scholar] [CrossRef]
- Liu, M.; Pereira, F.A.; Price, S.D.; Chu, M.J.; Shope, C.; Himes, D.; Eatock, R.A.; Brownell, W.E.; Lysakowski, A.; Tsai, M.J. Essential role of BETA2/NeuroD1 in development of the vestibular and auditory systems. Genes Dev. 2000, 14, 2839–2854. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.; McCluskey, M.; Wensel, T.G.; Naggert, J.K.; Nishina, P.M. New mouse models for recessive retinitis pigmentosa caused by mutations in the Pde6a gene. Hum. Mol. Genet. 2009, 18, 178–192. [Google Scholar] [CrossRef] [PubMed]
- Petersen-Jones, S.M.; Entz, D.D.; Sargan, D.R. CGMP phosphodiesterase-alpha mutation causes progressive retinal atrophy in the Cardigan Welsh corgi dog. Investig. Ophthalmol. Vis. Sci. 1999, 40, 1637–1644. [Google Scholar]
- Pichard, V.; Provost, N.; Mendes-Madeira, A.; Libeau, L.; Hulin, P.; Tshilenge, K.T.; Biget, M.; Ameline, B.; Deschamps, J.Y.; Weber, M.; et al. AAV-mediated Gene Therapy Halts Retinal Degeneration in PDE6beta-deficient Dogs. J. Am. Soc. Gene Ther. 2016, 24, 867–876. [Google Scholar] [CrossRef]
- Dvir, L.; Srour, G.; Abu-Ras, R.; Miller, B.; Shalev, S.A.; Ben-Yosef, T. Autosomal-recessive early-onset retinitis pigmentosa caused by a mutation in PDE6G, the gene encoding the gamma subunit of rod cGMP phosphodiesterase. Am. J. Hum. Genet. 2010, 87, 258–264. [Google Scholar] [CrossRef]
- Tsang, S.H.; Gouras, P.; Yamashita, C.K.; Kjeldbye, H.; Fisher, J.; Farber, D.B.; Goff, S.P. Retinal degeneration in mice lacking the gamma subunit of the rod cGMP phosphodiesterase. Science 1996, 272, 1026–1029. [Google Scholar] [CrossRef]
- Xu, M.; Yamada, T.; Sun, Z.; Eblimit, A.; Lopez, I.; Wang, F.; Manya, H.; Xu, S.; Zhao, L.; Li, Y. Mutations in POMGNT1 cause non-syndromic retinitis pigmentosa. Hum. Mol. Genet. 2016, 25, 1479–1488. [Google Scholar] [CrossRef]
- Liu, J.; Ball, S.L.; Yang, Y.; Mei, P.; Zhang, L.; Shi, H.; Kaminski, H.J.; Lemmon, V.P.; Hu, H. A genetic model for muscle-eye-brain disease in mice lacking protein O-mannose 1,2-N-acetylglucosaminyltransferase (POMGnT1). Mech. Dev. 2006, 123, 228–240. [Google Scholar] [CrossRef]
- Nevet, M.J.; Shalev, S.A.; Zlotogora, J.; Mazzawi, N.; Ben-Yosef, T. Identification of a prevalent founder mutation in an Israeli Muslim Arab village confirms the role of PRCD in the aetiology of retinitis pigmentosa in humans. J. Med. Genet. 2010, 47, 533–537. [Google Scholar] [CrossRef] [PubMed]
- Zangerl, B.; Goldstein, O.; Philp, A.R.; Lindauer, S.J.P.; Pearce-Kelling, S.E.; Mullins, R.F.; Graphodatsky, A.S.; Ripoll, D.; Felix, J.S.; Stone, E.M.; et al. Identical mutation in a novel retinal gene causes progressive rod-cone degeneration in dogs and retinitis pigmentosa in humans. Genomics 2006, 88, 551–563. [Google Scholar] [CrossRef] [PubMed]
- Permanyer, J.; Navarro, R.; Friedman, J.; Pomares, E.; Castro-Navarro, J.; Marfany, G.; Swaroop, A.; Gonzalez-Duarte, R. Autosomal Recessive Retinitis Pigmentosa with Early Macular Affectation Caused by Premature Truncation in PROM1. Investig. Ophthalmol. Vis. Sci. 2010, 51, 2656–2663. [Google Scholar] [CrossRef] [PubMed]
- Den Hollander, A.I.; McGee, T.L.; Ziviello, C.; Banfi, S.; Dryja, T.P.; Gonzalez-Fernandez, F.; Ghosh, D.; Berson, E.L. A Homozygous Missense Mutation in the IRBP Gene (RBP3) Associated with Autosomal Recessive Retinitis Pigmentosa. Investig. Ophthalmol. Vis. Sci. 2009, 50, 1864–1872. [Google Scholar] [CrossRef]
- Liou, G.I.; Fei, Y.J.; Peachey, N.S.; Matragoon, S.; Wei, S.H.; Blaner, W.S.; Wang, Y.X.; Liu, C.Y.; Gottesman, M.E.; Ripps, H. Early onset photoreceptor abnormalities induced by targeted disruption of the interphotoreceptor retinoid-binding protein gene. J. Neurosci. 1998, 18, 4511–4520. [Google Scholar] [CrossRef]
- Arno, G.; Agrawal, S.A.; Eblimit, A.; Bellingham, J.; Xu, M.; Wang, F.; Chakarova, C.; Parfitt, D.A.; Lane, A.; Burgoyne, T. Mutations in REEP6 Cause Autosomal-Recessive Retinitis Pigmentosa. Am. J. Hum. Genet. 2016, 99, 1305–1315. [Google Scholar] [CrossRef]
- Agrawal, S.A.; Burgoyne, T.; Eblimit, A.; Bellingham, J.; Parfitt, D.A.; Lane, A.; Nichols, R.; Asomugha, C.; Hayes, M.J.; Munro, P.M.; et al. REEP6 deficiency leads to retinal degeneration through disruption of ER homeostasis and protein trafficking. Hum. Mol. Genet. 2017, 26, 2667–2677. [Google Scholar] [CrossRef]
- Maeda, T.; van Hooser, J.P.; Driessen, C.A.G.G.; Filipek, S.; Janssen, J.J.M.; Palczewski, K. Evaluation of the role of the retinal G protein-coupled receptor (RGR) in the vertebrate retina in vivo. J. Neurochem. 2003, 85, 944–956. [Google Scholar] [CrossRef] [Green Version]
- Saari, J.C.; Nawrot, M.; Kennedy, B.N.; Garwin, G.G.; Hurley, J.B.; Huang, J.; Possin, D.E.; Crabb, J.W. Visual cycle impairment in cellular retinaldehyde binding protein (CRALBP) knockout mice results in delayed dark adaptation. Neuron 2001, 29, 739–748. [Google Scholar] [CrossRef]
- Davidson, A.E.; Sergouniotis, P.I.; Mackay, D.S.; Wright, G.A.; Waseem, N.H.; Michaelides, M.; Holder, G.E.; Robson, A.G.; Moore, A.T.; Plagnol, V.; et al. RP1L1 Variants are Associated with a Spectrum of Inherited Retinal Diseases Including Retinitis Pigmentosa and Occult Macular Dystrophy. Hum. Mutat. 2013, 34, 506–514. [Google Scholar] [CrossRef]
- Yamashita, T.; Liu, J.W.; Gao, J.G.; LeNoue, S.; Wang, C.G.; Kaminoh, J.; Bowne, S.J.; Sullivan, L.S.; Daiger, S.P.; Zhang, K.; et al. Essential and Synergistic Roles of RP1 and RP1L1 in Rod Photoreceptor Axoneme and Retinitis Pigmentosa. J. Neurosci. 2009, 29, 9748–9760. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Simon, M.I.; Matthes, M.T.; Yasumura, D.; LaVail, M.M. Increased susceptibility to light damage in an arrestin knockout mouse model of Oguchi disease (stationary night blindness). Investig. Ophthalmol. Vis. Sci. 1999, 40, 2978–2982. [Google Scholar]
- Goldstein, O.; Jordan, J.A.; Aguirre, G.D.; Acland, G.M. A non-stop S-antigen gene mutation is associated with late onset hereditary retinal degeneration in dogs. Mol. Vis. 2013, 19, 1871–1884. [Google Scholar] [PubMed]
- Corton, M.; Avila-Fernandez, A.; Campello, L.; Sanchez, M.; Benavides, B.; Lopez-Molina, M.I.; Fernandez-Sanchez, L.; Sanchez-Alcudia, R.; da Silva, L.R.J.; Reyes, N.; et al. Identification of the Photoreceptor Transcriptional Co-Repressor SAMD11 as Novel Cause of Autosomal Recessive Retinitis Pigmentosa. Sci. Rep. 2016, 6, 35370. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.B.; Huang, X.F.; Lv, J.N.; Xiang, L.; Li, D.Q.; Chen, J.F.; Huang, C.J.; Wu, J.Y.; Lu, F.; Qu, J. SLC7A14 linked to autosomal recessive retinitis pigmentosa. Nat. Commun. 2014, 5, 3517. [Google Scholar] [CrossRef] [PubMed]
- Kannabiran, C.; Palavalli, L.; Jalali, S. Mutation of SPATA7 in a family with autosomal recessive early-onset retinitis pigmentosa. J. Mol. Genet. Med. 2012, 6, 301–303. [Google Scholar] [CrossRef]
- Zhong, H.; Eblimit, A.; Moayedi, Y.; Boye, S.L.; Chiodo, V.A.; Chen, Y.; Li, Y.; Nichols, R.M.; Hauswirth, W.W.; Chen, R. AAV8(Y733F)-mediated gene therapy in a Spata7 knockout mouse model of Leber congenital amaurosis and retinitis pigmentosa. Gene. Ther. 2015, 22, 619–627. [Google Scholar] [CrossRef] [Green Version]
- Wedatilake, Y.; Niazi, R.; Fassone, E.; Powell, C.A.; Pearce, S.; Plagnol, V.; Saldanha, J.W.; Kleta, R.; Chong, W.K.; Footitt, E.; et al. TRNT1 deficiency: Clinical, biochemical and molecular genetic features. Orphanet J. Rare Dis. 2016, 11, 90. [Google Scholar] [CrossRef] [PubMed]
- DeLuca, A.P.; Whitmore, S.S.; Barnes, J.; Sharma, T.P.; Westfall, T.A.; Scott, C.A.; Weed, M.C.; Wiley, J.S.; Wiley, L.A.; Johnston, R.M.; et al. Hypomorphic mutations in TRNT1 cause retinitis pigmentosa with erythrocytic microcytosis. Hum. Mol. Genet. 2016, 25, 44–56. [Google Scholar] [CrossRef]
- Tadenev, A.L.; Kulaga, H.M.; May-Simera, H.L.; Kelley, M.W.; Katsanis, N.; Reed, R.R. Loss of Bardet-Biedl syndrome protein-8 (BBS8) perturbs olfactory function, protein localization, and axon targeting. Proc. Natl. Acad. Sci. USA 2011, 108, 10320–10325. [Google Scholar] [CrossRef]
- Downs, L.M.; Wallin-Hakansson, B.; Bergstrom, T.; Mellersh, C.S. A novel mutation in TTC8 is associated with progressive retinal atrophy in the golden retriever. Canine Genet. Epidemiol. 2014, 1, 4. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, S.; Shiva, N.; Ikeda, A.; Smith, R.S.; Nusinowitz, S.; Yan, G.; Lin, T.R.; Chu, S.; Heckenlively, J.R.; North, M.A.; et al. Retinal degeneration but not obesity is observed in null mutants of the tubby-like protein 1 gene. Hum. Mol. Genet. 2000, 9, 155–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.Q.; Bulgakov, O.V.; Darrow, K.N.; Pawlyk, B.; Adamian, M.; Liberman, M.C.; Li, T.S. Usherin is required for maintenance of retinal photoreceptors and normal development of cochlear hair cells. Proc. Natl. Acad. Sci. USA 2007, 104, 4413–4418. [Google Scholar] [CrossRef] [Green Version]
- Avila-Fernandez, A.; Perez-Carro, R.; Corton, M.; Lopez-Molina, M.I.; Campello, L.; Garanto, A.; Fernandez-Sanchez, L.; Duijkers, L.; Lopez-Martinez, M.A.; Riveiro-Alvarez, R.; et al. Whole-exome sequencing reveals ZNF408 as a new gene associated with autosomal recessive retinitis pigmentosa with vitreal alterations. Hum. Mol. Genet. 2015, 24, 4037–4048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collin, R.W.J.; Nikopoulos, K.; Dona, M.; Gilissen, C.; Hoischen, A.; Boonstra, F.N.; Poulter, J.A.; Kondo, H.; Berger, W.; Toomes, C.; et al. ZNF408 is mutated in familial exudative vitreoretinopathy and is crucial for the development of zebrafish retinal vasculature. Proc. Natl. Acad. Sci. USA 2013, 110, 9856–9861. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Nakaya, N.; Chavali, V.R.M.; Ma, Z.W.; Jiao, X.D.; Sieving, P.A.; Riazuddin, S.; Tomarev, S.I.; Ayyagari, R.; Riazuddin, S.A.; et al. A Mutation in ZNF513, a Putative Regulator of Photoreceptor Development, Causes Autosomal-Recessive Retinitis Pigmentosa. Am. J. Hum. Genet. 2010, 87, 400–409. [Google Scholar] [CrossRef] [Green Version]
- Webb, T.R.; Parfitt, D.A.; Gardner, J.C.; Martinez, A.; Bevilacqua, D.; Davidson, A.E.; Zito, I.; Thiselton, D.L.; Ressa, J.H.C.; Apergi, M.; et al. Deep intronic mutation in OFD1, identified by targeted genomic next-generation sequencing, causes a severe form of X-linked retinitis pigmentosa (RP23). Hum. Mol. Genet. 2012, 21, 3647–3654. [Google Scholar] [CrossRef] [Green Version]
- Ferrante, M.I.; Zullo, A.; Barra, A.; Bimonte, S.; Messaddeq, N.; Studer, M.; Dolle, P.; Franco, B. Oral-facial-digital type I protein is required for primary cilia formation and left-right axis specification. Nat. Genet. 2006, 38, 112–117. [Google Scholar] [CrossRef]
- Zullo, A.; Iaconis, D.; Barra, A.; Cantone, A.; Messaddeq, N.; Capasso, G.; Dolle, P.; Igarashi, P.; Franco, B. Kidney-specific inactivation of Ofd1 leads to renal cystic disease associated with upregulation of the mTOR pathway. Hum. Mol. Genet. 2010, 19, 2792–2803. [Google Scholar] [CrossRef] [Green Version]
- Mookherjee, S.; Hiriyanna, S.; Kaneshiro, K.; Li, L.; Li, Y.; Li, W.; Qian, H.; Li, T.; Khanna, H.; Colosi, P.; et al. Long-term rescue of cone photoreceptor degeneration in retinitis pigmentosa 2 (RP2)-knockout mice by gene replacement therapy. Hum. Mol. Genet. 2015, 24, 6446–6458. [Google Scholar] [CrossRef]
- Thompson, D.A.; Khan, N.W.; Othman, M.I.; Chang, B.; Jia, L.; Grahek, G.; Wu, Z.J.; Hiriyanna, S.; Nellissery, J.; Li, T.S.; et al. Rd9 Is a Naturally Occurring Mouse Model of a Common Form of Retinitis Pigmentosa Caused by Mutations in RPGR-ORF15. PLoS ONE 2012, 7, e35865. [Google Scholar] [CrossRef] [PubMed]
- Appelbaum, T.; Becker, D.; Santana, E.; Aguirre, G.D. Molecular studies of phenotype variation in canine RPGR-XLPRA1. Mol. Vis. 2016, 22, 319–331. [Google Scholar] [PubMed]
- Venter, J.C.; Adams, M.D.; Myers, E.W.; Li, P.W.; Mural, R.J.; Sutton, G.G.; Smith, H.O.; Yandell, M.; Evans, C.A.; Holt, R.A.; et al. The sequence of the human genome. Science 2001, 291, 1304–1351. [Google Scholar] [CrossRef] [PubMed]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. International Human Genome Sequencing Consortium. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar]
- Ellegren, H. Comparative genomics and the study of evolution by natural selection. Mol. Ecol. 2008, 17, 4586–4596. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Brum, J.R.; Dutilh, B.E.; Sunagawa, S.; Duhaime, M.B.; Loy, A.; Poulos, B.T.; Solonenko, N.; Lara, E.; Poulain, J.; et al. Ecogenomics and potential biogeochemical impacts of globally abundant ocean viruses. Nature 2016, 537, 689–693. [Google Scholar] [CrossRef] [Green Version]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [Green Version]
- Prufer, K.; Racimo, F.; Patterson, N.; Jay, F.; Sankararaman, S.; Sawyer, S.; Heinze, A.; Renaud, G.; Sudmant, P.H.; de Filippo, C.; et al. The complete genome sequence of a Neanderthal from the Altai Mountains. Nature 2014, 505, 43–49. [Google Scholar] [CrossRef]
- Slon, V.; Mafessoni, F.; Vernot, B.; de Filippo, C.; Grote, S.; Viola, B.; Hajdinjak, M.; Peyregne, S.; Nagel, S.; Brown, S.; et al. The genome of the offspring of a Neanderthal mother and a Denisovan father. Nature 2018, 561, 113–116. [Google Scholar] [CrossRef]
- Achilli, A.; Olivieri, A.; Semino, O.; Torroni, A. Ancient human genomes-keys to understanding our past. Science 2018, 360, 964–965. [Google Scholar] [CrossRef]
- Haak, W.; Lazaridis, I.; Patterson, N.; Rohland, N.; Mallick, S.; Llamas, B.; Brandt, G.; Nordenfelt, S.; Harney, E.; Stewardson, K.; et al. Massive migration from the steppe was a source for Indo-European languages in Europe. Nature 2015, 522, 207–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skoglund, P.; Malmstrom, H.; Omrak, A.; Raghavan, M.; Valdiosera, C.; Gunther, T.; Hall, P.; Tambets, K.; Parik, J.; Sjogren, K.G.; et al. Genomic diversity and admixture differs for Stone-Age Scandinavian foragers and farmers. Science 2014, 344, 747–750. [Google Scholar] [CrossRef] [PubMed]
- Gibbons, A. DNA reveals European roots of the ancient Philistines. Science 2019, 365, 17. [Google Scholar] [CrossRef] [PubMed]
- Feuda, R.; Rota-Stabelli, O.; Oakley, T.H.; Pisani, D. The comb jelly opsins and the origins of animal phototransduction. Genome Biol. Evol. 2014, 6, 1964–1971. [Google Scholar] [CrossRef] [PubMed]
- Ryan, J.F.; Pang, K.; Schnitzler, C.E.; Nguyen, A.D.; Moreland, R.T.; Simmons, D.K.; Koch, B.J.; Francis, W.R.; Havlak, P.; Program, N.C.; et al. The genome of the ctenophore Mnemiopsis leidyi and its implications for cell type evolution. Science 2013, 342, 1242592. [Google Scholar] [CrossRef] [PubMed]
- Picciani, N.; Kerlin, J.R.; Sierra, N.; Swafford, A.J.M.; Ramirez, M.D.; Roberts, N.G.; Cannon, J.T.; Daly, M.; Oakley, T.H. Prolific Origination of Eyes in Cnidaria with Co-option of Non-visual Opsins. Curr. Biol. 2018, 28, 2413–2419. [Google Scholar] [CrossRef]
- Srivastava, M.; Simakov, O.; Chapman, J.; Fahey, B.; Gauthier, M.E.; Mitros, T.; Richards, G.S.; Conaco, C.; Dacre, M.; Hellsten, U.; et al. The Amphimedon queenslandica genome and the evolution of animal complexity. Nature 2010, 466, 720–726. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, J.; Jiao, W.; Li, J.; Xun, X.; Sun, Y.; Guo, X.; Huan, P.; Dong, B.; Zhang, L.; et al. Scallop genome provides insights into evolution of bilaterian karyotype and development. Nat. Ecol. Evol. 2017, 1, 120. [Google Scholar] [CrossRef]
- Richter, D.J.; King, N. The genomic and cellular foundations of animal origins. Annu. Rev. Genet. 2013, 47, 509–537. [Google Scholar] [CrossRef]
- Dunn, C.W.; Leys, S.P.; Haddock, S.H. The hidden biology of sponges and ctenophores. Trends Ecol. Evol. 2015, 30, 282–291. [Google Scholar] [CrossRef] [Green Version]
- Weiner, M.P.; Gabriel, S.B.; Stephens, J.C. Genetic Variation: A Laboratory Manual; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2007. [Google Scholar]
- Reich, D.E.; Cargill, M.; Bolk, S.; Ireland, J.; Sabeti, P.C.; Richter, D.J.; Lavery, T.; Kouyoumjian, R.; Farhadian, S.F.; Ward, R.; et al. Linkage disequilibrium in the human genome. Nature 2001, 411, 199–204. [Google Scholar] [CrossRef]
- Swaroop, A.; Chew, E.Y.; Rickman, C.B.; Abecasis, G.R. Unraveling a multifactorial late-onset disease: from genetic susceptibility to disease mechanisms for age-related macular degeneration. Annu. Rev. Genom. Hum. Genet. 2009, 10, 19–43. [Google Scholar] [CrossRef] [PubMed]
- Klein, R.J.; Zeiss, C.; Chew, E.Y.; Tsai, J.Y.; Sackler, R.S.; Haynes, C.; Henning, A.K.; SanGiovanni, J.P.; Mane, S.M.; Mayne, S.T.; et al. Complement factor H polymorphism in age-related macular degeneration. Science 2005, 308, 385–389. [Google Scholar] [CrossRef] [PubMed]
- Haines, J.L.; Hauser, M.A.; Schmidt, S.; Scott, W.K.; Olson, L.M.; Gallins, P.; Spencer, K.L.; Kwan, S.Y.; Noureddine, M.; Gilbert, J.R.; et al. Complement factor H variant increases the risk of age-related macular degeneration. Science 2005, 308, 419–421. [Google Scholar] [CrossRef] [PubMed]
- Sofat, R.; Casas, J.P.; Webster, A.R.; Bird, A.C.; Mann, S.S.; Yates, J.R.; Moore, A.T.; Sepp, T.; Cipriani, V.; Bunce, C. Complement factor H genetic variant and age-related macular degeneration: Effect size, modifiers and relationship to disease subtype. Int. J. Epidemiol. 2012, 41, 250–262. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Atmaca-Sonmez, P.; Othman, M.; Branham, K.E.; Khanna, R.; Wade, M.S.; Li, Y.; Liang, L.; Zareparsi, S.; Swaroop, A.; et al. CFH haplotypes without the Y402H coding variant show strong association with susceptibility to age-related macular degeneration. Nat. Genet. 2006, 38, 1049–1054. [Google Scholar] [CrossRef] [Green Version]
- Rivera, A.; Fisher, S.A.; Fritsche, L.G.; Keilhauer, C.N.; Lichtner, P.; Meitinger, T.; Weber, B.H. Hypothetical LOC387715 is a second major susceptibility gene for age-related macular degeneration, contributing independently of complement factor H to disease risk. Hum. Mol. Genet. 2005, 14, 3227–3236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dewan, A.; Liu, M.; Hartman, S.; Zhang, S.S.; Liu, D.T.; Zhao, C.; Tam, P.O.; Chan, W.M.; Lam, D.S.; Snyder, M.; et al. HTRA1 promoter polymorphism in wet age-related macular degeneration. Science 2006, 314, 989–992. [Google Scholar] [CrossRef]
- Devlin, B.; Roeder, K. Genomic control for association studies. Biometrics 1999, 55, 997–1004. [Google Scholar] [CrossRef]
- Fritsche, L.G.; Chen, W.; Schu, M.; Yaspan, B.L.; Yu, Y.; Thorleifsson, G.; Zack, D.J.; Arakawa, S.; Cipriani, V.; Ripke, S.; et al. Seven new loci associated with age-related macular degeneration. Nat. Genet. 2013, 45, 433. [Google Scholar]
- Fritsche, L.G.; Igl, W.; Bailey, J.N.; Grassmann, F.; Sengupta, S.; Bragg-Gresham, J.L.; Burdon, K.P.; Hebbring, S.J.; Wen, C.; Gorski, M.; et al. A large genome-wide association study of age-related macular degeneration highlights contributions of rare and common variants. Nat. Genet. 2016, 48, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Leveillard, T.; Philp, N.J.; Sennlaub, F. Is Retinal Metabolic Dysfunction at the Center of the Pathogenesis of Age-related Macular Degeneration? Int. J. Mol. Sci. 2019, 20, 762. [Google Scholar] [CrossRef] [PubMed]
- Seddon, J.M.; Reynolds, R.; Maller, J.; Fagerness, J.A.; Daly, M.J.; Rosner, B. Prediction model for prevalence and incidence of advanced age-related macular degeneration based on genetic, demographic, and environmental variables. Investig. Ophthalmol. Vis. Sci. 2009, 50, 2044–2053. [Google Scholar] [CrossRef] [PubMed]
- Bowes, C.; Danciger, M.; Kozak, C.A.; Farber, D.B. Isolation of a candidate cDNA for the gene causing retinal degeneration in the rd mouse. Proc. Natl. Acad. Sci. USA 1989, 86, 9722–9726. [Google Scholar] [CrossRef] [PubMed]
- Livesey, F.J.; Furukawa, T.; Steffen, M.A.; Church, G.M.; Cepko, C.L. Microarray analysis of the transcriptional network controlled by the photoreceptor homeobox gene Crx. Curr. Biol. 2000, 10, 301–310. [Google Scholar] [CrossRef] [Green Version]
- Sharon, D.; Blackshaw, S.; Cepko, C.L.; Dryja, T.P. Profile of the genes expressed in the human peripheral retina, macula, and retinal pigment epithelium determined through serial analysis of gene expression (SAGE). Proc. Natl. Acad. Sci. USA 2002, 99, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Michaut, L.; Flister, S.; Neeb, M.; White, K.P.; Certa, U.; Gehring, W.J. Analysis of the eye developmental pathway in Drosophila using DNA microarrays. Proc. Natl. Acad. Sci. USA 2003, 100, 4024–4029. [Google Scholar] [CrossRef]
- Roesch, K.; Jadhav, A.P.; Trimarchi, J.M.; Stadler, M.B.; Roska, B.; Sun, B.B.; Cepko, C.L. The transcriptome of retinal Muller glial cells. J. Comp. Neurol. 2008, 509, 225–238. [Google Scholar] [CrossRef]
- Dorrell, M.I.; Aguilar, E.; Weber, C.; Friedlander, M. Global gene expression analysis of the developing postnatal mouse retina. Investig. Ophthalmol. Vis. Sci. 2004, 45, 1009–1019. [Google Scholar] [CrossRef]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [Green Version]
- Brazma, A.; Parkinson, H.; Sarkans, U.; Shojatalab, M.; Vilo, J.; Abeygunawardena, N.; Holloway, E.; Kapushesky, M.; Kemmeren, P.; Lara, G.G.; et al. ArrayExpress—A public repository for microarray gene expression data at the EBI. Nucleic Acids Res. 2003, 31, 68–71. [Google Scholar] [CrossRef] [PubMed]
- Sanfilippo, P.G.; Hewitt, A.W. Translating the ENCyclopedia Of DNA Elements Project findings to the clinic: ENCODE’s implications for eye disease. Clin. Exp. Ophthalmol. 2014, 42, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Consortium, M.; Shi, L.; Reid, L.H.; Jones, W.D.; Shippy, R.; Warrington, J.A.; Baker, S.C.; Collins, P.J.; de Longueville, F.; Kawasaki, E.S.; et al. The MicroArray Quality Control (MAQC) project shows inter- and intraplatform reproducibility of gene expression measurements. Nat. Biotechnol. 2006, 24, 1151–1161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, Y.; Yan, K.; Lin, F.; Anderson, K.; Sotiriou, C.; Andre, F.; Holmes, F.A.; Valero, V.; Booser, D.; Pippen, J.E., Jr.; et al. Determination of oestrogen-receptor status and ERBB2 status of breast carcinoma: A gene-expression profiling study. Lancet Oncol. 2007, 8, 203–211. [Google Scholar] [CrossRef]
- Hackam, A.S.; Strom, R.; Liu, D.; Qian, J.; Wang, C.; Otteson, D.; Gunatilaka, T.; Farkas, R.H.; Chowers, I.; Kageyama, M.; et al. Identification of gene expression changes associated with the progression of retinal degeneration in the rd1 mouse. Investig. Ophthalmol. Vis. Sci. 2004, 45, 2929–2942. [Google Scholar] [CrossRef] [PubMed]
- Rattner, A.; Nathans, J. The genomic response to retinal disease and injury: Evidence for endothelin signaling from photoreceptors to glia. J. Neurosci. 2005, 25, 4540–4549. [Google Scholar] [CrossRef] [PubMed]
- Thiersch, M.; Raffelsberger, W.; Frigg, R.; Samardzija, M.; Wenzel, A.; Poch, O.; Grimm, C. Analysis of the retinal gene expression profile after hypoxic preconditioning identifies candidate genes for neuroprotection. BMC Genom. 2008, 9, 73. [Google Scholar] [CrossRef] [PubMed]
- Cronin, T.; Raffelsberger, W.; Lee-Rivera, I.; Jaillard, C.; Niepon, M.L.; Kinzel, B.; Clerin, E.; Petrosian, A.; Picaud, S.; Poch, O.; et al. The disruption of the rod-derived cone viability gene leads to photoreceptor dysfunction and susceptibility to oxidative stress. Cell Death Differ. 2010, 17, 1199–1210. [Google Scholar] [CrossRef]
- Chen, Y.; Brooks, M.J.; Gieser, L.; Swaroop, A.; Palczewski, K. Transcriptome profiling of NIH3T3 cell lines expressing opsin and the P23H opsin mutant identifies candidate drugs for the treatment of retinitis pigmentosa. Pharm. Res. 2017, 115, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Nasonkin, I.O.; Merbs, S.L.; Lazo, K.; Oliver, V.F.; Brooks, M.; Patel, K.; Enke, R.A.; Nellissery, J.; Jamrich, M.; Le, Y.Z.; et al. Conditional knockdown of DNA methyltransferase 1 reveals a key role of retinal pigment epithelium integrity in photoreceptor outer segment morphogenesis. Development 2013, 140, 1330–1341. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Wang, D.; Dongye, M.; Zhu, Y.; Chen, C.; Wang, R.; Long, E.; Liu, Z.; Wu, X.; Lin, D.; et al. Loss-of-function mutations in FREM2 disrupt eye morphogenesis. Exp. Eye Res. 2019, 181, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Punzo, C.; Kornacker, K.; Cepko, C.L. Stimulation of the insulin/mTOR pathway delays cone death in a mouse model of retinitis pigmentosa. Nat. Neurosci. 2009, 12, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Siegert, S.; Cabuy, E.; Scherf, B.G.; Kohler, H.; Panda, S.; Le, Y.Z.; Fehling, H.J.; Gaidatzis, D.; Stadler, M.B.; Roska, B. Transcriptional code and disease map for adult retinal cell types. Nat. Neurosci. 2012, 15, 487–495. [Google Scholar] [CrossRef] [PubMed]
- Kalathur, R.K.; Gagniere, N.; Berthommier, G.; Poidevin, L.; Raffelsberger, W.; Ripp, R.; Leveillard, T.; Poch, O. RETINOBASE: A web database, data mining and analysis platform for gene expression data on retina. BMC Genom. 2008, 9, 208. [Google Scholar] [CrossRef] [PubMed]
- Leveillard, T.D. Knowledge Base for sensory systems for biologists and clinicians. Investig. Ophthalmol. Vis. Sci. 2016, 57, 12. [Google Scholar]
- Craig, P.; Cannon, A.; Kennedy, J.; Kukla, R. Pattern browsing and query adjustment for the exploratory analysis and cooperative visualisation of microarray time-course data. In Cooperative Design, Visualization, and Engineering; Springer International Publishing AG Adresse: Gewerbestrasse, Cham, Suisse, 2010; pp. 199–206. [Google Scholar]
- Becirovic, E.; Bohm, S.; Nguyen, O.N.; Riedmayr, L.M.; Koch, M.A.; Schulze, E.; Kohl, S.; Borsch, O.; Santos-Ferreira, T.; Ader, M.; et al. In Vivo Analysis of Disease-Associated Point Mutations Unveils Profound Differences in mRNA Splicing of Peripherin-2 in Rod and Cone Photoreceptors. PLoS Genet. 2016, 12, e1005811. [Google Scholar] [CrossRef] [PubMed]
- Buskin, A.; Zhu, L.; Chichagova, V.; Basu, B.; Mozaffari-Jovin, S.; Dolan, D.; Droop, A.; Collin, J.; Bronstein, R.; Mehrotra, S.; et al. Disrupted alternative splicing for genes implicated in splicing and ciliogenesis causes PRPF31 retinitis pigmentosa. Nat. Commun. 2018, 9, 4234. [Google Scholar] [CrossRef]
- Gamsiz, E.D.; Ouyang, Q.; Schmidt, M.; Nagpal, S.; Morrow, E.M. Genome-wide transcriptome analysis in murine neural retina using high-throughput RNA sequencing. Genomics 2012, 99, 44–51. [Google Scholar] [CrossRef] [Green Version]
- Brooks, M.J.; Rajasimha, H.K.; Roger, J.E.; Swaroop, A. Next-generation sequencing facilitates quantitative analysis of wild-type and Nrl(-/-) retinal transcriptomes. Mol. Vis. 2011, 17, 3034–3054. [Google Scholar]
- Farkas, M.H.; Grant, G.R.; White, J.A.; Sousa, M.E.; Consugar, M.B.; Pierce, E.A. Transcriptome analyses of the human retina identify unprecedented transcript diversity and 3.5 Mb of novel transcribed sequence via significant alternative splicing and novel genes. BMC Genom. 2013, 14, 486. [Google Scholar] [CrossRef]
- Xu, X.; Zhang, Y.; Williams, J.; Antoniou, E.; McCombie, W.R.; Wu, S.; Zhu, W.; Davidson, N.O.; Denoya, P.; Li, E. Parallel comparison of Illumina RNA-Seq and Affymetrix microarray platforms on transcriptomic profiles generated from 5-aza-deoxy-cytidine treated HT-29 colon cancer cells and simulated datasets. BMC Bioinform. 2013, 14, 1. [Google Scholar] [CrossRef] [PubMed]
- Mustafi, D.; Kevany, B.M.; Genoud, C.; Bai, X.; Palczewski, K. Photoreceptor phagocytosis is mediated by phosphoinositide signaling. FASEB J. 2013, 27, 4585–4595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cech, T.R.; Steitz, J.A. The noncoding RNA revolution-trashing old rules to forge new ones. Cell 2014, 157, 77–94. [Google Scholar] [CrossRef] [PubMed]
- Busskamp, V.; Krol, J.; Nelidova, D.; Daum, J.; Szikra, T.; Tsuda, B.; Juttner, J.; Farrow, K.; Scherf, B.G.; Alvarez, C.P.; et al. miRNAs 182 and 183 are necessary to maintain adult cone photoreceptor outer segments and visual function. Neuron 2014, 83, 586–600. [Google Scholar] [CrossRef] [PubMed]
- Krol, J.; Krol, I.; Alvarez, C.P.; Fiscella, M.; Hierlemann, A.; Roska, B.; Filipowicz, W. A network comprising short and long noncoding RNAs and RNA helicase controls mouse retina architecture. Nat. Commun. 2015, 6, 7305. [Google Scholar] [CrossRef]
- Karali, M.; Peluso, I.; Gennarino, V.A.; Bilio, M.; Verde, R.; Lago, G.; Dolle, P.; Banfi, S. miRNeye: A microRNA expression atlas of the mouse eye. BMC Genom. 2010, 11, 715. [Google Scholar] [CrossRef]
- Lewin, A.S.; Drenser, K.A.; Hauswirth, W.W.; Nishikawa, S.; Yasumura, D.; Flannery, J.G.; LaVail, M.M. Ribozyme rescue of photoreceptor cells in a transgenic rat model of autosomal dominant retinitis pigmentosa. Nat. Med. 1998, 4, 967–971. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, M.; Palfi, A.; Chadderton, N.; Millington-Ward, S.; Ader, M.; Cronin, T.; Tuohy, T.; Auricchio, A.; Hildinger, M.; Tivnan, A.; et al. RNA interference-mediated suppression and replacement of human rhodopsin in vivo. Am. J. Hum. Genet. 2007, 81, 127–135. [Google Scholar] [CrossRef]
- Enright, J.M.; Lawrence, K.A.; Hadzic, T.; Corbo, J.C. Transcriptome profiling of developing photoreceptor subtypes reveals candidate genes involved in avian photoreceptor diversification. J. Comp. Neurol. 2015, 523, 649–668. [Google Scholar] [CrossRef]
- Shekhar, K.; Lapan, S.W.; Whitney, I.E.; Tran, N.M.; Macosko, E.Z.; Kowalczyk, M.; Adiconis, X.; Levin, J.Z.; Nemesh, J.; Goldman, M.; et al. Comprehensive Classification of Retinal Bipolar Neurons by Single-Cell Transcriptomics. Cell 2016, 166, 1308–1323. [Google Scholar] [CrossRef]
- Quadrato, G.; Nguyen, T.; Macosko, E.Z.; Sherwood, J.L.; Min Yang, S.; Berger, D.R.; Maria, N.; Scholvin, J.; Goldman, M.; Kinney, J.P.; et al. Cell diversity and network dynamics in photosensitive human brain organoids. Nature 2017, 545, 48–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heng, J.S.; Rattner, A.; Stein-O’Brien, G.L.; Winer, B.L.; Jones, B.W.; Vernon, H.J.; Goff, L.A.; Nathans, J. Hypoxia tolerance in the Norrin-deficient retina and the chronically hypoxic brain studied at single-cell resolution. Proc. Natl. Acad. Sci. USA 2019, 116, 9103–9114. [Google Scholar] [CrossRef] [Green Version]
- Clark, B.S.; Stein-O’Brien, G.L.; Shiau, F.; Cannon, G.H.; Davis-Marcisak, E.; Sherman, T.; Santiago, C.P.; Hoang, T.V.; Rajaii, F.; James-Esposito, R.E.; et al. Single-Cell RNA-Seq Analysis of Retinal Development Identifies NFI Factors as Regulating Mitotic Exit and Late-Born Cell Specification. Neuron 2019, 102, 1111–1126. [Google Scholar] [CrossRef]
- Voigt, A.P.; Whitmore, S.S.; Flamme-Wiese, M.J.; Riker, M.J.; Wiley, L.A.; Tucker, B.A.; Stone, E.M.; Mullins, R.F.; Scheetz, T.E. Molecular characterization of foveal versus peripheral human retina by single-cell RNA sequencing. Exp. Eye Res. 2019, 184, 234–242. [Google Scholar] [CrossRef]
- Lubec, G.; Afjehi-Sadat, L.; Yang, J.W.; John, J.P. Searching for hypothetical proteins: Theory and practice based upon original data and literature. Prog. Neurobiol. 2005, 77, 90–127. [Google Scholar] [CrossRef]
- Nookala, S.; Gandrakota, R.; Wohabrebbi, A.; Wang, X.; Howell, D.; Giorgianni, F.; Beranova-Giorgianni, S.; Desiderio, D.M.; Jablonski, M.M. In search of the identity of the XAP-1 antigen: A protein localized to cone outer segments. Investig. Ophthalmol. Vis. Sci. 2010, 51, 2736–2743. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, H.; Komori, N. Ocular proteomics: Cataloging photoreceptor proteins by two-dimensional gel electrophoresis and mass spectrometry. Methods Enzym. 2000, 316, 492–511. [Google Scholar]
- Vizcaino, J.A.; Csordas, A.; del-Toro, N.; Dianes, J.A.; Griss, J.; Lavidas, I.; Mayer, G.; Perez-Riverol, Y.; Reisinger, F.; Ternent, T.; et al. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 2016, 44, D447–D456. [Google Scholar] [CrossRef]
- Garin-Muga, A.; Odriozola, L.; Martinez-Val, A.; del Toro, N.; Martinez, R.; Molina, M.; Cantero, L.; Rivera, R.; Garrido, N.; Dominguez, F.; et al. Detection of Missing Proteins Using the PRIDE Database as a Source of Mass Spectrometry Evidence. J. Proteome Res. 2016, 15, 4101–4115. [Google Scholar] [CrossRef] [PubMed]
- Semba, R.D.; Enghild, J.J.; Venkatraman, V.; Dyrlund, T.F.; van Eyk, J.E. The Human Eye Proteome Project: Perspectives on an emerging proteome. Proteomics 2013, 13, 2500–2511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavusoglu, N.; Thierse, D.; Mohand-Said, S.; Chalmel, F.; Poch, O.; Van-Dorsselaer, A.; Sahel, J.A.; Leveillard, T. Differential proteomic analysis of the mouse retina: the induction of crystallin proteins by retinal degeneration in the rd1 mouse. Mol. Cell Proteom. 2003, 2, 494–505. [Google Scholar] [CrossRef] [PubMed]
- Ly, A.; Merl-Pham, J.; Priller, M.; Gruhn, F.; Senninger, N.; Ueffing, M.; Hauck, S.M. Proteomic Profiling Suggests Central Role Of STAT Signaling during Retinal Degeneration in the rd10 Mouse Model. J. Proteome Res. 2016, 15, 1350–1359. [Google Scholar] [CrossRef] [PubMed]
- Stettler, O.; Joshi, R.L.; Wizenmann, A.; Reingruber, J.; Holcman, D.; Bouillot, C.; Castagner, F.; Prochiantz, A.; Moya, K.L. Engrailed homeoprotein recruits the adenosine A1 receptor to potentiate ephrin A5 function in retinal growth cones. Development 2012, 139, 215–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dona, M.; Bachmann-Gagescu, R.; Texier, Y.; Toedt, G.; Hetterschijt, L.; Tonnaer, E.L.; Peters, T.A.; van Beersum, S.E.; Bergboer, J.G.; Horn, N.; et al. NINL and DZANK1 Co-function in Vesicle Transport and Are Essential for Photoreceptor Development in Zebrafish. PLoS Genet. 2015, 11, e1005574. [Google Scholar] [CrossRef]
- Ramirez, J.; Martinez, A.; Lectez, B.; Lee, S.Y.; Franco, M.; Barrio, R.; Dittmar, G.; Mayor, U. Proteomic Analysis of the Ubiquitin Landscape in the Drosophila Embryonic Nervous System and the Adult Photoreceptor Cells. PLoS ONE 2015, 10, e0139083. [Google Scholar] [CrossRef]
- Crabb, J.W.; Miyagi, M.; Gu, X.; Shadrach, K.; West, K.A.; Sakaguchi, H.; Kamei, M.; Hasan, A.; Yan, L.; Rayborn, M.E.; et al. Drusen proteome analysis: An approach to the etiology of age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2002, 99, 14682–14687. [Google Scholar] [CrossRef] [Green Version]
- Umeda, S.; Suzuki, M.T.; Okamoto, H.; Ono, F.; Mizota, A.; Terao, K.; Yoshikawa, Y.; Tanaka, Y.; Iwata, T. Molecular composition of drusen and possible involvement of anti-retinal autoimmunity in two different forms of macular degeneration in cynomolgus monkey (Macaca fascicularis). FASEB J. 2005, 19, 1683–1685. [Google Scholar] [CrossRef]
- Rogers, R.S.; Dharsee, M.; Ackloo, S.; Sivak, J.M.; Flanagan, J.G. Proteomics analyses of human optic nerve head astrocytes following biomechanical strain. Mol. Cell Proteom. 2012, 11, M111.012302. [Google Scholar] [CrossRef]
- Mallikarjuna, K.; Sundaram, C.S.; Sharma, Y.; Deepa, P.R.; Khetan, V.; Gopal, L.; Biswas, J.; Sharma, T.; Krishnakumar, S. Comparative proteomic analysis of differentially expressed proteins in primary retinoblastoma tumors. Proteom. Clin. Appl. 2010, 4, 449–463. [Google Scholar] [CrossRef]
- Danda, R.; Ganapathy, K.; Sathe, G.; Madugundu, A.K.; Ramachandran, S.; Krishnan, U.M.; Khetan, V.; Rishi, P.; Keshava Prasad, T.S.; Pandey, A.; et al. Proteomic profiling of retinoblastoma by high resolution mass spectrometry. Clin. Proteom. 2016, 13, 29. [Google Scholar] [CrossRef]
- Torok, Z.; Peto, T.; Csosz, E.; Tukacs, E.; Molnar, A.M.; Berta, A.; Tozser, J.; Hajdu, A.; Nagy, V.; Domokos, B.; et al. Combined Methods for Diabetic Retinopathy Screening, Using Retina Photographs and Tear Fluid Proteomics Biomarkers. J. Diabetes Res. 2015, 2015, 623619. [Google Scholar] [CrossRef] [PubMed]
- Nobl, M.; Reich, M.; Dacheva, I.; Siwy, J.; Mullen, W.; Schanstra, J.P.; Choi, C.Y.; Kopitz, J.; Kretz, F.T.; Auffarth, G.U.; et al. Proteomics of vitreous in neovascular age-related macular degeneration. Exp. Eye Res. 2016, 146, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Velez, G.; Tang, P.H.; Cabral, T.; Cho, G.Y.; Machlab, D.A.; Tsang, S.H.; Bassuk, A.G.; Mahajan, V.B. Personalized Proteomics for Precision Health: Identifying Biomarkers of Vitreoretinal Disease. Transl. Vis. Sci. Technol. 2018, 7, 12. [Google Scholar] [CrossRef] [PubMed]
- Velez, G.; Machlab, D.A.; Tang, P.H.; Sun, Y.; Tsang, S.H.; Bassuk, A.G.; Mahajan, V.B. Proteomic analysis of the human retina reveals region-specific susceptibilities to metabolic- and oxidative stress-related diseases. PLoS ONE 2018, 13, e0193250. [Google Scholar] [CrossRef] [PubMed]
- Hauck, S.M.; Suppmann, S.; Ueffing, M. Proteomic profiling of primary retinal Muller glia cells reveals a shift in expression patterns upon adaptation to in vitro conditions. Glia 2003, 44, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Tan, G.; Levenkova, N.; Li, T.; Pugh, E.N., Jr.; Rux, J.J.; Speicher, D.W.; Pierce, E.A. The proteome of the mouse photoreceptor sensory cilium complex. Mol. Cell Proteom. 2007, 6, 1299–1317. [Google Scholar] [CrossRef] [PubMed]
- Kwok, M.C.; Holopainen, J.M.; Molday, L.L.; Foster, L.J.; Molday, R.S. Proteomics of photoreceptor outer segments identifies a subset of SNARE and Rab proteins implicated in membrane vesicle trafficking and fusion. Mol. Cell Proteom. 2008, 7, 1053–1066. [Google Scholar] [CrossRef]
- Song, H.; Sokolov, M. Analysis of protein expression and compartmentalization in retinal neurons using serial tangential sectioning of the retina. J. Proteome Res. 2009, 8, 346–351. [Google Scholar] [CrossRef]
- Reidel, B.; Thompson, J.W.; Farsiu, S.; Moseley, M.A.; Skiba, N.P.; Arshavsky, V.Y. Proteomic profiling of a layered tissue reveals unique glycolytic specializations of photoreceptor cells. Mol. Cell Proteom. 2011, 10, M110.002469. [Google Scholar] [CrossRef]
- Merl, J.; Ueffing, M.; Hauck, S.M.; von Toerne, C. Direct comparison of MS-based label-free and SILAC quantitative proteome profiling strategies in primary retinal Muller cells. Proteomics 2012, 12, 1902–1911. [Google Scholar] [CrossRef]
- Von Toerne, C.; Menzler, J.; Ly, A.; Senninger, N.; Ueffing, M.; Hauck, S.M. Identification of a novel neurotrophic factor from primary retinal Muller cells using stable isotope labeling by amino acids in cell culture (SILAC). Mol. Cell Proteom. 2014, 13, 2371–2381. [Google Scholar] [CrossRef] [PubMed]
- Crabb, J.W.; Yuan, X.; Dvoriantchikova, G.; Ivanov, D.; Crabb, J.S.; Shestopalov, V.I. Preliminary quantitative proteomic characterization of glaucomatous rat retinal ganglion cells. Exp. Eye Res. 2010, 91, 107–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, L.; Zhou, X.Y.; Jylha, A.; Aapola, U.; Liu, D.N.; Koh, S.K.; Tian, D.; Quah, J.; Uusitalo, H.; Beuerman, R.W.; et al. Quantitation of 47 human tear proteins using high resolution multiple reaction monitoring (HR-MRM) based-mass spectrometry. J. Proteom. 2015, 115, 36–48. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Linton, J.D.; Hurley, J.B. Probing Metabolism in the Intact Retina Using Stable Isotope Tracers. Methods Enzym. 2015, 561, 149–170. [Google Scholar]
- Du, J.; Cleghorn, W.M.; Contreras, L.; Lindsay, K.; Rountree, A.M.; Chertov, A.O.; Turner, S.J.; Sahaboglu, A.; Linton, J.; Sadilek, M.; et al. Inhibition of mitochondrial pyruvate transport by zaprinast causes massive accumulation of aspartate at the expense of glutamate in the retina. J. Biol. Chem. 2013, 288, 36129–36140. [Google Scholar] [CrossRef] [PubMed]
- Kanow, M.A.; Giarmarco, M.M.; Jankowski, C.S.; Tsantilas, K.; Engel, A.L.; Du, J.; Linton, J.D.; Farnsworth, C.C.; Sloat, S.R.; Rountree, A.; et al. Biochemical adaptations of the retina and retinal pigment epithelium support a metabolic ecosystem in the vertebrate eye. Elife 2017, 6, e28899. [Google Scholar] [CrossRef]
- Weiss, E.R.; Osawa, S.; Xiong, Y.; Dhungana, S.; Carlson, J.; McRitchie, S.; Fennell, T.R. Broad spectrum metabolomics for detection of abnormal metabolic pathways in a mouse model for retinitis pigmentosa. Exp. Eye Res. 2019, 184, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Chertov, A.O.; Holzhausen, L.; Kuok, I.T.; Couron, D.; Parker, E.; Linton, J.D.; Sadilek, M.; Sweet, I.R.; Hurley, J.B. Roles of glucose in photoreceptor survival. J. Biol. Chem. 2011, 286, 34700–34711. [Google Scholar] [CrossRef]
- Ly, A.; Schone, C.; Becker, M.; Rattke, J.; Meding, S.; Aichler, M.; Suckau, D.; Walch, A.; Hauck, S.M.; Ueffing, M. High-resolution MALDI mass spectrometric imaging of lipids in the mammalian retina. Histochem. Cell Biol. 2015, 143, 453–462. [Google Scholar] [CrossRef]
- Bowrey, H.E.; Anderson, D.M.; Pallitto, P.; Gutierrez, D.B.; Fan, J.; Crouch, R.K.; Schey, K.L.; Ablonczy, Z. Imaging mass spectrometry of the visual system: Advancing the molecular understanding of retina degenerations. Proteom. Clin. Appl. 2016, 10, 391–402. [Google Scholar] [CrossRef]
- Berg, J.M.; Tymoczko, J.L.; Stryer, L. Biochemistry, 5th edition. N.Y. WH Freeman 2006, 38, 76. [Google Scholar]
- Tohge, T.; de Souza, L.P.; Fernie, A.R. Genome-enabled plant metabolomics. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2014, 966, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Molday, L.L.; Wu, W.W.; Molday, R.S. Retinoschisin (RS1), the protein encoded by the X-linked retinoschisis gene, is anchored to the surface of retinal photoreceptor and bipolar cells through its interactions with a Na/K ATPase-SARM1 complex. J. Biol. Chem. 2007, 282, 32792–32801. [Google Scholar] [CrossRef] [PubMed]
- Fathinajafabadi, A.; Perez-Jimenez, E.; Riera, M.; Knecht, E.; Gonzalez-Duarte, R. CERKL, a retinal disease gene, encodes an mRNA-binding protein that localizes in compact and untranslated mRNPs associated with microtubules. PLoS ONE 2014, 9, e87898. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.Y.; Agyekum, B.; Venkatesan, A.; Hall, D.R.; Keightley, A.; Bjes, E.S.; Bouyain, S.; Price, J.L. Noncanonical FK506-binding protein BDBT binds DBT to enhance its circadian function and forms foci at night. Neuron 2013, 80, 984–996. [Google Scholar] [CrossRef] [PubMed]
- Orlandi, C.; Posokhova, E.; Masuho, I.; Ray, T.A.; Hasan, N.; Gregg, R.G.; Martemyanov, K.A. GPR158/179 regulate G protein signaling by controlling localization and activity of the RGS7 complexes. J. Cell Biol. 2012, 197, 711–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marmorstein, L.Y.; McLaughlin, P.J.; Stanton, J.B.; Yan, L.; Crabb, J.W.; Marmorstein, A.D. Bestrophin interacts physically and functionally with protein phosphatase 2A. J. Biol. Chem. 2002, 277, 30591–30597. [Google Scholar] [CrossRef]
- Boldt, K.; Mans, D.A.; Won, J.; van Reeuwijk, J.; Vogt, A.; Kinkl, N.; Letteboer, S.J.; Hicks, W.L.; Hurd, R.E.; Naggert, J.K.; et al. Disruption of intraflagellar protein transport in photoreceptor cilia causes Leber congenital amaurosis in humans and mice. J. Clin. Investig. 2011, 121, 2169–2180. [Google Scholar] [CrossRef] [PubMed]
- Zulliger, R.; Conley, S.M.; Mwoyosvi, M.L.; Stuck, M.W.; Azadi, S.; Naash, M.I. SNAREs Interact with Retinal Degeneration Slow and Rod Outer Segment Membrane Protein-1 during Conventional and Unconventional Outer Segment Targeting. PLoS ONE 2015, 10, e0138508. [Google Scholar] [CrossRef]
- Fridlich, R.; Delalande, F.; Jaillard, C.; Lu, J.; Poidevin, L.; Cronin, T.; Perrocheau, L.; Millet-Puel, G.; Niepon, M.L.; Poch, O.; et al. The thioredoxin-like protein rod-derived cone viability factor (RdCVFL) interacts with TAU and inhibits its phosphorylation in the retina. Mol. Cell Proteom. 2009, 8, 1206–1218. [Google Scholar] [CrossRef]
- Nawrot, M.; West, K.; Huang, J.; Possin, D.E.; Bretscher, A.; Crabb, J.W.; Saari, J.C. Cellular retinaldehyde-binding protein interacts with ERM-binding phosphoprotein 50 in retinal pigment epithelium. Investig. Ophthalmol. Vis. Sci. 2004, 45, 393–401. [Google Scholar] [CrossRef] [PubMed]
- Boylan, J.P.; Wright, A.F. Identification of a novel protein interacting with RPGR. Hum. Mol. Genet. 2000, 9, 2085–2093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roepman, R.; Letteboer, S.J.; Arts, H.H.; van Beersum, S.E.; Lu, X.; Krieger, E.; Ferreira, P.A.; Cremers, F.P. Interaction of nephrocystin-4 and RPGRIP1 is disrupted by nephronophthisis or Leber congenital amaurosis-associated mutations. Proc. Natl. Acad. Sci. USA 2005, 102, 18520–18525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mick, D.U.; Rodrigues, R.B.; Leib, R.D.; Adams, C.M.; Chien, A.S.; Gygi, S.P.; Nachury, M.V. Proteomics of Primary Cilia by Proximity Labeling. Dev. Cell 2015, 35, 497–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsybovsky, Y.; Wang, B.; Quazi, F.; Molday, R.S.; Palczewski, K. Posttranslational modifications of the photoreceptor-specific ABC transporter ABCA4. Biochemistry 2011, 50, 6855–6866. [Google Scholar] [CrossRef]
- Soldi, M.; Bremang, M.; Bonaldi, T. Biochemical systems approaches for the analysis of histone modification readout. Biochim. Biophys. Acta 2014, 1839, 657–668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spencer, W.J.; Pearring, J.N.; Salinas, R.Y.; Loiselle, D.R.; Skiba, N.P.; Arshavsky, V.Y. Progressive Rod-Cone Degeneration (PRCD) Protein Requires N-Terminal S-Acylation and Rhodopsin Binding for Photoreceptor Outer Segment Localization and Maintaining Intracellular Stability. Biochemistry 2016, 55, 5028–5037. [Google Scholar] [CrossRef]
- Chiang, C.K.; Tworak, A.; Kevany, B.M.; Xu, B.; Mayne, J.; Ning, Z.; Figeys, D.; Palczewski, K. Quantitative phosphoproteomics reveals involvement of multiple signaling pathways in early phagocytosis by the retinal pigmented epithelium. J. Biol. Chem. 2017, 292, 19826–19839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wakeham, C.M.; Wilmarth, P.A.; Cunliffe, J.M.; Klimek, J.E.; Ren, G.; David, L.L.; Morgans, C.W. Identification of PKCalpha-dependent phosphoproteins in mouse retina. J. Proteom. 2019, 206, 103423. [Google Scholar] [CrossRef]
- Sanidas, I.; Morris, R.; Fella, K.A.; Rumde, P.H.; Boukhali, M.; Tai, E.C.; Ting, D.T.; Lawrence, M.S.; Haas, W.; Dyson, N.J. A Code of Mono-phosphorylation Modulates the Function of RB. Mol. Cell 2019, 73, 985–1000. [Google Scholar] [CrossRef]
- Thingholm, T.E.; Jensen, O.N. Enrichment and characterization of phosphopeptides by immobilized metal affinity chromatography (IMAC) and mass spectrometry. Methods Mol. Biol. 2009, 527, 47–56. [Google Scholar] [PubMed]
- Kapphahn, R.J.; Giwa, B.M.; Berg, K.M.; Roehrich, H.; Feng, X.; Olsen, T.W.; Ferrington, D.A. Retinal proteins modified by 4-hydroxynonenal: Identification of molecular targets. Exp. Eye Res. 2006, 83, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Meer, S.G.; Miyagi, M.; Rayborn, M.E.; Hollyfield, J.G.; Crabb, J.W.; Salomon, R.G. Carboxyethylpyrrole protein adducts and autoantibodies, biomarkers for age-related macular degeneration. J. Biol. Chem. 2003, 278, 42027–42035. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Zou, L.; Zhang, X.; Branco, V.; Wang, J.; Carvalho, C.; Holmgren, A.; Lu, J. Redox Signaling Mediated by Thioredoxin and Glutathione Systems in the Central Nervous System. Antioxid. Redox Signal. 2017, 27, 989–1010. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, M.; Mata-Cabana, A.; Kieselbach, T. The disulfide proteome and other reactive cysteine proteomes: Analysis and functional significance. Antioxid Redox Signal. 2011, 14, 2581–2642. [Google Scholar] [CrossRef] [PubMed]
- Leveillard, T.; Sahel, J.A. Metabolic and redox signaling in the retina. Cell Mol. Life Sci. 2017, 74, 3649–3665. [Google Scholar] [CrossRef]
- Yano, H.; Wong, J.H.; Lee, Y.M.; Cho, M.J.; Buchanan, B.B. A strategy for the identification of proteins targeted by thioredoxin. Proc. Natl. Acad. Sci. USA 2001, 98, 4794–4799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurd, T.R.; Prime, T.A.; Harbour, M.E.; Lilley, K.S.; Murphy, M.P. Detection of reactive oxygen species-sensitive thiol proteins by redox difference gel electrophoresis: Implications for mitochondrial redox signaling. J. Biol. Chem. 2007, 282, 22040–22051. [Google Scholar] [CrossRef]
- Leichert, L.I.; Gehrke, F.; Gudiseva, H.V.; Blackwell, T.; Ilbert, M.; Walker, A.K.; Strahler, J.R.; Andrews, P.C.; Jakob, U. Quantifying changes in the thiol redox proteome upon oxidative stress in vivo. Proc. Natl. Acad. Sci. USA 2008, 105, 8197–8202. [Google Scholar] [CrossRef] [PubMed]
- Fu, C.; Hu, J.; Liu, T.; Ago, T.; Sadoshima, J.; Li, H. Quantitative analysis of redox-sensitive proteome with DIGE and ICAT. J. Proteome Res. 2008, 7, 3789–3802. [Google Scholar] [CrossRef]
- Wang, B.; Hom, G.; Zhou, S.; Guo, M.; Li, B.; Yang, J.; Monnier, V.M.; Fan, X. The oxidized thiol proteome in aging and cataractous mouse and human lens revealed by ICAT labeling. Aging Cell 2017, 16, 244–261. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Grueso, M.J.; Gonzalez-Ojeda, R.; Requejo-Aguilar, R.; McDonagh, B.; Fuentes-Almagro, C.A.; Muntane, J.; Barcena, J.A.; Padilla, C.A. Thioredoxin and glutaredoxin regulate metabolism through different multiplex thiol switches. Redox Biol. 2019, 21, 101049. [Google Scholar] [CrossRef] [PubMed]


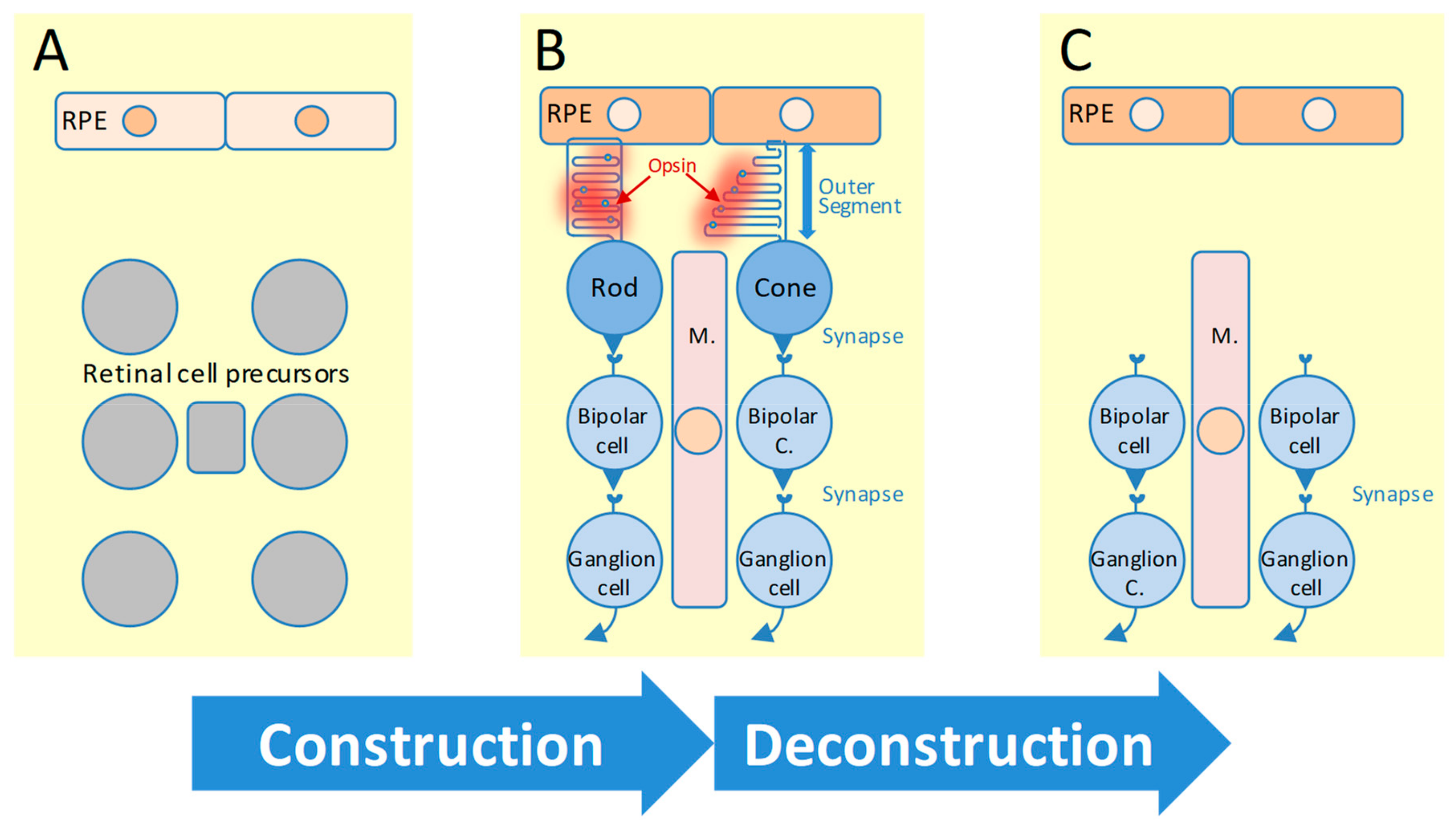{kind=link}
| N | Inheritance | Gene | Rd1 [71] | RD [72] | Mouse Tissue | RPE/PR [73] | Freq. | Other Ret. Dis. | Other OMIM | Mouse Models | Other Models. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Autosomal dominant | ARL3 | Rod-like | PR-death | Neurons | NF | Rare [83] | No | No | [84] | No |
| 2 | ADIPOR1 | Rod-like | PR-death | Bone mar. | Ret./RPE | Rare [85] | One | No | [86] | No | |
| 3 | BEST1 | Hom | Infl. | Testis | NF | Rare [87] | Four | No | [88] | Dog [89,90] | |
| 4 | CA4 | NE | NOp | Intestine | NF | Rare [91] | No | No | No | No | |
| 5 | CRX | Rod-like | PR-death | Ret./RPE | Ret./RPE | 1% [54] | Four | No | [92] | Cat [93] | |
| 6 | FSCN2 | Rod-like | NOp | Ret./RPE | Retina | 3% [94] | One | No | [95] | No | |
| 7 | GUCA1B | Rod-like | PR-death | Ret./RPE | Ret./RPE | 0-5% [94] | One | No | [96] | No | |
| 8 | HK1 | Rod-like | Hom | Testis | Retina | Rare [97] | No | 142600 | No | No | |
| 9 | IMPDH1 | Rod-like | PR-death | Ret./RPE | Retina | 2–3% [94] | One | No | [98] | No | |
| 10 | IMPG1 | Rod-like | PR-death | Ret./RPE | Retina | Rare [99] | Two | No | No | No | |
| 11 | KLHL7 | Hom | Hom | Ub | Ret./RPE | 1–2% [100] | No | 611119 | No | No | |
| 12 | NR2E3 | Rod-like | PR-death | Ret./RPE | Retina | 3.5% [101] | Four | No | [102] | No | |
| 13 | NRL | Rod-like | PR-death | Ret./RPE | Retina | 2% [94] | One | No | [25] | No | |
| 14 | PRPF3 | Hom | PR-death | Ub | Ret./RPE | 1% [94] | No | No | [103] | No | |
| 15 | PRPF4 | Hom | Hom | Ub | RPE | Rare [104] | No | No | No | ZebF [105] | |
| 16 | PRPF6 | Hom | Hom | Ub | Ret./RPE | Rare [104] | No | No | No | No | |
| 17 | PRPF8 | Hom | PR-death | Ub | Ret./RPE | 2% [94] | No | No | [103] | No | |
| 18 | PRPF31 | Rod-like | Hom | Ub | NF | 2–4% [94] | No | No | [106] | No | |
| 19 | PRPH2 | Rod-like | PR-death | Ret./RPE | Ret./RPE | 1–8% [94] | Six | No | [107,108] | No | |
| 20 | RDH12 | Rod-like | PR-death | Ret./RPE | Ret./RPE | Rare [109] | One | No | [110] | No | |
| 21 | RHO | Rod-like | PR-death | Ret./RPE | Ret./RPE | 2–26% [94] | Two | No | [111,112,113] | [114,115] | |
| 22 | ROM1 | Rod-like | PR-death | Ret./RPE | Ret./RPE | 1% [94] | One | No | [116] | No | |
| 23 | RP1 | Rod-like | PR-death | Ret./RPE | RPE | 4–8% [94] | No | No | [117,118] | No | |
| 24 | RP9 | Cone-L. | PR-death | Ub | Retina | Rare [94] | No | No | No | No | |
| 25 | RPE65 | Hom | NE | RPE | NF | Rare | Two | No | [104] | Dog [76] | |
| 26 | SEMA4A | Cone-L. | NOp | Ub | Ret./RPE | Rare [119] | One | No | [120] | No | |
| 27 | SNRNP200 | Hom | Hom | Ub | NF | <2% [121] | No | No | No | No | |
| 28 | SPP2 | NE | NOp | Kidn. Liv. | NF | Rare [122] | No | No | No | No | |
| 29 | TOPORS | Hom | Hom | Ub | RPE | Rare [123] | No | No | [124] | No | |
| 30 | Autosomal recessive | ABCA4 | Rod-like | PR-death | Ret./RPE | Ret./RPE | 5–6% [54] | Four | No | [125] | Dog [126] |
| 31 | ADIPOR1 | Rod-like | PR-death | Bone Mar. | Ret./RPE | Rare | One | No | [86] | No | |
| 32 | AGBL5 | Hom | Hom | Testis | Retina | Rare [127] | No | No | No | No | |
| 33 | AHR | Rod-like | Hom | Mast cells / Ret. | NF | Rare [128] | No | No | [129] | No | |
| 34 | ARHGEF18 | Hom | Hom | White cells / RPE | Ret./RPE | Rare [130] | No | No | No | No | |
| 35 | ARL6 | Rod-like | PR-death | Ub | Retina | 1% [54] | One | No | [131] | No | |
| 36 | ARL2BP | Hom | Hom | Testis | Ret./RPE | Rare [132] | No | No | No | No | |
| 37 | BBS1 | Rod-like | Hom | Ret./RPE | RPE | 2-3% [54] | One | No | [133] | No | |
| 38 | BBS2 | Rod-like | Hom | Ub | Ret./RPE | 0.8% [54] | One | No | [134] | No | |
| 39 | BEST1 | Hom | Infl. | Testis | NF | Rare [87] | Four | No | [88] | Dog [89,90] | |
| 40 | C2orf71 | NF | PR-death | NF | NF | 1% [135] | No | No | [136] | No | |
| 41 | C8orf37 | No orth. | NOp | No ortho | No ortho | Rare [137] | Three | No | No | No | |
| 42 | CERKL | Rod-like | NF | Ub | NF | 1% [54] | One | No | [138] | No | |
| 43 | CLCC1 | Hom | Hom | Ub | Retina | Rare [139] | No | No | [139] | No | |
| 44 | CLRN1 | NOp | Infl. | NOp | NF | 1% [54] | One | No | [140] | ZebF [141] | |
| 45 | CNGA1 | Rod-like | PR-death | Ret./RPE | Retina | 1% [54] | No | No | No | Xen [142] | |
| 46 | CNGB1 | NF | PR-death | NF | Ret./RPE | 4% [54] | No | No | [143] | Dog [144] | |
| 47 | CRB1 | Rod-like | Hom | Ret./RPE | NF | 1% [54] | Three | No | [145] | Rat [146] | |
| 48 | CYP4V2 | Cone-L. | Hom | Liver | NF | Rare [147] | One | No | [148] | No | |
| 49 | DHDDS | Hom | PR-death | Ub | Ret./RPE | 1–8% [149] | No | No | No | ZebF [150] | |
| 50 | DHX38 | Hom | Hom | Ub | Retina | Rare [151] | One | No | No | No | |
| 51 | EMC1 | NF | Hom | NF | NF | Rare [152] | No | 616846 | No | No | |
| 52 | EYS | No orth. | NOp | No ortho | No ortho | [153,154] | No | No | No ortho | ZebF [155] | |
| 53 | FAM161A | Rod-like | PR-death | Ret./RPE | NF | 2% [156] | No | No | [157] | Dog [158] | |
| 54 | GPR125 | Hom | Hom | Epidermis | NF | Rare [152] | No | No | No | No | |
| 55 | HGSNAT | Cone-L. | Hom | Microglia | Retina | Rare [159] | No | 610453 | No | No | |
| 56 | IDH3B | Hom | Hom | Adipo. | Ret./RPE | Rare [160] | No | No | No | No | |
| 57 | IFT140 | Hom | Hom | Bone mar. | NF | Rare [161] | Two | 614620 | [162] | No | |
| 58 | IFT172 | Hom | PR-death | Testis | NF | Rare [163] | One | 607386 | [164] | No | |
| 59 | IMPG2 | NF | Hom | NF | RPE | Rare [165] | One | No | No | No | |
| 60 | KIAA1549 | No orth. | PR-death | No ortho | NF | Rare [152] | No | No | No | No | |
| 61 | KIZ | Rod-like | NF | Testis | NF | Rare [166] | No | No | No | No | |
| 62 | LRAT | Infl. | NOp | RPE | RPE | 1% [54,167] | One | No | [168] | No | |
| 63 | MAK | Rod-like | PR-death | Ret./RPE | RPE | [149,169] | No | No | [170] | No | |
| 64 | MERTK | Cone-L. | Hom | Ub | RPE | 1% [54] | No | No | [171] | [172] | |
| 65 | MVK | Hom | Hom | Ub | Retina | Rare [173] | No | 251170 | [174] | No | |
| 66 | NEK2 | Cone-L. | NOp | Ub | NF | Rare [175] | No | No | [176] | ZebF [175] | |
| 67 | NEUROD1 | Rod-like | Hom | Cerebel. | NF | Rare [177] | No | 601724 | [178] | No | |
| 68 | NRL | Rod-like | PR-death | Ret./RPE | Retina | 2% [94] | One | No | [25] | No | |
| 69 | PDE6A | Rod-like | PR-death | Ret./RPE | Retina | 3–4% [54] | No | No | [179] | Dog [180] | |
| 70 | PDE6B | Rod-like | PR-death | Ret./RPE | RPE | 4–5% [54] | One | No | rd1, rd10 | Dog [181] | |
| 71 | PDE6G | Rod-like | PR-death | Ret./RPE | NF | Rare [182] | No | No | [183] | No | |
| 72 | POMGNT1 | Hom | Hom | Saliv. gl. | RPE | Rare [184] | No | 606822 | [185] | No | |
| 73 | PRCD | No orth. | PR-death | No ortho | No ortho | Rare [186] | No | No | No ortho | Dog [187] | |
| 74 | PROM1 | Rod-like | PR-death | Ub | Ret./RPE | Rare [188] | Four | No | [186] | No | |
| 75 | RBP3 | Rod-like | PR-death | Ret./RPE | Ret./RPE | Rare [189] | No | No | [190] | No | |
| 76 | REEP6 | Rod-like | PR-death | Liver/Ret./RPE/Testis | RPE | Rare [191] | No | No | [192] | No | |
| 77 | RGR | Infl. | PR-death | RPE | RPE | 0.5% [54] | One | No | [193] | No | |
| 78 | RHO | Rod-like | PR-death | Ret./RPE | Ret./RPE | 1% [54] | Two | No | Rho-/- | No | |
| 79 | RLBP1 | Cone-L. | PR-death | Ret./RPE | Ret./RPE | 1% [54] | Three | No | [194] | No | |
| 80 | RP1L1 | Rod-like | PR-death | Ret./RPE | NF | 0.5% [195] | One | No | [196] | No | |
| 81 | RPE65 | Hom | NE | RPE | NF | 2% [54] | Two | No | [104] | Dog [76] | |
| 82 | SAG | Rod-like | PR-death | Ret./RPE | Ret./RPE | >1% [54] | One | No | [197] | Dog [198] | |
| 83 | SAMD11 | Rod-like | Hom | Bone mar. Ret./RPE | RPE | Rare [199] | No | No | No | No | |
| 84 | SLC7A14 | Cone-L. | PR-death | Brain | NF | Rare [200] | No | No | [200] | No | |
| 85 | SPATA7 | Hom | Hom | Testis | RPE | Rare [201] | One | No | [202] | No | |
| 86 | TRNT1 | Hom | NOp | Ub | NF | Rare [203] | No | 612907 | No | ZebF [204] | |
| 87 | TTC8 | Rod-like | PR-death | Ub | NF | >1% [54] | One | No | [205] | Dog [206] | |
| 88 | TULP1 | Rod-like | PR-death | Ret./RPE | Retina | 1% [54] | One | No | [207] | No | |
| 89 | USH2A | Rod-like | PR-death | Ub | NF | 17% [54] | One | No | [208] | No | |
| 90 | ZNF408 | No orth. | Infl. | No ortho | No ortho | Rare [209] | One | No | No | ZebF [210] | |
| 91 | ZNF513 | No orth. | Hom | No ortho | No ortho | Rare [211] | No | No | No | ZebF [211] | |
| 92 | X-linked | OFD1 | Hom | PR-death | Ub | RPE | Rare [212] | Two | 300170 | [213] | ZebF [214] |
| 93 | RP2 | Hom | NOp | Ub | NF | 7–10% [12] | No | No | [215] | No | |
| 94 | RPGR | NF | PR-death | NF | NF | 80% [12] | Three | No | [216] | Dog [217] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blond, F.; Léveillard, T. Functional Genomics of the Retina to Elucidate its Construction and Deconstruction. Int. J. Mol. Sci. 2019, 20, 4922. https://doi.org/10.3390/ijms20194922
Blond F, Léveillard T. Functional Genomics of the Retina to Elucidate its Construction and Deconstruction. International Journal of Molecular Sciences. 2019; 20(19):4922. https://doi.org/10.3390/ijms20194922
Chicago/Turabian StyleBlond, Frédéric, and Thierry Léveillard. 2019. "Functional Genomics of the Retina to Elucidate its Construction and Deconstruction" International Journal of Molecular Sciences 20, no. 19: 4922. https://doi.org/10.3390/ijms20194922
APA StyleBlond, F., & Léveillard, T. (2019). Functional Genomics of the Retina to Elucidate its Construction and Deconstruction. International Journal of Molecular Sciences, 20(19), 4922. https://doi.org/10.3390/ijms20194922