Molecular Network-Based Drug Prediction in Thyroid Cancer

 ,
,
Abstract
:1. Introduction
2. Result
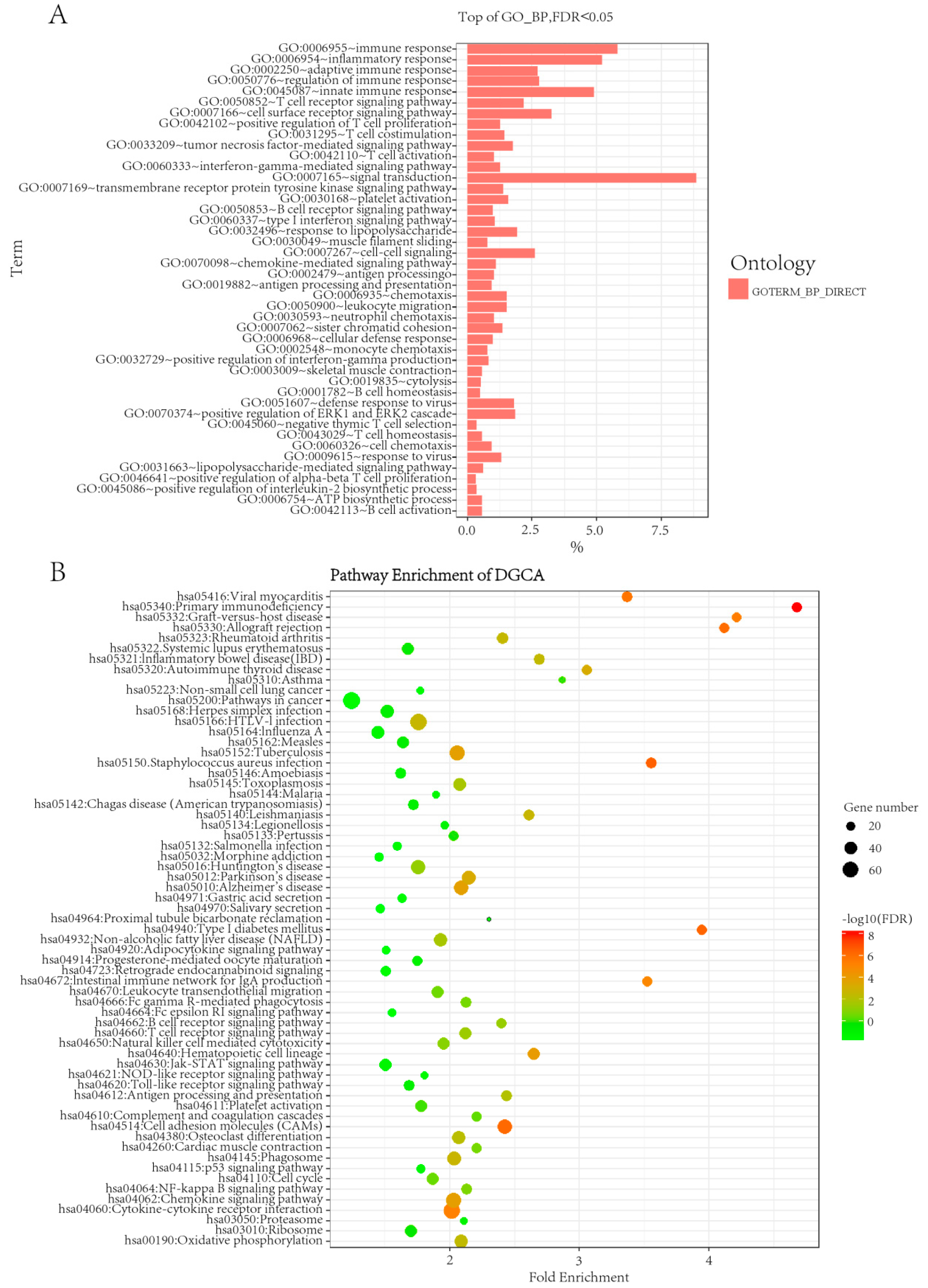
2.1. Differential Gene Expressions and Their Functions Between Thyroid Carcinoma and Normal Samples
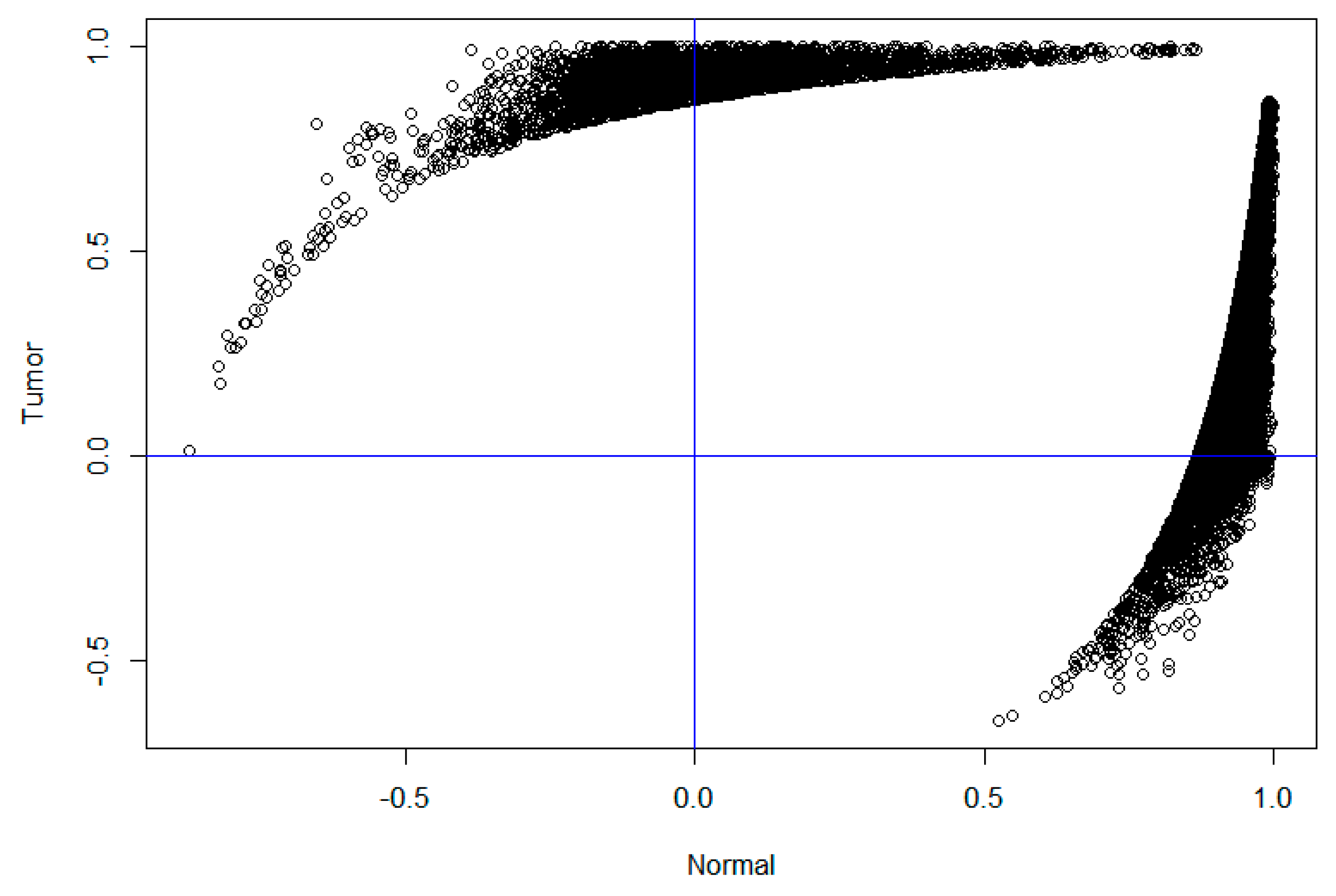
2.2. Analysis of Differentially Co-Expressed Gene Pairs between Normal and Diseased Samples
2.3. Prioritize Drug and Gene Targets Using Online Pharmacogenomics Methods
3. Discussion
4. Materials and Methods
4.1. Gene Co-Expression Network Construction of Normal and Diseased Samples
4.2. Differential Connection Analysis
4.3. Drugs and Disturbing Gene Labels
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Nikiforov, Y.E.; Nikiforova, M.N. Molecular genetics and diagnosis of thyroid cancer. Nat. Rev. Endocrinol. 2011, 7, 569–580. [Google Scholar] [CrossRef]
- LiVolsi, V.A. Papillary thyroid carcinoma: An update. Mod. Pathol. 2011, 24, S1–S9. [Google Scholar] [CrossRef] [PubMed]
- Lamb, J. The Connectivity Map: A new tool for biomedical research. Nat. Rev. Cancer 2007, 7, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Salvatore, G.; De Falco, V.; Salerno, P.; Nappi, T.C.; Pepe, S.; Troncone, G.; Carlomagno, F.; Melillo, R.M.; Wilhelm, S.M.; Santoro, M. BRAF is a therapeutic target in aggressive thyroid carcinoma. Clin. Cancer Res. 2006, 12, 1623–1629. [Google Scholar] [CrossRef] [PubMed]
- Duan, Q.; Flynn, C.; Niepel, M.; Hafner, M.; Muhlich, J.L.; Fernandez, N.F.; Rouillard, A.D.; Tan, C.M.; Chen, E.Y.; Golub, T.R.; et al. LINCS Canvas Browser: Interactive web app to query, browse and interrogate LINCS L1000 gene expression signatures. Nucl. Acids Res. 2014, 42, W449–W460. [Google Scholar] [CrossRef] [PubMed]
- Dudley, J.T.; Sirota, M.; Shenoy, M.; Pai, R.K.; Roedder, S.; Chiang, A.P.; Morgan, A.A.; Sarwal, M.M.; Pasricha, P.J.; Butte, A.J. Computational repositioning of the anticonvulsant topiramate for inflammatory bowel disease. Sci. Transl. Med. 2011, 3, 96ra76. [Google Scholar] [CrossRef] [PubMed]
- Calvert, S.; Tacutu, R.; Sharifi, S.; Teixeira, R.; Ghosh, P.; de Magalhaes, J.P. A network pharmacology approach reveals new candidate caloric restriction mimetics in C. elegans. Aging Cell 2016, 15, 256–266. [Google Scholar] [CrossRef] [PubMed]
- Mirza, N.; Sills, G.J.; Pirmohamed, M.; Marson, A.G. Identifying new antiepileptic drugs through genomics-based drug repurposing. Hum. Mol. Genet. 2017, 26, 527–537. [Google Scholar] [CrossRef] [PubMed]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehar, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Horvath, S. A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol. 2005, 4, Article17. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Monteiro, C.D.; Jagodnik, K.M.; Fernandez, N.F.; Gundersen, G.W.; Rouillard, A.D.; Jenkins, S.L.; Feldmann, A.S.; Hu, K.S.; McDermott, M.G.; et al. Extraction and analysis of signatures from the Gene Expression Omnibus by the crowd. Nat. Commun. 2016, 7, 12846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The GTEx Consortium. The Genotype-Tissue Expression (GTEx) pilot analysis: Multitissue gene regulation in humans. Science 2015, 348, 648–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucl. Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef] [PubMed]
- McKenzie, A.T.; Katsyv, I.; Song, W.M.; Wang, M.; Zhang, B. DGCA: A comprehensive R package for Differential Gene Correlation Analysis. BMC Syst. Biol. 2016, 10, 106. [Google Scholar] [CrossRef]
- Adam, J.K.; Odhav, B.; Bhoola, K.D. Immune responses in cancer. Pharmacol. Ther. 2003, 99, 113–132. [Google Scholar] [CrossRef]
- Elias, A.N.; Szekeres, A.V.; Stone, S.; Weathersbee, P.; Valenta, L.J.; Haw, T. Gaba-ergic and dopaminergic regulation of thyroid stimulating hormone. Effects of baclofen and metoclopramide. Horm. Res. 1984, 19, 171–175. [Google Scholar] [CrossRef]
- Aoun, E.G.; Lee, M.R.; Haass-Koffler, C.L.; Swift, R.M.; Addolorato, G.; Kenna, G.A.; Leggio, L. Relationship between the thyroid axis and alcohol craving. Alcohol Alcoholism 2015, 50, 24–29. [Google Scholar] [CrossRef]
- Catalani, S.; Palma, F.; Battistelli, S.; Nuvoli, B.; Galati, R.; Benedetti, S. Reduced cell viability and apoptosis induction in human thyroid carcinoma and mesothelioma cells exposed to cidofovir. Toxicol. In Vitro 2017, 41, 49–55. [Google Scholar] [CrossRef]
- Pradeep, P.V.; Jayashree, B. Soap bubble type of calcification in thyroid: A radiological surprise! Otolaryngol. Head Neck Surg. 2011, 144, 642–643. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Integrated genomic characterization of papillary thyroid carcinoma. Cell 2014, 159, 676–690. [Google Scholar] [CrossRef] [PubMed]
- Vannucchi, G.; Covelli, D.; Perrino, M.; De Leo, S.; Fugazzola, L. Ultrasound-guided percutaneous ethanol injection in papillary thyroid cancer metastatic lymph-nodes. Endocrine 2014, 47, 648–651. [Google Scholar] [CrossRef] [PubMed]
- Ayroldi, E.; Petrillo, M.G.; Marchetti, M.C.; Cannarile, L.; Ronchetti, S.; Ricci, E.; Cari, L.; Avenia, N.; Moretti, S.; Puxeddu, E.; et al. Long glucocorticoid-induced leucine zipper regulates human thyroid cancer cell proliferation. Cell Death Dis. 2018, 9, 305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, K.G.; Bhatt, H.V.; Choudhury, A.R. Alteration in thyroid after formaldehyde (HCHO) treatment in rats. Ind. Health 2003, 41, 295–297. [Google Scholar] [CrossRef] [PubMed]
- Higashiyama, T.; Nishida, Y.; Morino, K.; Ugi, S.; Nishio, Y.; Maegawa, H.; Ohji, M. Use of MRI signal intensity of extraocular muscles to evaluate methylprednisolone pulse therapy in thyroid-associated ophthalmopathy. Jpn. J. Ophthalmol. 2015, 59, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Guo, F. Identification of drug-target interactions via multiple information integration. Inf. Sci. 2017, 418, 546–560. [Google Scholar] [CrossRef]
- Zeng, X.; Liao, Y.; Liu, Y.; Zou, Q. Prediction and Validation of Disease Genes Using HeteSim Scores. IEEE/ACM Trans. Comput. Biol. Bioinform. 2017, 14, 687–695. [Google Scholar] [CrossRef]
- Zeng, X.; Ding, N.; Rodríguez-Patón, A.; Zou, Q. Probability-based collaborative filtering model for predicting gene–disease associations. BMC Med. Genom. 2017, 10, 76. [Google Scholar] [CrossRef]
- Zeng, X.; Lin, W.; Guo, M.; Zou, Q. A comprehensive overview and evaluation of circular RNA detection tools. PLoS Comput. Biol. 2017, 13, e1005420. [Google Scholar] [CrossRef]
- Liu, Y.; Zeng, X.; He, Z.; Zou, Q. Inferring MicroRNA-Disease Associations by Random Walk on a Heterogeneous Network with Multiple Data Sources. IEEE/ACM Trans. Comput. Biol. Bioinform. 2017, 14, 905–915. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zou, Q.; Rodruguez-Paton, A.; Zeng, X. Meta-path methods for prioritizing candidate disease miRNAs. IEEE/ACM Trans. Comput. Biol. Bioinform. 2018. [Google Scholar] [CrossRef] [PubMed]
- Liao, Z.J.; Li, D.P.; Wang, X.R.; Li, L.S.; Zou, Q. Cancer Diagnosis Through IsomiR Expression with Machine Learning Method. Curr. Bioinform. 2018, 13, 57–63. [Google Scholar] [CrossRef]
- Cabarle, F.G.C.; Adorna, H.N.; Jiang, M.; Zeng, X. Spiking Neural P Systems With Scheduled Synapses. IEEE Trans. Nanobiosci. 2017, 16, 792–801. [Google Scholar] [CrossRef] [PubMed]
- Song, T.; Rodríguez-Patón, A.; Zheng, P.; Zeng, X. Spiking Neural P Systems with Colored Spikes. IEEE Trans. Cogn. Dev. Syst. 2018. [Google Scholar] [CrossRef]
- Song, T.; Zeng, X.; Zheng, P.; Jiang, M.; Rodríguez-Patón, A. A parallel workflow pattern modelling using spiking neural P systems with colored spikes. IEEE Trans. Nanobiosci. 2018, 17, 474–484. [Google Scholar] [CrossRef]
- Yip, L.; Kelly, L.; Shuai, Y.; Armstrong, M.J.; Nikiforov, Y.E.; Carty, S.E.; Nikiforova, M.N. MicroRNA signature distinguishes the degree of aggressiveness of papillary thyroid carcinoma. Ann. Surg. Oncol. 2011, 18, 2035–2041. [Google Scholar] [CrossRef]
- He, H.; Jazdzewski, K.; Li, W.; Liyanarachchi, S.; Nagy, R.; Volinia, S.; Calin, G.A.; Liu, C.G.; Franssila, K.; Suster, S.; et al. The role of microRNA genes in papillary thyroid carcinoma. Proc. Natl. Acad. Sci. USA 2005, 102, 19075–19080. [Google Scholar] [CrossRef] [Green Version]
- Pallante, P.; Visone, R.; Ferracin, M.; Ferraro, A.; Berlingieri, M.T.; Troncone, G.; Chiappetta, G.; Liu, C.G.; Santoro, M.; Negrini, M.; et al. MicroRNA deregulation in human thyroid papillary carcinomas. Endocr. Relat. Cancer 2006, 13, 497–508. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Guo, Q.; Zhao, Y.; Chen, J.; Wang, S.; Hu, J.; Sun, Y. BRAF-activated long non-coding RNA contributes to cell proliferation and activates autophagy in papillary thyroid carcinoma. Oncol. Lett. 2014, 8, 1947–1952. [Google Scholar] [CrossRef] [Green Version]
- Jendrzejewski, J.; He, H.; Radomska, H.S.; Li, W.; Tomsic, J.; Liyanarachchi, S.; Davuluri, R.V.; Nagy, R.; de la Chapelle, A. The polymorphism rs944289 predisposes to papillary thyroid carcinoma through a large intergenic noncoding RNA gene of tumor suppressor type. Proc. Natl. Acad. Sci. USA 2012, 109, 8646–8651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amaral, P.P.; Clark, M.B.; Gascoigne, D.K.; Dinger, M.E.; Mattick, J.S. lncRNAdb: A reference database for long noncoding RNAs. Nucl. Acids Res. 2011, 39, D146–D151. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Xu, X.; Xi, B.; Dai, Q.; Li, C.; Su, L.; Zhou, X.; Tang, M.; Yao, Y.; Yang, J. Molecular network-based identification of competing endogenous RNAs in thyroid carcinoma. Genes 2018, 9, 44. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Gene1_Sym. | Gene2_Sym. | Normal_cor | Normal_pVal | Tumor_cor | Tumor_pVal | pValDiff |
|---|---|---|---|---|---|---|
| NOTUM | PLD5 | −0.388 | 0.003 | 0.992 | 0 | 1.40 × 10−102 |
| NKAIN1 | MATN1 | −0.242 | 0.068 | 0.999 | 0 | 3.75 × 10−92 |
| MPPED1 | NKAIN1 | −0.196 | 0.141 | 0.998 | 0 | 3.62 × 10−89 |
| SOX14 | PRAP1 | −0.186 | 0.162 | 0.993 | 0 | 1.44 × 10−88 |
| SOX14 | MATN1 | −0.171 | 0.200 | 0.994 | 0 | 1.33 × 10−87 |
| SLC13A5 | IHH | −0.167 | 0.210 | 0.995 | 0 | 2.32 × 10−87 |
| PRAP1 | IHH | −0.163 | 0.220 | 1.000 | 0 | 3.82 × 10−87 |
| DHRS2 | DPYS | −0.161 | 0.228 | 0.998 | 0 | 5.51 × 10−87 |
| GLIS1 | KCNT1 | −0.157 | 0.239 | 0.997 | 0 | 9.36 × 10−87 |
| KLHL1 | SOHLH1 | −0.153 | 0.250 | 0.990 | 0 | 3.15 × 10−87 |
| Module | Gene No | Function | DEGs_overlap | ||
|---|---|---|---|---|---|
| Term | FDR | ||||
| Normal | Brown | 4469 | GO:0006954~inflammatory response | 3.98 × 10−24 | 237 |
| Blue | 4240 | GO:0043087~regulation of GTPase activity | 7.96 × 10−5 | 102 | |
| Turquoise | 3850 | GO:0006412~translation | 1.92 × 10−51 | 50 | |
| Black | 3615 | GO:0008380~RNA splicing | 3.85 × 10−5 | 64 | |
| Pink | 1190 | GO:0006355~regulation of transcription, DNA-templated | 2.97 × 10−106 | 4 | |
| Tumor | Turquoise | 4346 | GO:0006351~transcription, DNA-templated | 1.31 × 10−57 | 29 |
| Brown | 2550 | GO:0006355~regulation of transcription, DNA-templated | 9.43 × 10−6 | 29 | |
| Blue | 2458 | GO:0070125~mitochondrial translational elongation | 2.75 × 10−9 | 30 | |
| Thistle2 | 1973 | GO:0006955~immune response | 1.78 × 10−61 | 30 | |
| 1025 Differential Genes | 219 Differential Genes Involved in Differential Co-Expressions | ||
|---|---|---|---|
| Drug ID | Drug Name | Drug ID | Drug Name |
| drug:P4898 | glucocorticoid|dexamethasone | drug:P5684 | cidofovir(2−) |
| drug:P4171 | ethanol|6alpha-methylprednisolone | drug:P5683 | cidofovir(2−) |
| drug:P5683 | cidofovir(2−) | drug:P4396 | baclofen |
| drug:P4401 | baclofen | drug:P4401 | baclofen |
| drug:P4409 | baclofen | drug:P4391 | baclofen |
| drug:P4562 | formaldehyde | drug:P4397 | baclofen |
| drug:P4566 | formaldehyde | drug:P3977 | dimethyl sulfide|dimethyl sulfoxide|solvent |
| drug:P2096 | Erlotinib|dimethyl sulfoxide | drug:P4392 | baclofen |
| drug:P5684 | cidofovir(2−) | drug:P3986 | dimethyl sulfide|dimethyl sulfoxide|solvent |
| drug:P4563 | formaldehyde | drug:P4438 | oxygen atom|2-butoxyethanol |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, X.; Long, H.; Xi, B.; Ji, B.; Li, Z.; Dang, Y.; Jiang, C.; Yao, Y.; Yang, J. Molecular Network-Based Drug Prediction in Thyroid Cancer. Int. J. Mol. Sci. 2019, 20, 263. https://doi.org/10.3390/ijms20020263
Xu X, Long H, Xi B, Ji B, Li Z, Dang Y, Jiang C, Yao Y, Yang J. Molecular Network-Based Drug Prediction in Thyroid Cancer. International Journal of Molecular Sciences. 2019; 20(2):263. https://doi.org/10.3390/ijms20020263
Chicago/Turabian StyleXu, Xingyu, Haixia Long, Baohang Xi, Binbin Ji, Zejun Li, Yunyue Dang, Caiying Jiang, Yuhua Yao, and Jialiang Yang. 2019. "Molecular Network-Based Drug Prediction in Thyroid Cancer" International Journal of Molecular Sciences 20, no. 2: 263. https://doi.org/10.3390/ijms20020263
APA StyleXu, X., Long, H., Xi, B., Ji, B., Li, Z., Dang, Y., Jiang, C., Yao, Y., & Yang, J. (2019). Molecular Network-Based Drug Prediction in Thyroid Cancer. International Journal of Molecular Sciences, 20(2), 263. https://doi.org/10.3390/ijms20020263