1. Introduction
Despite relentless efforts in developing anticancer therapeutics, cancer still remains as the leading cause of death worldwide [
1]. Although a wide variety of chemotherapy, radiotherapy and recently introduced immunotherapies are expected to cure cancer, there have been numerous reports of failure of these therapeutic modalities. One of the major reasons behind failure of anticancer therapies is the heterogeneous nature of the disease. Heterogeneity involves alterations, such as mutations, overexpression, loss or under-expression of various genes, especially those related to cell cycle regulation. For example, the impact of alterations in cell cycle checkpoint proteins, p16
INK4a, p21
WAF1/Cip1 and retinoblastoma (Rb) in tumor development and progression have been well investigated [
2,
3]. The p21
WAF1/Cip1 and p16
INK4a act as tumor suppressors and arrest cell cycle progression by interfering with the interaction between cyclin-dependent kinases (Cdk) and cyclins [
4]. Persistent intra-tumoral hypoxia is a characteristic feature of many solid tumors. The expression of both p21
WAF1/Cip1 and p16
INK4a is increased in the early time point (~6 h) and then decreased in the later time point in various cancer cells under hypoxia condition [
5]. The loss of p16
INK4a in primary tumors is attributed to various genetic and epigenetic alterations including mutation of the gene (CDKN2) encoding p16
INK4a protein or promoter hypermethylation [
3]. Intra-tumoral hypoxia drives tumor cells to invade and migrate through stromal matrix and metastasize to distant locations, resulting in malignant carcinomas. The invasion and migration of tumor cells require alterations in cell polarization, activation of a group of matrix degrading enzymes, such as matrix metalloproteinases (MMP)-2 and -9. The transcriptional activation of MMP-2 is partly mediated by the binding of transcription factor specificity protein-1 (SP1) at the promoter site of MMP-2 and the activation of SP-1 is mediated via phosphorylation of SP-1 by cyclin-Cdks. The p16
INK4a as an inhibitor of Cdks have been shown to diminish SP-1 binding at MMP-2 promoter and reduced migration of cancer cells via downregulation of MMP-2 [
6]. We have recently reported that the expression of p16
INK4a is negatively regulated by hYSK1, also known as a serine-threonine protein kinase (STK)-25, which plays a critical role in cell polarity, invasion, and migration. The hYSK1-mediated loss or downregulation of p16
INK4a has been shown to result in an increase in the expression of MMP-2 through transcriptional activation of SP-1, and in the enhancement of the migration and invasion of tumor cells including those of the melanoma and fibrosarcoma [
7].
The p21
WAF1/Cip1 is another Cdk inhibitor that suppresses tumor growth by inhibiting proliferation, inducing apoptosis, and blocking DNA synthesis in many cancers. Although p21
WAF1/Cip1 being a p53-regulated gene is thought to play a tumor suppressive role, several studies have reported a pro-tumorigenic role of p21
WAF1/Cip1 [
8]. Zhang et al. [
9] recently reported that the level of p21
WAF1/Cip1is reduced in breast cancer tissues as well as in cultured cell lines, and the patients having high p21
WAF1/Cip1 level showed longer survival than the patients having a low p21
WAF1/Cip1 level. The micro-RNA (miR-3619)-mediated activation of p21
WAF1/Cip1 induced growth arrest and inhibited metastasis of breast cancer cells [
9]. Others have also shown that restoration of p21
WAF1/Cip1 inhibits migration and invasion of various cancer cells [
10,
11]. Since hYSK1 enhanced tumor cells proliferation and migration by down-regulation of p16
INK4a [
7], we have been interested to examine if p21
WAF1/Cip1 also plays a role in hYSK1-induced tumor cells migration under hypoxia. The hYSK1 is an oxidative stress response protein. According to the Cancer Genome Atlas (TCGA) dataset, mRNA level of hYSK1 is high in cancers, such as glioma and endometrial cancer. In liver and colorectal cancers, a high protein expression of hYSK1 is associated with a decrease in five years survival rate as compared to those having a low expression of hYSK1 [
12]. We have recently reported that hYSK1 interacted with 20–40 a.a. and 140–200 a.a. of p16
INK4a, thereby decreasing the translocation of p16
INK4a from cytosol to nucleus in hypoxic condition [
7]. The reduced p16
INK4a nuclear localization by overexpression of hYSK1 leads to the activation of SP-1 transcriptional activity, resulting in increased MMP-2 expression and enhanced cancer cell proliferation and migration. Since p21
WAF1/Cip1 inhibits cyclin-Cdks, we were interested to see if hYSK1 also targets p21
WAF1/Cip1 to excel tumor cell migration via activation of MMP-2. Here we report that hYSK1 directly interacted with p21
WAF1/Cip1, thereby resulting in SP-1-mediated suppression of p16
INK4a promoter activity and increased MMP-2 expression under hypoxic condition leading to increased proliferation and migration of cancer cells.
3. Discussion
The perturbation of cell cycle checkpoint proteins, especially cyclin-Cdk inhibitor’s function is common in many cancers. Among the cyclin-Cdk inhibitors, the p16
INK4a and p21
WAF1/Cip1 have been extensively studied for their role in neoplastic transformation of cells. The p16
INK4a alteration has caused by genetic and epigenetic regulation such as promoter hyper-methylations and histone modifications [
13,
14]. The p16
INK4a is involved in arresting cell cycle at G0 and early G1 phase by disrupting cyclin-Cdk4/6 interaction. The inactivation of Cdk4/6 activity results in reduced phosphorylation of pRb and compromised interaction between phospho-pRb and E2F transcription factor, thereby reducing the transcription of genes encoding proteins involved in cell proliferation [
15]. Besides blocking cyclin-Cdk4/6-mediated pRb phosphorylation, p16
INK4a also causes cell cycle arrest by binding with transcription factor TFIIH, resulting in reduced phosphorylation of RNA polymerase II [
16]. It is also reported that p16
INK4a interacted with c-Jun N-terminal kinases (JNKs) and inhibits the UV-induced cell transformation by activation of c-Jun transcription factor [
17]. We have previously reported that p16
INK4a function is lost by its interaction with eukaryotic elongation factor 1A2 and hYSK1, thereby increasing proliferation and migration of various cancer cells [
7,
18]. The p21
WAF1/Cip1 is another cell cycle regulatory protein that also inhibits cyclin dependent kinases by working as a downstream signaling molecule to p53. Although initially identified as a kinase inhibitor to induce apoptosis, there is paradoxical findings that p21
WAF1/Cip1 allows cell cycle exit and promotes cell proliferation [
8]. It has been reported that p21
WAF1/Cip1 regulates the expression of p16
INK4a by binding of SP-1 transcription factor on GC region (−449~−459) of p16
INK4a promoter [
19].
The hYSK1, also known as STK25, is a member of the germinal center kinase III (GCK III) subfamily of the sterile 20 (STE20) kinase superfamily [
20]. Although much is known about the role of this protein in targeting Golgi apparatus, in the regulation of cellular polarization, program cell death and cell migration under stress conditions [
21,
22], role of this protein in cancer has not fully elucidated. We have recently reported that hYSK1 promotes migration of melanoma and fibrosarcoma cells by repressing p16
INK4a nuclear translocation via direct interaction, thereby resulting in increased SP-1-mediated MMP2 transcription [
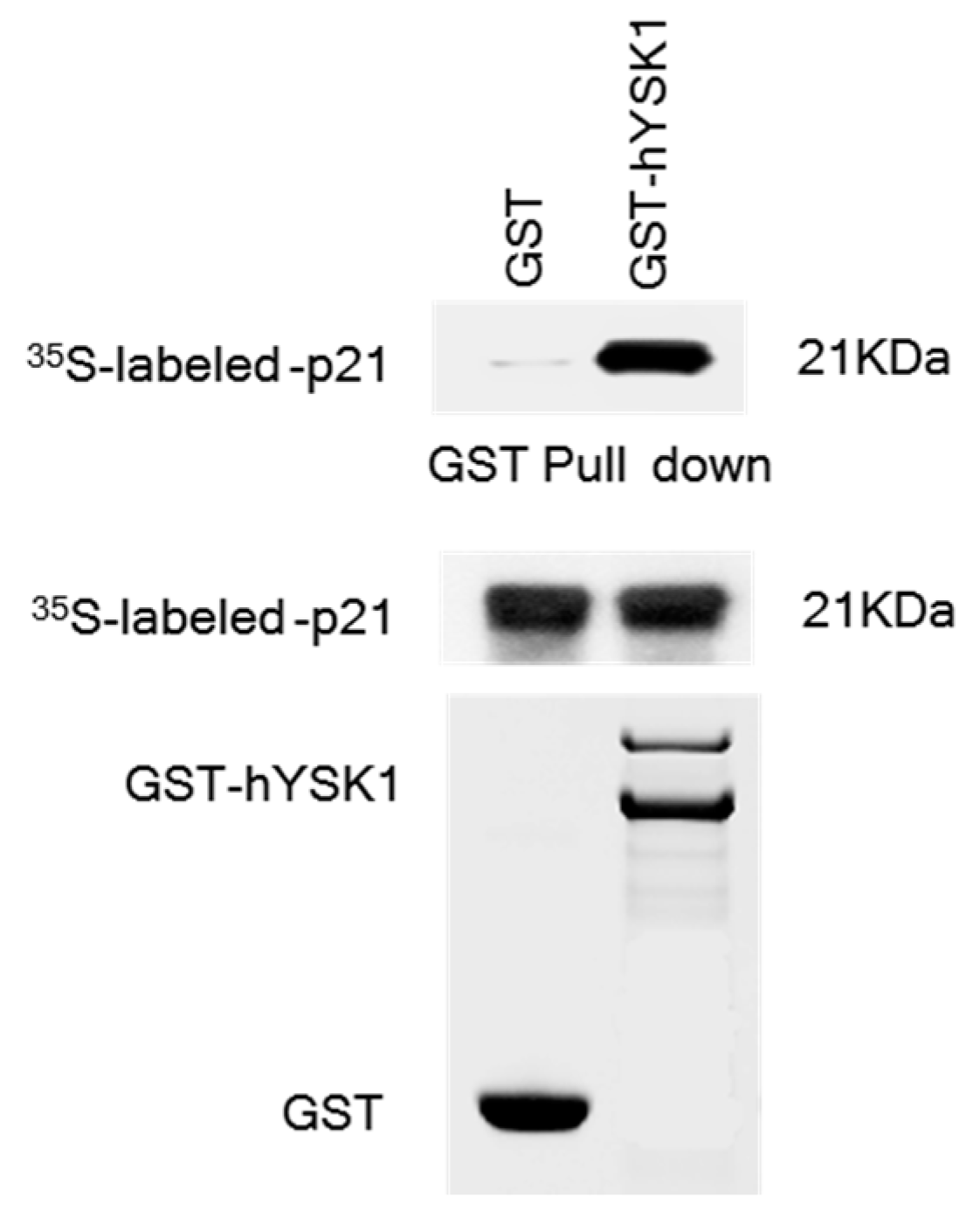
7]. In the present study, we sought to interrogate if hYSK1 follows additional mechanisms in promoting tumor cell proliferation and migration. We have identified p21
WAF1/Cip1 as a novel binding partner of hYSK1 (
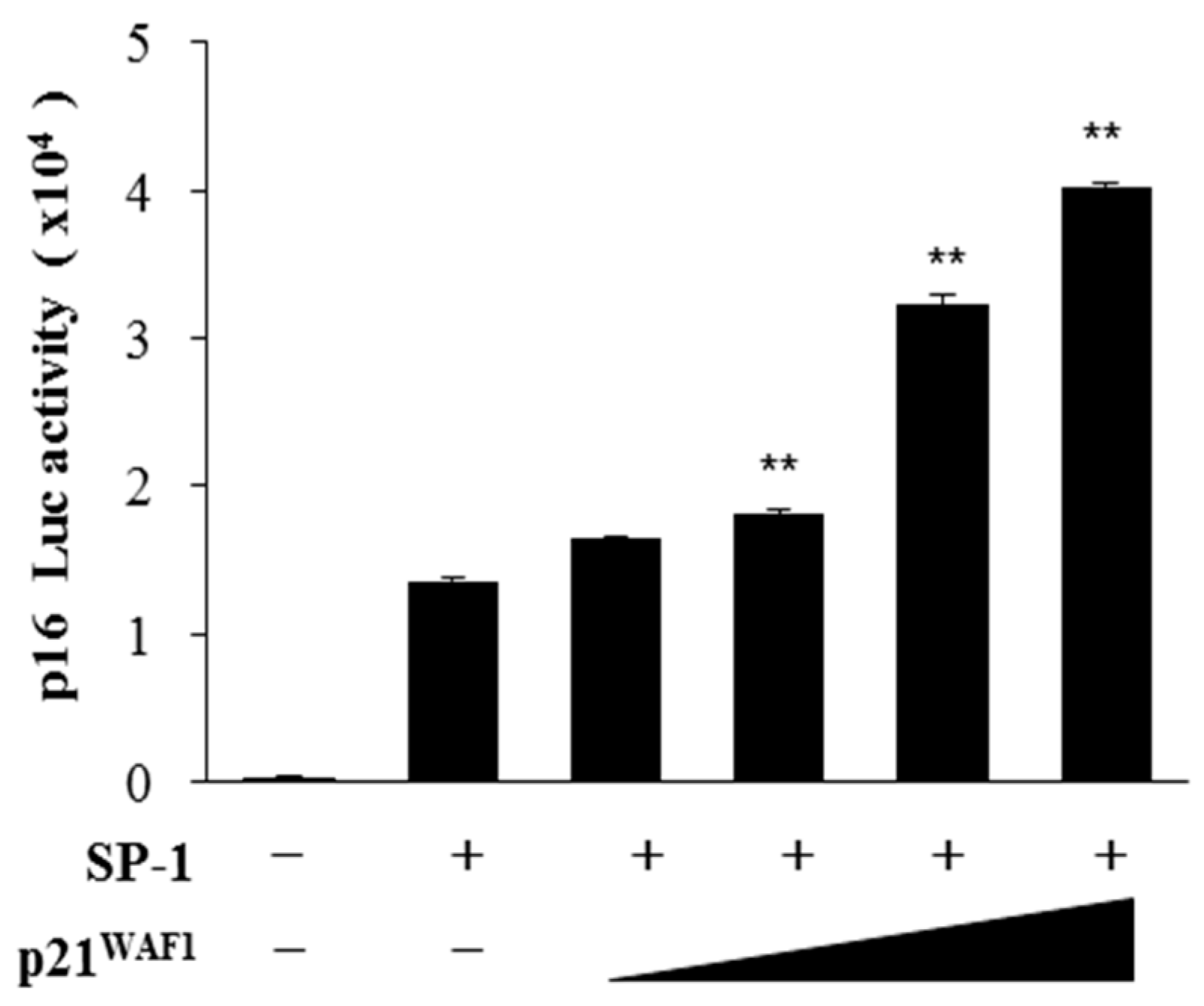
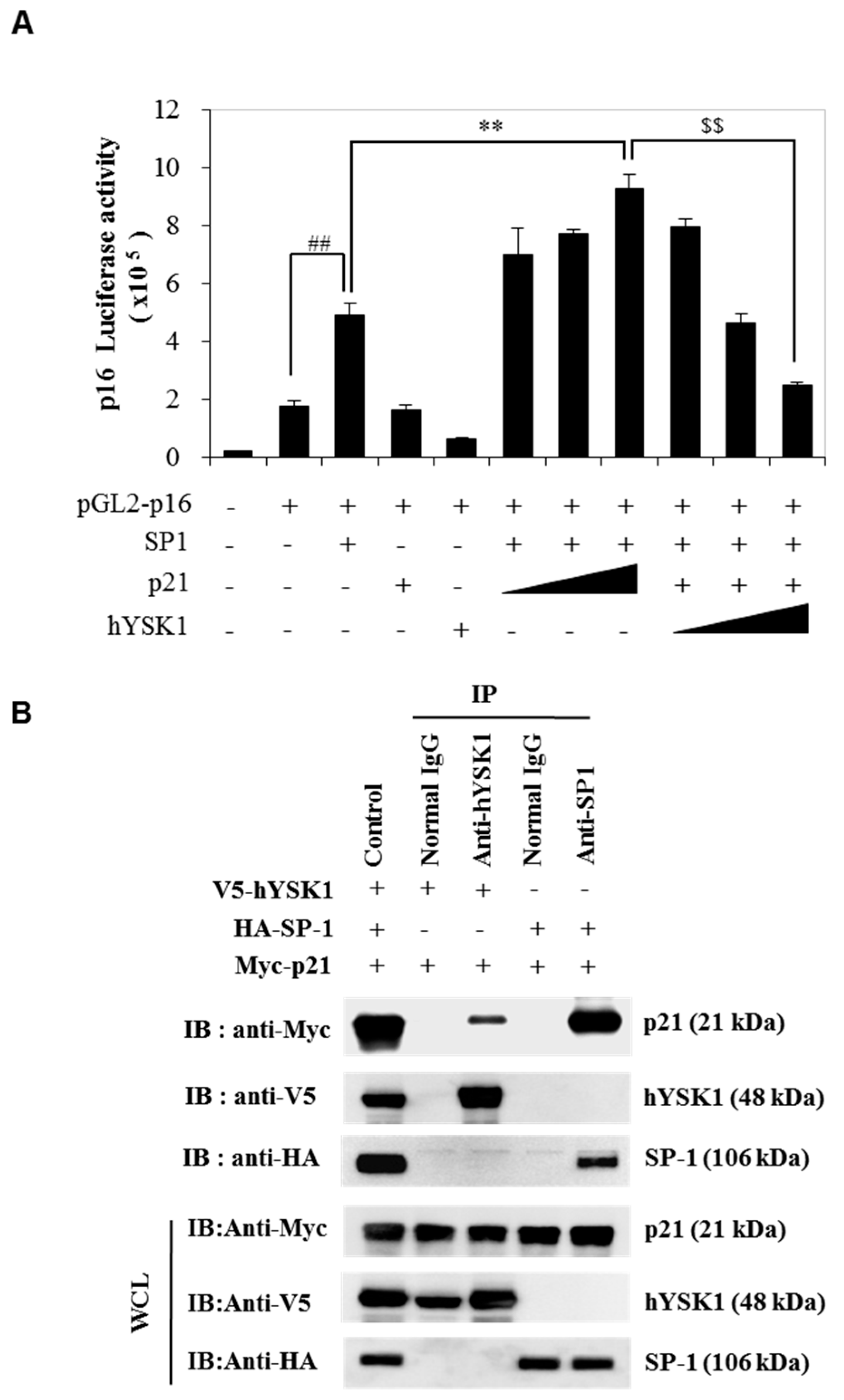
Figure 1). Moreover, by increasing the amount of p21
WAF1/Cip1, we noticed the enhancement of SP-1-mediated p16
INK4a transactivation (
Figure 2). Furthermore, the p16
INK4a expression was inhibited by increasing amount of hYSK1 because hYSK1 has interacted with p21
WAF1/Cip1 but not SP-1, and reduced the binding of SP-1 on the p16
INK4a promoter (
Figure 3).
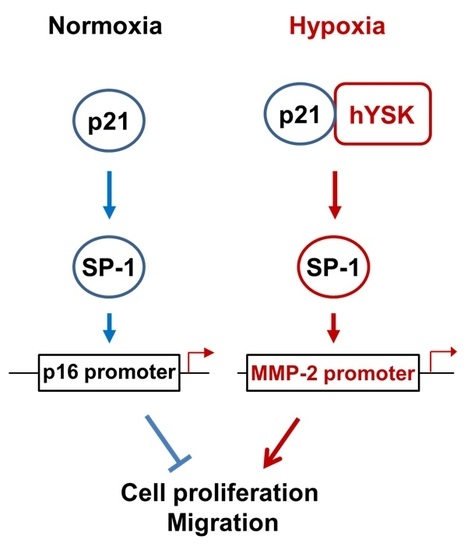
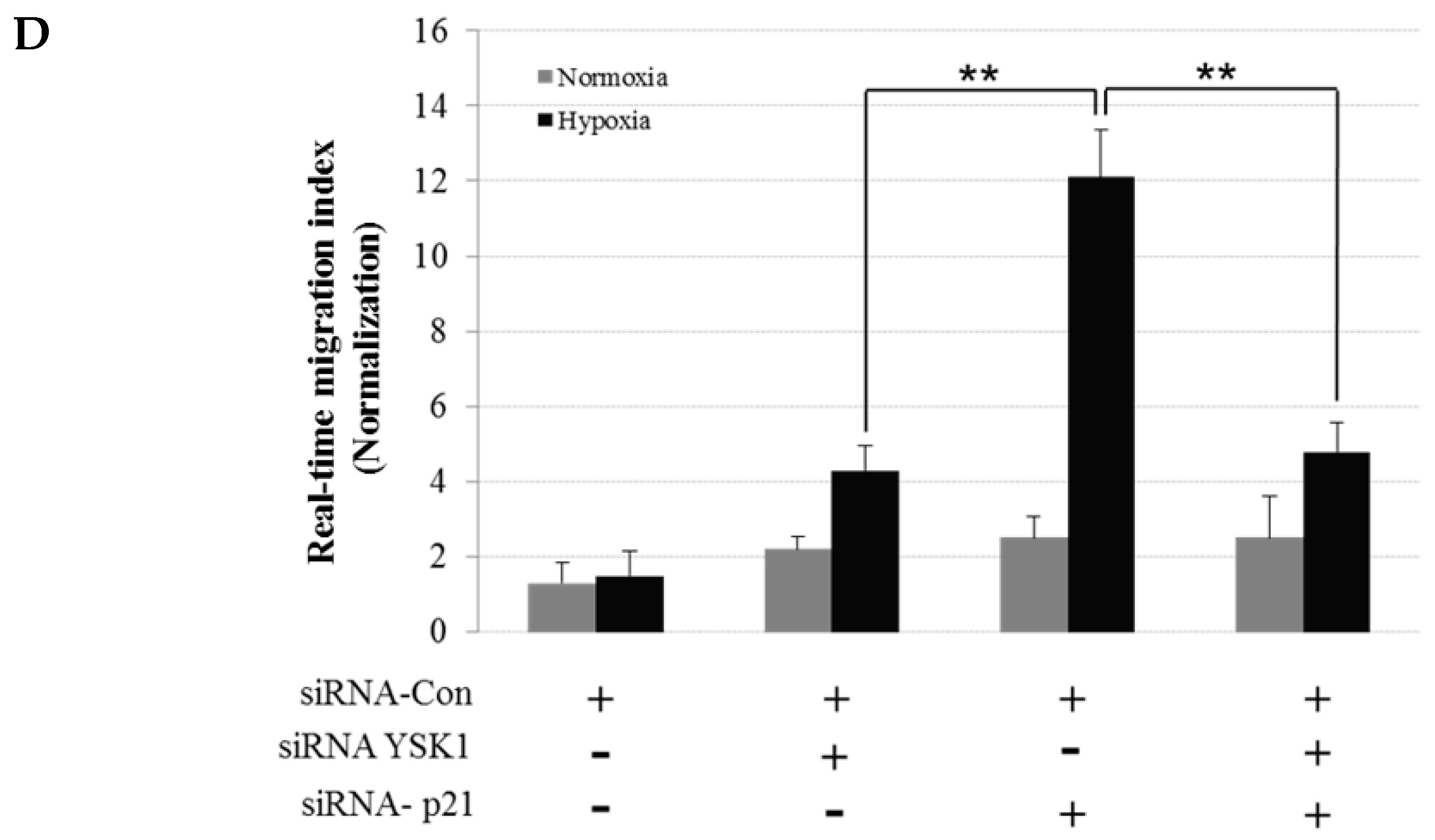
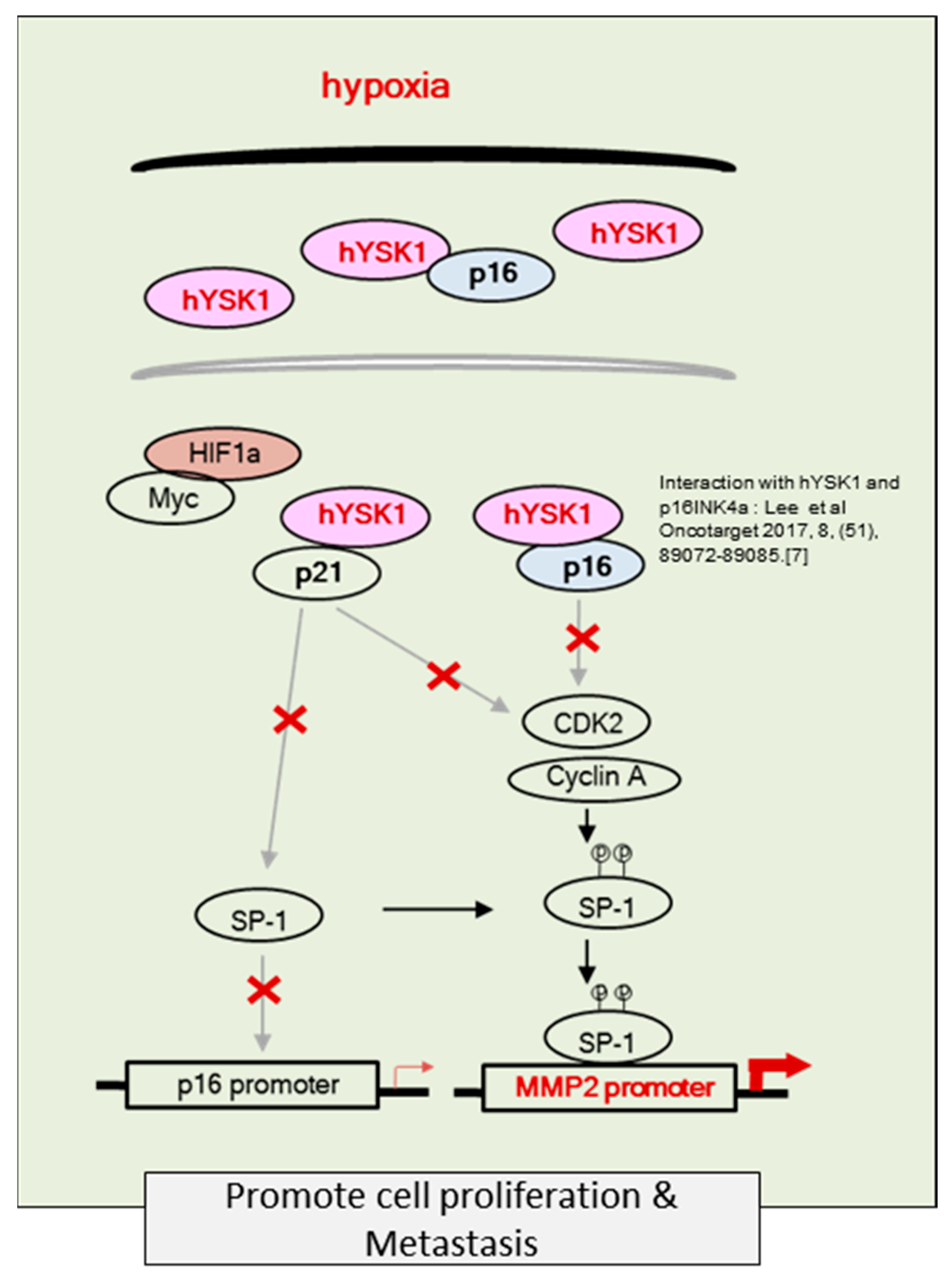
Hypoxia condition is the main signature of cancer mass and promotes epithelial-mesenchymal transition, migration and invasion of cancer cells [
23,
24]. Among the many different signaling pathways involved in hypoxia-triggered tumor cell migration, hYSK1 has recently been identified as a potential regulator, which diminishes p16
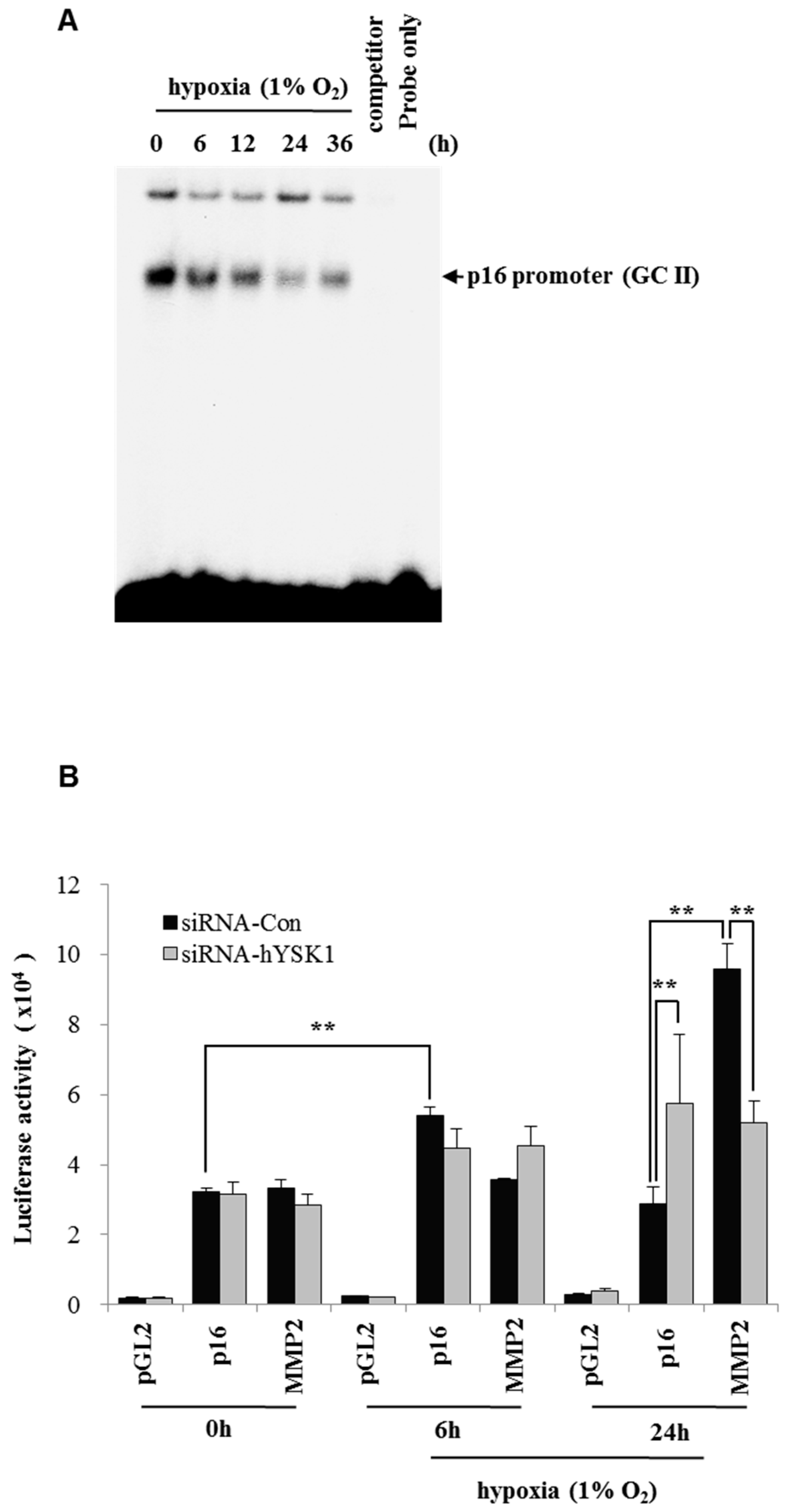
INK4a function. Our study revealed that the binding activity of SP-1 on p16
INK4a promoter was decreased in hypoxia condition by time course (
Figure 4). We also showed that the hYSK1 inhibited p16
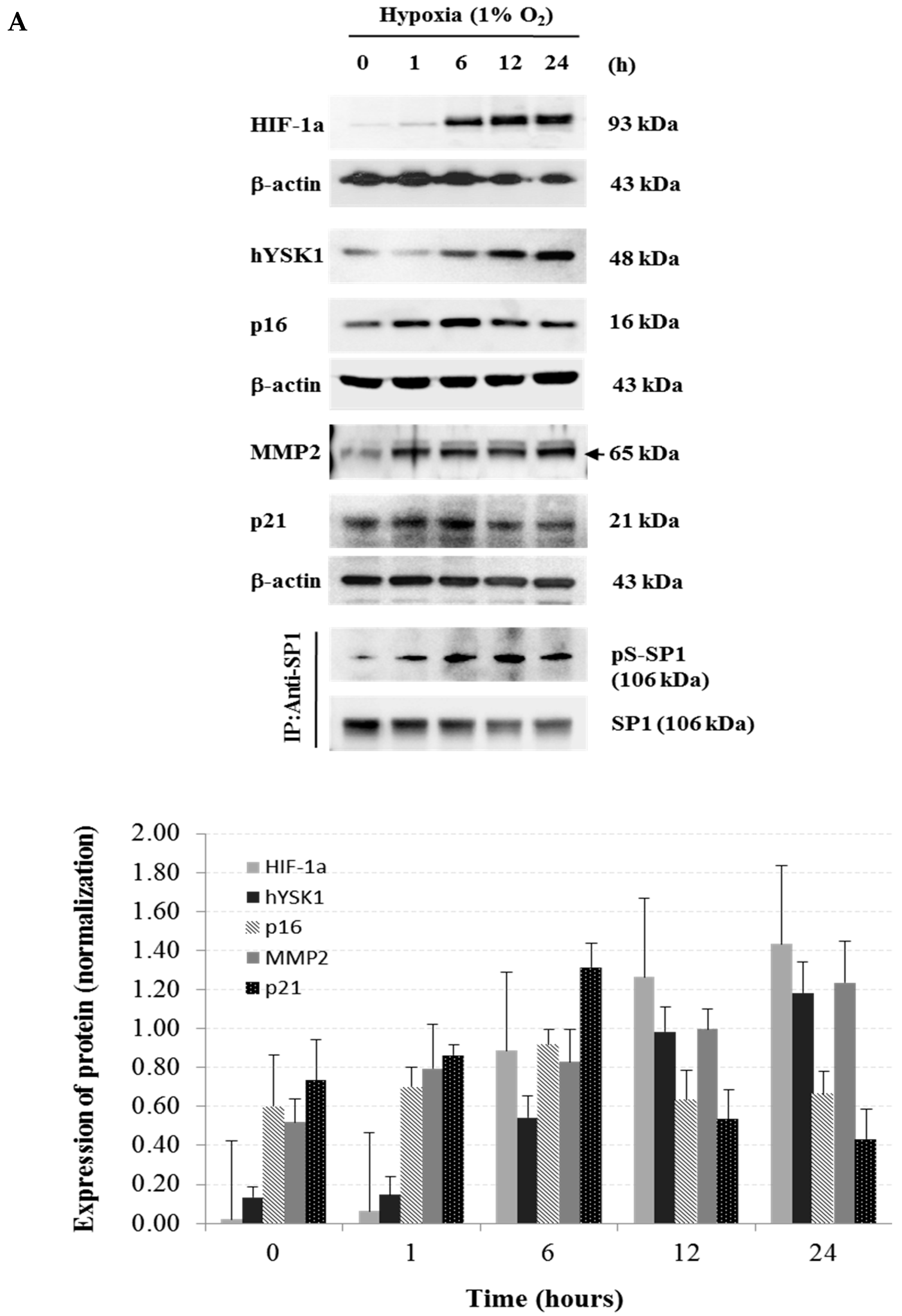
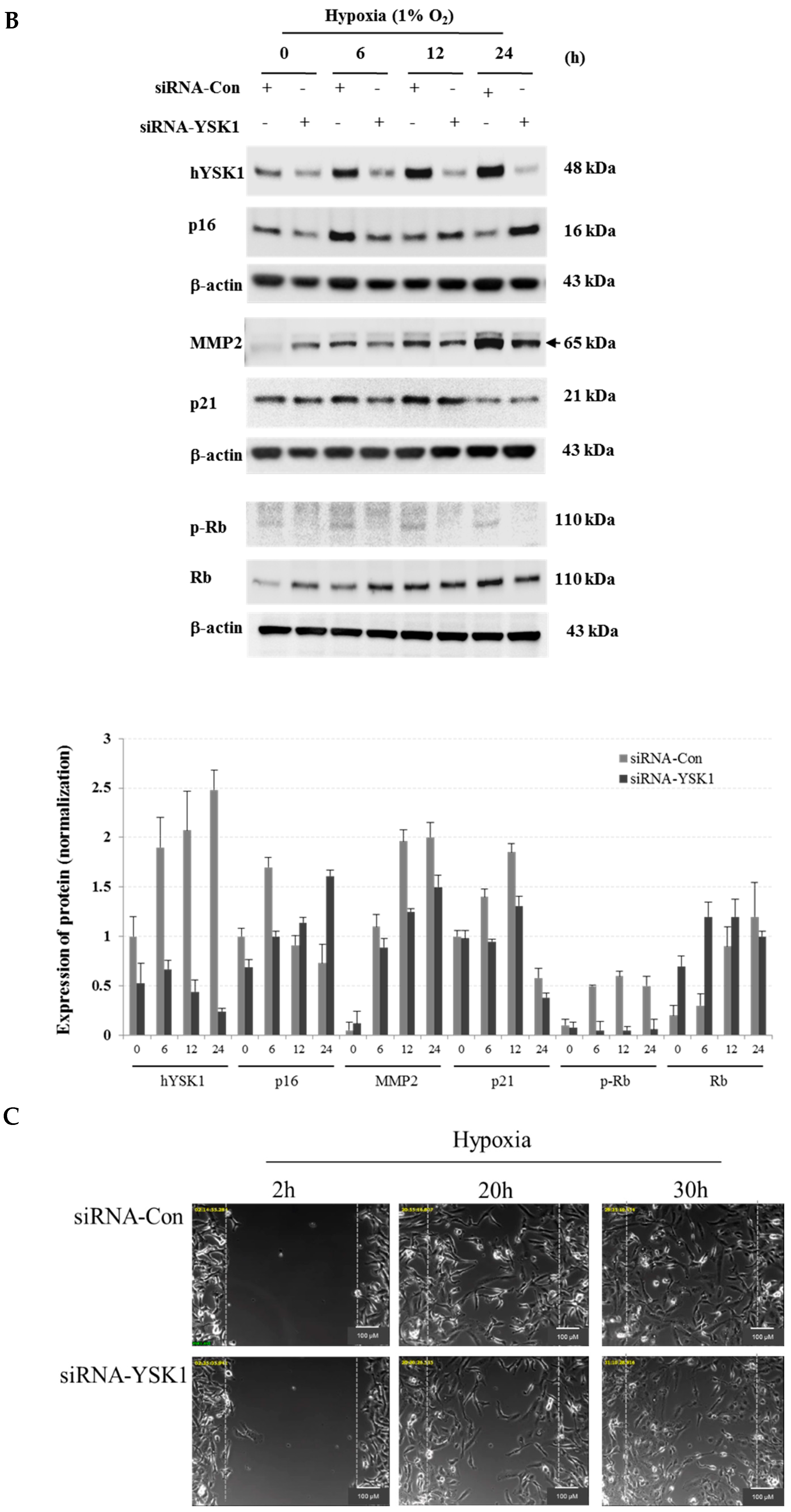
INK4a expression but induced MMP-2 expression and cell migration in hypoxia condition (
Figure 5). Taken together, we identified a potential regulatory mechanism of p16
INK4a/MMP-2 expression in cancer by loss of function of p21
WAF1/Cip1 through the interaction with hYSK1 in hypoxia (
Figure 6). Conversely, Wu et al. [
25] have demonstrated that STK25 (a.k.a hYSK1) suppressed the proliferation of human colorectal cancer cells and silencing STK25 enhanced xenograft tumor growth in mice. Thus, additional studies are warranted to define the exact role of hYSK1 in cancer. However, based on the result of present study, the antagonist development of hYSK1 would be considered as a new therapeutic avenue for developing anticancer drugs.
4. Materials and Methods
4.1. Cell Culture, Plasmids, and Transfection
SK-MEL-28 (human melanoma), HT-1080 (fibrosarcoma) and COS-7 (African green monkey kidney) cells were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). SK-MEL-28 cells and HT-1080 were cultured in MEM containing penicillin (100 units/mL), streptomycin (100 μg/mL), sodium pyruvate (1 mM), and 10% fetal bovine serum (FBS) (Invitrogen, Gibco, MA, USA). COS-7 cells were grown in DMEM containing penicillin (100 units/mL), streptomycin (100 µg/mL), and 10% FBS. Cells were maintained at 37 °C in a humidified atmosphere of 95% air/5% CO2.
pcDNA3.1-v5-hYSK1 was generated by PCR using the human cDNA clone-hYSK1 (Origene Technologies, Rockville, MD, USA) as a template. The PCR product was purified, digested with EcoRI/XhoI, and cloned into the EcoRI/XhoI sites of pcDNA3.1-v5-HisA (Invitrogen). The GST-hYSK1 was inserted in-frame into the BamHI/XhoI site of the pGEX-5X-1 vector (Amersham Biosciences, Buckinghamshire, UK). The HA-SP-1 and Myc-p21WAF1/Cip1 were cloned into the EcoRI/XhoI site of the pCMV-HA and pCMV-Myc vectors (Clontech Laboratories, Mountain View, CA, USA). The pGL2-p16INK4a-luc vector was a gift from Dr. Gordon Peters (Cancer Research UK, London, UK) and the pGL2-MMP-2-luc vector was a gift from Dr. Etty N. Benveniste (The University of Alabama at Birmingham, Birmingham, AL, USA). Various expression vectors were amplified in E. coli XL1-blue or BL21 cells and plasmids were purified using a Qiagen midi kit (Qiagen, Hilden, Germany). The DNA sequences of all plasmids were confirmed by sequencing (Dye Terminator ABI Type Seq., Bionex, NJ, USA).
COS-7 cells were transfected with pGL2-p16INK4a-luc and, pcDNA3.1- p21WAF1/Cip1, HA-SP-1 or pcDNA3.1-v5-hisA-hYSK plasmids using the jetPEI poly transfection reagent (Polyplus, Illkirch, France). For transient gene silencing, SK-MEL-28 cells were transfected using si-control and si-hYSK1 (Dharmacon, Lafayette, CO, USA). For the reporter gene assay, COS-7 cells were seeded in 24-well plates and incubated for 24 h followed by transfection with, pGL2-p16-luc, or pGL2-MMP-2-luc reporter plasmids using the jetPEI poly transfection reagent based on the manufacturer’s instructions. The pRL-TK reporter plasmid was used as an internal control. Cells were harvested after 24 h and disrupted with 5× lysis buffer. Luciferase activity was determined after normalization to pRL-TK activity (Promega, Madison, WI, USA).
4.2. GST Pull-Down Assay
Full length of p21WAF1/Cip1 was translated in vitro with l-[35S] methionine using the TNT Quick Coupled Transcription/Translation System (Promega). Full-length YSK1 proteins were produced in E. coli BL21 as GST-fusion proteins and then purified on GST-Sepharose 4B beads. The 35S-Met-labeled p21WAF1/Cip1 proteins were incubated with either GST or GST-YSK1 fusion proteins bound to Sepharose beads and precipitated in a pull-down assay. The bound proteins were washed three times and boiled with 2.5× sample buffer for 3 min, centrifuged, and then the supernatant fraction was examined by 15% SDS-PAGE analysis. The binding was detected by autoradiography.
4.3. Transfection and Luciferase Activity
The pcDNA3.1-p16INK4a or pcDNA3.1-v5-hisA-hYSK1 plasmid was transfected using the jetPEI poly transfection reagent (Polyplus, Illkirch, France) into HT-1080, COS-1, or COS-7 cells to generate p16INK4a- or p16INK4a-hYSK1-expressing cells. For transient gene silencing, si-control and si-hYSK1 (Dharmacon) were transfected into HT-1080, SK-MEL-28, and A2058 cells. shRNA-control and shRNA-hYSK1 were transfected into HT-1080 and SK-MEL-28 cells. For the reporter gene assay, COS-7 cells were seeded in 24-well plates and incubated for 24 h followed by transfection with pcDNA3-p16INK4a, HA-SP-1, cyclin A, pGL2-p16-luc, or pGL2-MMP-2-luc reporter plasmids using the jetPEI poly transfection reagent based on the manufacturer’s instructions. The pRL-TK reporter plasmid was used as an internal control. Cells were harvested after 24 h and disrupted with 5× lysis buffer. Luciferase activity was determined after normalization to pRL-TK activity (Promega).
4.4. Electrophoretic Mobility Shift Assay (EMSA)
EMSA for SP-1 DNA binding was performed using a DNA–protein binding detection kit, according to the manufacturer’s protocol (GIBCO BRL, Grand Island, NY, USA). Briefly, the SP-1 oligonucleotide probe 5′–GACCGAGTGCGCTCGGCGGCTGCGGAGAGGGGTAGAGCAGGCAGCGGGCGGCGGGGAGCAGC–3′ (SP-1 binding site in p16
INK4a promoter) [
26] was labeled with [γ-
32P]ATP by T4 polynucleotide kinase and purified on a Nick column (Amersham Pharmacia Biotech, Buckinghamshire, UK). The binding reaction was carried out in 25 mL of the mixture containing 5 mL of incubation buffer (10 mM Tris–HCl, pH 7.5, 100 mM NaCl, 1 mM DTT, 1 mM EDTA and 4% glycerol), 10 mg of nuclear extracts and 100,000 c.p.m. of [γ-
32P] ATP-end labeled oligonucleotide. After 50 min incubation at room temperature, 2 mL of 0.1% bromophenol blue was added, and samples were separated through 6% non-denaturing polyacrylamide gel at 150 V in a cold room for 2 h. Finally, the gel was dried and exposed to an X-ray film.
4.5. Immunoprecipitation
Transfected COS-7 cells or SK-MEL-28 cells were harvested in NET-NL lysis buffer containing 50 mM Tris (pH 7.5), 5 mM EDTA, 150 mM NaCl, 1 mM DTT, 0.01% NP-40, 0.2 mM PMSF, and a mixture of protease inhibitors (Roche Diagnostics, Basel, Switzerland). Cell lysates (200 μg) were clarified by centrifugation before overnight incubation at 4 °C with hYSK1 and SP-1 (Santa Cruz Biotechnology, Dallas, TX, USA) antibodies in NET-NR buffer (50 mM Tris (pH 7.5), 5 mM EDTA, 150 mM NaCl, 1 mM DTT, 0.01% NP-40, 2 μg/mL BSA, 0.2 mM PMSF), and a mixture of protease inhibitors (Roche Diagnostics, Basel, Switzerland). An aliquot of 50 μL pre-washed protein A/G-agarose beads (Roche Diagnostics; 50% slurry) was then added to the mixture and incubated for 2 h at 4 °C. Immunoprecipitates were recovered by centrifugation, washed three times in NET-NW buffer (50 mM Tris (pH 7.5), 5 mM EDTA, 150 mM NaCl, 1 mM DTT, 0.01% NP-40, and 0.2 mM PMSF) and resolved by sodium dodecylsulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) and western blotting.
4.6. Western Blot Analysis
Cells were disrupted on ice for 30 min in cell lysis buffer which contains 20 mM Tris (pH 7.5), 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM sodium vanadate, 1 μg/mL leupeptin, and 1 mM phenylmethylsulfonyl fluoride. After centrifuged at 14,000 rpm for 20 min, the cell lysates were collected, and protein concentration was determined by using a protein assay reagent (Bio-Rad labs, Hercules, CA, USA). The total cellular protein extracts were separated by SDS-PAGE and transferred to polyvinylidene fluoride (PVDF) membranes in 20 mM Tris-HCl (pH 8.0), containing 150 mM glycine and 20% (v/v) methanol. Membranes were blocked with 5% nonfat dry milk in 1× Tris-buffered saline (TBS) containing 0.05% Tween 20 (TBS-T) and incubated with antibodies against p16INK4a, p21WAF1/Cip1, hYSK1, pRb, Rb, HIF-1a, pSP-1, SP-1, MMP-2, and β-actin. The blots were washed three times in 1× TBS-T buffer followed by incubation with the appropriate HRP-linked IgG. The specific proteins in the blots were visualized using an enhanced chemiluminescence (ECL) detection kit (GE Healthcare Bioscience, Pittsburgh, PA, USA).
4.7. Cell Migration Assay
Wound migration of cells was measured using Culture-Inserts (Ibidi GmbH, Martinsried, Germany). The Culture-Inserts were placed in 30 mm dish, and siRNA-control and siRNA-hYSK1-transfected SK-EML-28 cells were seeded at a density of 5 × 104 cells in each well with the Culture-Inserts. After 24 h of incubation, the Culture-Inserts were removed, a cell-free gap of 500 μm was created and then transferred to a hypoxic (1% CO2) chamber. The movement of cells was detected by an inverted microscope (BX50, Olympus, Tokyo, Japan) by time course.
Cell movement was analyzed using AVI meta imaging software. For real-time cell migration, we used the xCELLigence RTCA DP system and fibronectin-coated CIM plates (Roche Diagnostics, Basel, Switzerland). Cells were seeded at 10,000 cells per well and the migratory behavior of each cell line was monitored for 30 h. The assay was performed based on the manufacturer’s instructions (Roche Diagnostics, Basel, Switzerland).
4.8. Statistical Analysis
Values were expressed as means ± S.E.M. from at least three independent experiments. Statistical significance was determined by Student’s t-test and a p-value less than 0.05 was considered statistically significant.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







