Evaluation of Genomic Prediction for Pasmo Resistance in Flax



Abstract
:1. Introduction
2. Results
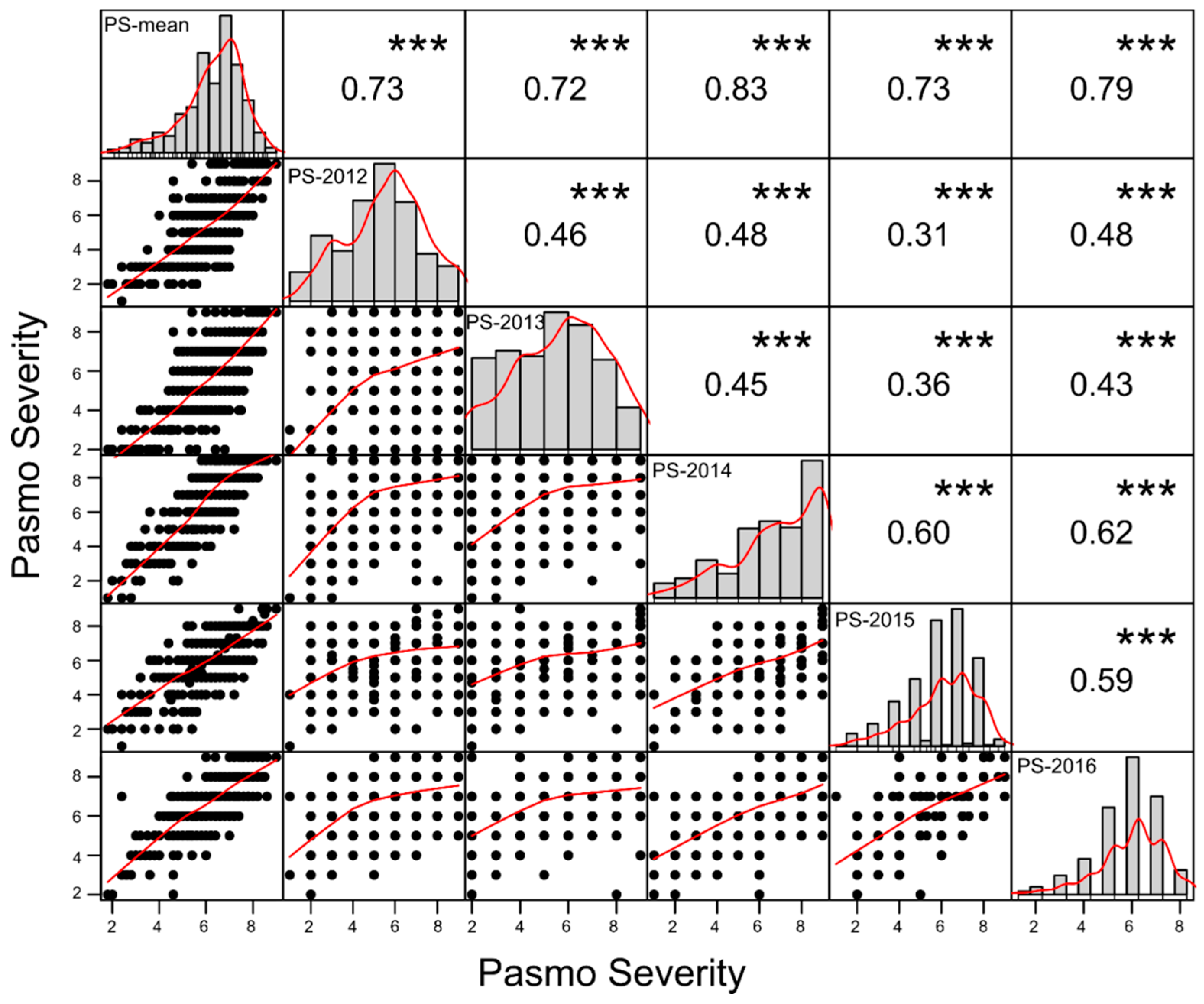
2.1. Evaluation of Pasmo Resistance
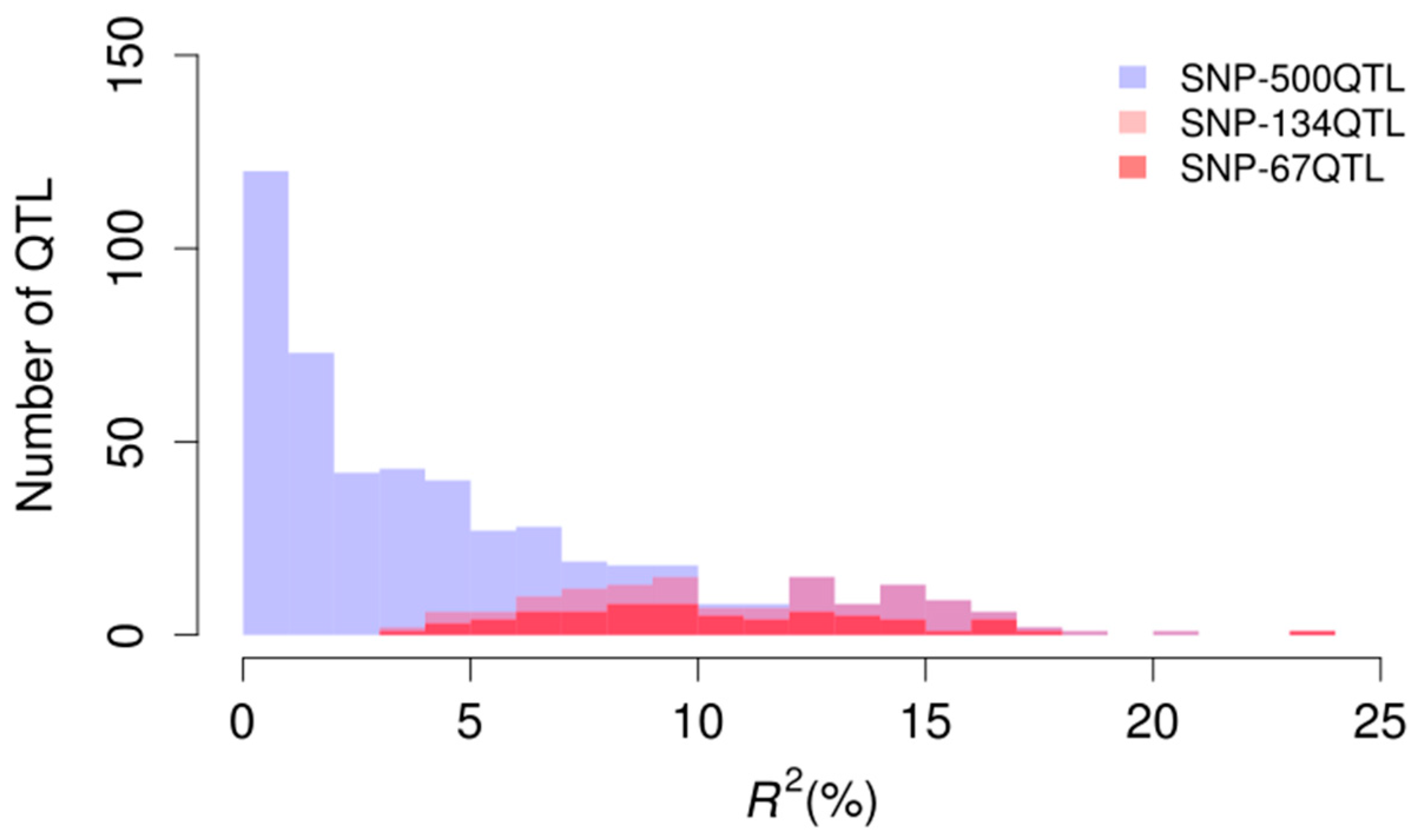
2.2. Evaluation of Marker Sets Used in Genomic Prediction
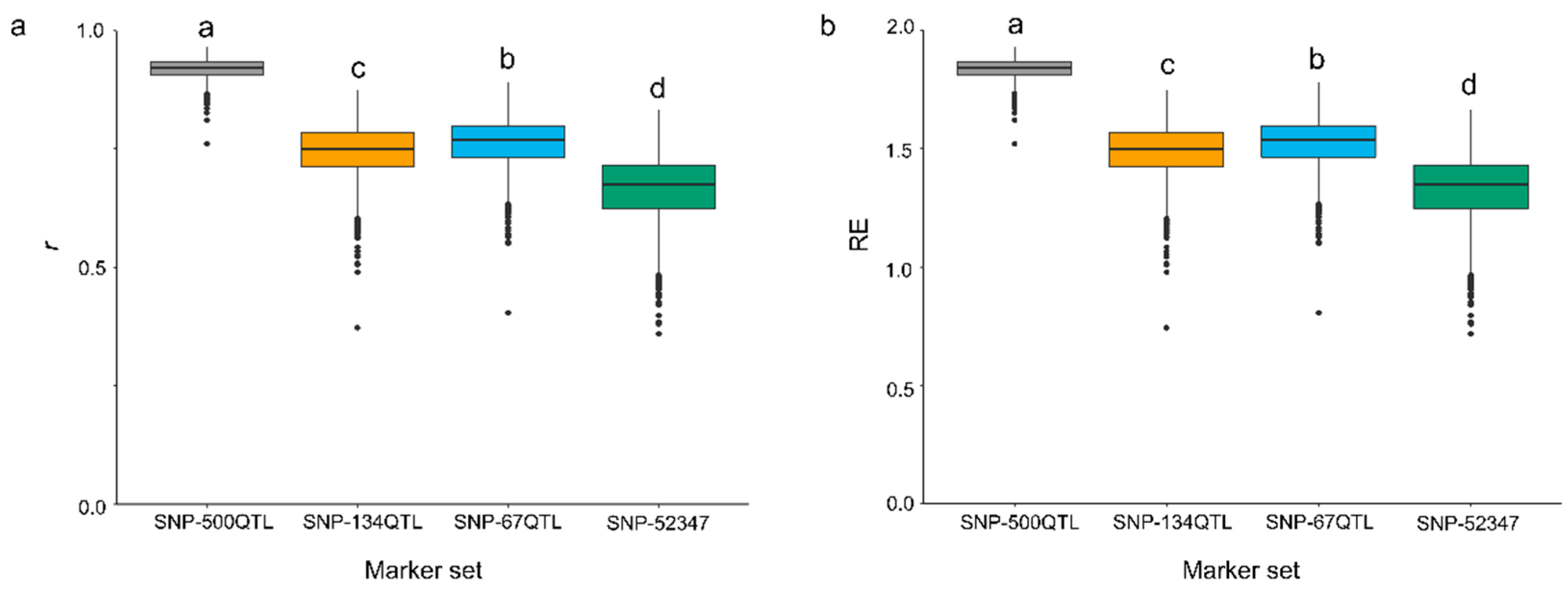
2.3. Accuracy of Genomic Prediction in Relation to Marker Sets and Pasmo Severity Datasets
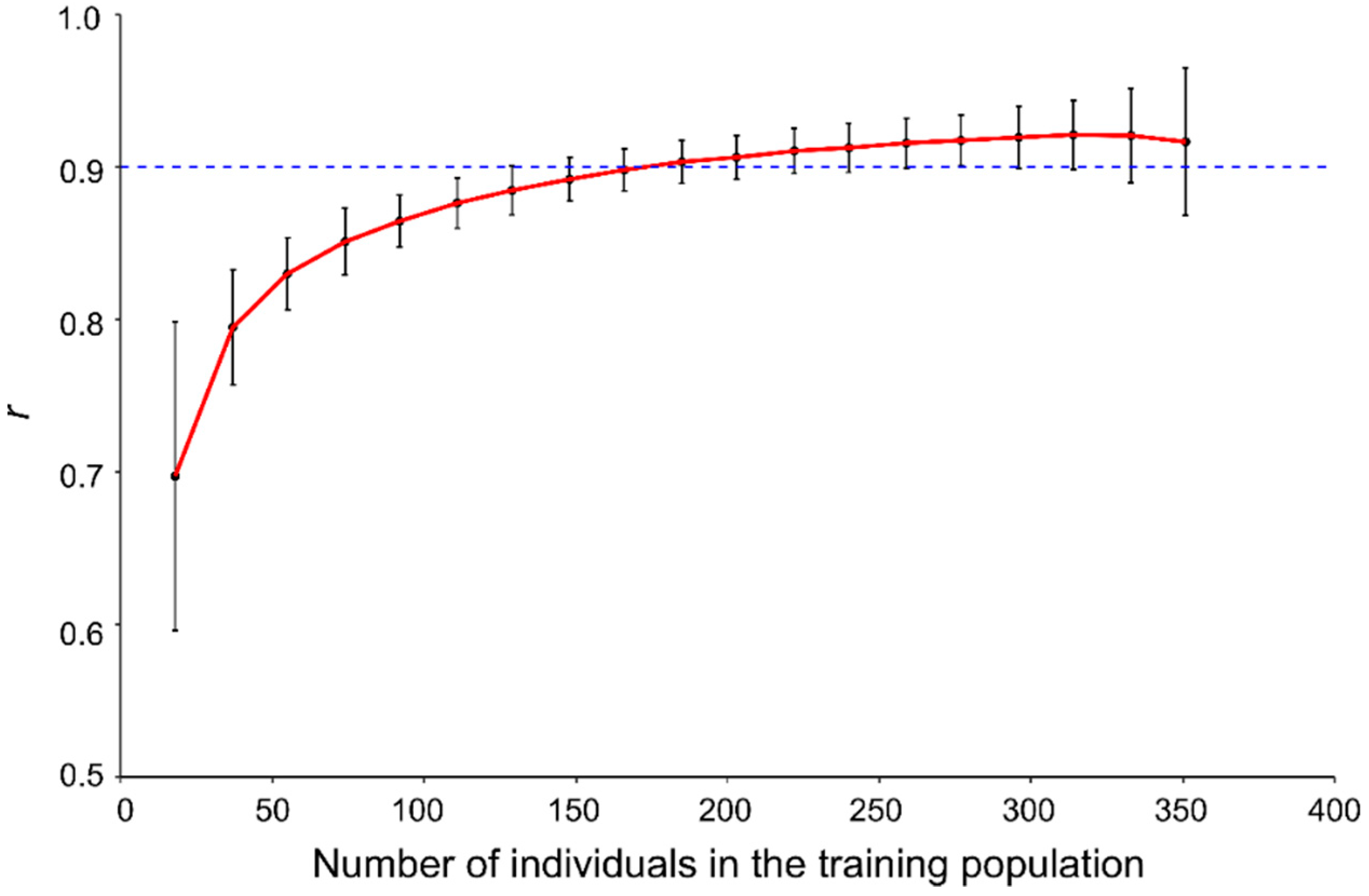
2.4. Sample Size of Training Populations versus Genomic Prediction Accuracy
2.5. Prediction Models of Pasmo Resistance
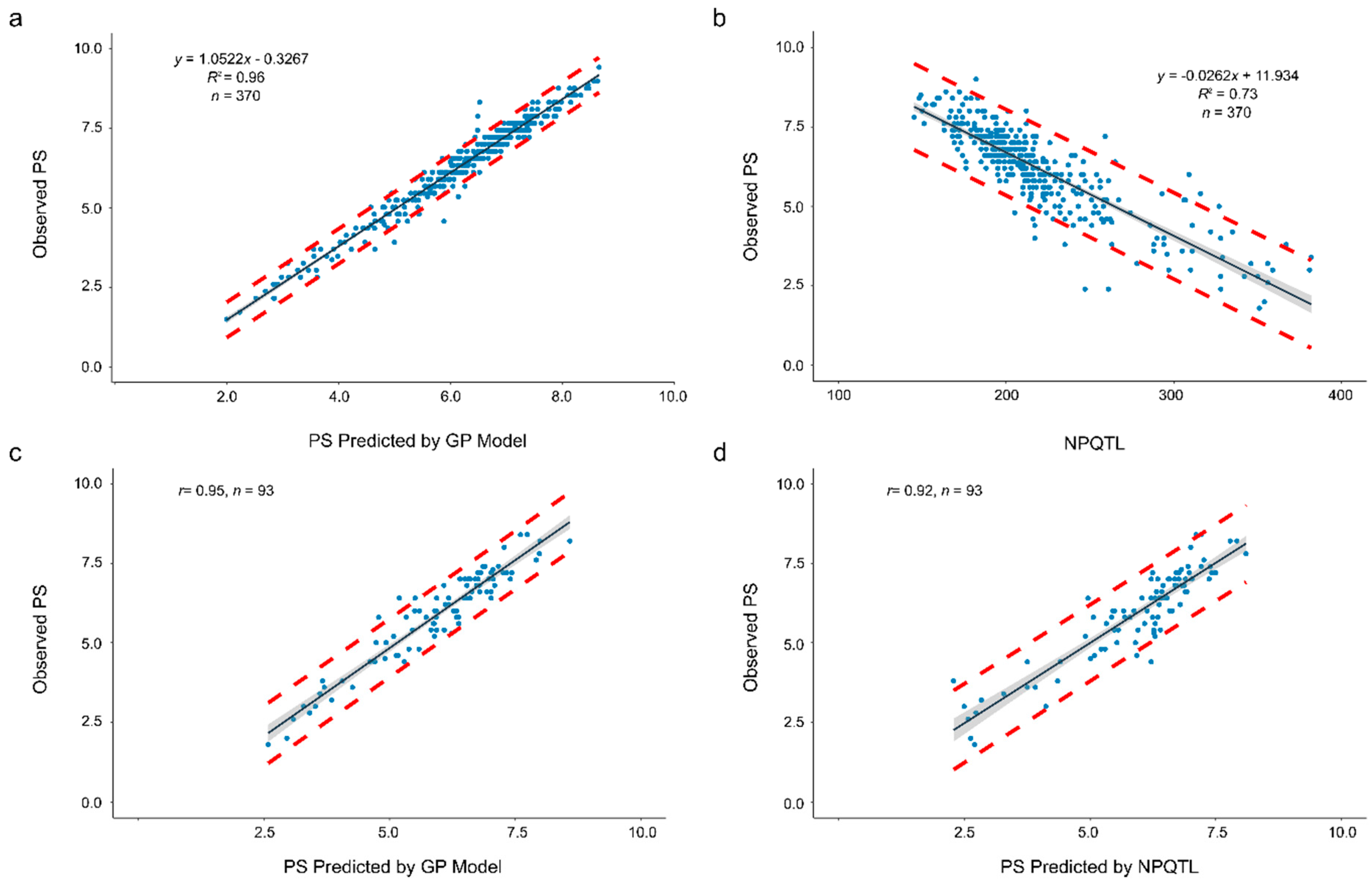
2.6. A Case Study of Genomic Prediction
3. Discussion
3.1. All Detected QTL Used as Markers in Genomic Prediction
3.2. Superior Performance of Genomic Prediction Combined with GWAS
3.3. Accuracy of GP Modelling by Environment, Training Population and Statistical Methods
3.4. Pasmo Severity Prediction Using Number of Positive-Effect QTL
3.5. Breeding Application of Genomic Prediction
4. Materials and Methods
4.1. Population
4.2. Pasmo Resistance Data
4.3. Genomic Data
4.4. Genomic Prediction Models
4.5. Evaluation of Prediction Models
4.6. Phenotypic Variation Explained by Markers
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ANOVA | Analysis of variance |
| BL | Bayesian LASSO |
| BRR | Bayesian ridge regression |
| DON | Deoxynivalenol |
| FHB | Fusarium head blight |
| G × E | Genotype by environment interaction |
| GBS | Genotyping by sequencing |
| GEBV | Genomic estimated breeding value |
| GP | Genomic prediction |
| GS | Genomic selection |
| GWAS | Genome-wide association study |
| MARS | Marker-assisted recurrent selection |
| MAS | Marker-assisted selection |
| NPQTL | Number of QTL with positive-effect alleles |
| PGRC | Plant Gene Resources of Canada |
| PP | Test/prediction population |
| PR | Pasmo resistance |
| PS | Pasmo severity |
| QTL | Quantitative trait locus/loci |
| RE | Relative efficiency |
| RR-BLUP | Ridge regression best linear unbiased prediction |
| SNPs | Single nucleotide polymorphisms |
| TP | Training population |
| VP | Validation population |
References
- Diederichsen, A.; Kusters, P.M.; Kessler, D.; Bainas, Z.; Gugel, R.K. Assembling a core collection from the flax world collection maintained by Plant Gene Resources of Canada. Genet. Resour. Crop Evol. 2012, 60, 1479–1485. [Google Scholar] [CrossRef]
- Vera, C.L.; Irvine, R.B.; Duguid, S.D.; Rashid, K.Y.; Clarke, F.R.; Slaski, J.J. Pasmo disease and lodging in flax as affected by pyraclostrobin fungicide, N fertility and year. Can. J. Plant Sci. 2014, 94, 119–126. [Google Scholar] [CrossRef]
- You, F.M.; Jia, G.; Xiao, J.; Duguid, S.D.; Rashid, K.Y.; Booker, H.M.; Cloutier, S. Genetic variability of 27 traits in a core collection of flax (Linum usitatissimum L.). Front. Plant Sci. 2017, 8, 1636. [Google Scholar] [CrossRef]
- He, L.; Xiao, J.; Rashid, K.Y.; Yao, Z.; Li, P.; Jia, G.; Wang, X.; Cloutier, S.; You, F.M. Genome-wide association studies for pasmo resistance in flax (Linum usitatissimum L.). Front. Plant Sci. 2019, 9, 1982. [Google Scholar] [CrossRef]
- Diederichsen, A.; Rozhmina, T.A.; Kudrjavceva, L.P. Variation patterns within 153 flax (Linum usitatissimum L.) genebank accessions based on evaluation for resistance to fusarium wilt, anthracnose and pasmo. Plant Genet. Resour. 2008, 6, 22–32. [Google Scholar] [CrossRef]
- Collard, B.C.Y.; Mackill, D.J. Marker-assisted selection: An approach for precision plant breeding in the twenty-first century. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2008, 363, 557–572. [Google Scholar] [CrossRef]
- Heslot, N.; Jannink, J.L.; Sorrells, M.E. Perspectives for genomic selection applications and research in plants. Crop Sci. 2015, 55, 1–12. [Google Scholar] [CrossRef]
- Xu, Y.; Crouch, J.H. Marker-assisted selection in plant breeding: From publications to practice. Crop Sci. 2008, 48, 391–407. [Google Scholar] [CrossRef]
- Meuwissen, T.H.; Hayes, B.J.; Goddard, M.E. Prediction of total genetic value using genome-wide dense marker maps. Genetics 2001, 157, 1819–1829. [Google Scholar]
- Lipka, A.E.; Kandianis, C.B.; Hudson, M.E.; Yu, J.M.; Drnevich, J.; Bradbury, P.J.; Gore, M.A. From association to prediction: Statistical methods for the dissection and selection of complex traits in plants. Curr. Opin. Plant Biol. 2015, 24, 110–118. [Google Scholar] [CrossRef]
- Arruda, M.P.; Brown, P.J.; Lipka, A.E.; Krill, A.M.; Thurber, C.; Kolb, F.L. Genomic selection for predicting Fusarium head blight resistance in a wheat breeding program. Plant Genome 2015, 8. [Google Scholar] [CrossRef]
- Daetwyler, H.D.; Bansal, U.K.; Bariana, H.S.; Hayden, M.J.; Hayes, B.J. Genomic prediction for rust resistance in diverse wheat landraces. Theor. Appl. Genet. 2014, 127, 1795–1803. [Google Scholar] [CrossRef]
- Technow, F.; Burger, A.; Melchinger, A.E. Genomic prediction of northern corn leaf blight resistance in maize with combined or separated training sets for heterotic groups. G3 Genes Genomes Genet. 2013, 3, 197–203. [Google Scholar] [CrossRef]
- Poland, J.; Rutkoski, J. Advances and challenges in genomic selection for disease resistance. Annu. Rev. Phytopathol. 2016, 54, 79–98. [Google Scholar] [CrossRef]
- Gianola, D. Priors in whole-genome regression: The Bayesian alphabet returns. Genetics 2013, 194, 573–596. [Google Scholar] [CrossRef]
- Desta, Z.A.; Ortiz, R. Genomic selection: Genome-wide prediction in plant improvement. Trends Plant Sci. 2014, 19, 592–601. [Google Scholar] [CrossRef]
- Whittaker, J.C.; Thompson, R.; Denham, M.C. Marker-assisted selection using ridge regression. Genet. Res. 2000, 75, 249–252. [Google Scholar] [CrossRef]
- Tibshirani, R. Regression shrinkage and selection via the Lasso. J. R. Stat. Soc. Ser. B Methodol. 1996, 58, 267–288. [Google Scholar] [CrossRef]
- Jiang, Y.; Zhao, Y.; Rodemann, B.; Plieske, J.; Kollers, S.; Korzun, V.; Ebmeyer, E.; Argillier, O.; Hinze, M.; Ling, J.; et al. Potential and limits to unravel the genetic architecture and predict the variation of Fusarium head blight resistance in European winter wheat (Triticum aestivum L.). Heredity 2015, 114, 318–326. [Google Scholar] [CrossRef]
- Spindel, J.; Begum, H.; Akdemir, D.; Virk, P.; Collard, B.; Redona, E.; Atlin, G.; Jannink, J.L.; McCouch, S.R. Genomic selection and association mapping in rice (Oryza sativa): Effect of trait genetic architecture, training population composition, marker number and statistical model on accuracy of rice genomic selection in elite, tropical rice breeding lines. PLoS Genet. 2015, 11, e1004982. [Google Scholar]
- Zhang, J.; Song, Q.; Cregan, P.B.; Jiang, G.L. Genome-wide association study, genomic prediction and marker-assisted selection for seed weight in soybean (Glycine max). Theor. Appl. Genet. 2016, 129, 117–130. [Google Scholar] [CrossRef]
- Li, Y.; Ruperao, P.; Batley, J.; Edwards, D.; Khan, T.; Colmer, T.D.; Pang, J.; Siddique, K.H.M.; Sutton, T. Investigating drought tolerance in chickpea using genome-wide association mapping and genomic selection based on whole-genome resequencing data. Front. Plant Sci. 2018, 9, 190. [Google Scholar] [CrossRef]
- Yu, J.; Buckler, E.S. Genetic association mapping and genome organization of maize. Curr. Opin. Biotechnol. 2006, 17, 155–160. [Google Scholar] [CrossRef]
- Arojju, S.K.; Conaghan, P.; Barth, S.; Milbourne, D.; Casler, M.D.; Hodkinson, T.R.; Michel, T.; Byrne, S.L. Genomic prediction of crown rust resistance in Lolium perenne. BMC Genet. 2018, 19, 35. [Google Scholar] [CrossRef]
- You, F.M.; Booker, H.M.; Duguid, S.D.; Jia, G.; Cloutier, S. Accuracy of genomic selection in biparental populations of flax (Linum usitatissimum L.). Crop J. 2016, 4, 290–303. [Google Scholar] [CrossRef]
- Dekkers, J.C. Prediction of response to marker-assisted and genomic selection using selection index theory. J. Anim. Breed. Genet. 2007, 124, 331–341. [Google Scholar] [CrossRef]
- Ziyomo, C.; Bernardo, R. Drought tolerance in maize: Indirect selection through secondary traits versus genomewide selection. Crop Sci. 2013, 53, 1269–1275. [Google Scholar] [CrossRef]
- Crossa, J.; Perez-Rodriguez, P.; Cuevas, J.; Montesinos-Lopez, O.; Jarquin, D.; de Los Campos, G.; Burgueno, J.; Gonzalez-Camacho, J.M.; Perez-Elizalde, S.; Beyene, Y.; et al. Genomic selection in plant breeding: Methods, models, and perspectives. Trends. Plant Sci. 2017, 22, 961–975. [Google Scholar] [CrossRef]
- Gowda, M.; Das, B.; Makumbi, D.; Babu, R.; Semagn, K.; Mahuku, G.; Olsen, M.S.; Bright, J.M.; Beyene, Y.; Prasanna, B.M. Genome-wide association and genomic prediction of resistance to maize lethal necrosis disease in tropical maize germplasm. Theor. Appl. Genet. 2015, 128, 1957–1968. [Google Scholar] [CrossRef] [Green Version]
- Rutkoski, J.; Benson, J.; Jia, Y.; Brown-Guedira, G.; Jannink, J.-L.; Sorrells, M. Evaluation of genomic prediction methods for Fusarium head blight resistance in wheat. Plant Genome 2012, 5, 51–61. [Google Scholar] [CrossRef]
- Deshmukh, R.; Sonah, H.; Patil, G.; Chen, W.; Prince, S.; Mutava, R.; Vuong, T.; Valliyodan, B.; Nguyen, H.T. Integrating omic approaches for abiotic stress tolerance in soybean. Front. Plant Sci. 2014, 5, 244. [Google Scholar] [CrossRef]
- Spindel, J.E.; Begum, H.; Akdemir, D.; Collard, B.; Redona, E.; Jannink, J.L.; McCouch, S. Genome-wide prediction models that incorporate de novo GWAS are a powerful new tool for tropical rice improvement. Heredity 2016, 116, 395–408. [Google Scholar] [CrossRef] [Green Version]
- Kayondo, S.I.; Pino del Carpio, D.; Lozano, R.; Ozimati, A.; Wolfe, M.; Baguma, Y.; Gracen, V.; Offei, S.; Ferguson, M.; Kawuki, R.; et al. Genome-wide association mapping and genomic prediction for CBSD resistance in Manihot esculenta. Sci. Rep. 2018, 8, 1549. [Google Scholar] [CrossRef]
- Wang, X.; Xu, Y.; Hu, Z.L.; Xu, C.W. Genomic selection methods for crop improvement: Current status and prospects. Crop J. 2018, 6, 330–340. [Google Scholar] [CrossRef]
- Jarquin, D.; Kocak, K.; Posadas, L.; Hyma, K.; Jedlicka, J.; Graef, G.; Lorenz, A. Genotyping by sequencing for genomic prediction in a soybean breeding population. BMC Genom. 2014, 15, 740. [Google Scholar] [CrossRef]
- Asoro, F.G.; Newell, M.A.; Beavis, W.D.; Scott, M.P.; Jannink, J.-L. Accuracy and training population design for genomic selection on quantitative traits in elite North American oats. Plant Genome 2011, 4, 132–144. [Google Scholar] [CrossRef]
- Lorenzana, R.E.; Bernardo, R. Accuracy of genotypic value predictions for marker-based selection in biparental plant populations. Theor. Appl. Genet. 2009, 120, 151–161. [Google Scholar] [CrossRef]
- Goddard, M. Genomic selection: Prediction of accuracy and maximisation of long term response. Genetica 2009, 136, 245–257. [Google Scholar] [CrossRef]
- Nielsen, H.M.; Sonesson, A.K.; Yazdi, H.; Meuwissen, T.H.E. Comparison of accuracy of genome-wide and BLUP breeding value estimates in sib based aquaculture breeding schemes. Aquaculture 2009, 289, 259–264. [Google Scholar] [CrossRef]
- Lorenz, A.J.; Chao, S.; Asoro, F.G.; Heffner, E.L.; Hayashi, T.; Iwata, H.; Smith, K.P.; Sorrells, M.E.; Jannink, J.L. Genomic selection in plant breeding. Adv. Agron. 2011, 110, 77–123. [Google Scholar]
- Cuevas, J.; Crossa, J.; Montesinos-Lopez, O.A.; Burgueno, J.; Perez-Rodriguez, P.; de los Campos, G. Bayesian genomic prediction with genotype x environment interaction kernel models. G3 Genes Genomes Genet. 2017, 7, 41–53. [Google Scholar]
- Dong, H.; Wang, R.; Yuan, Y.; Anderson, J.; Pumphrey, M.; Zhang, Z.; Chen, J. Evaluation of the potential for genomic selection to improve spring wheat resistance to Fusarium head blight in the Pacific Northwest. Front. Plant Sci. 2018, 9, 911. [Google Scholar] [CrossRef]
- Isidro, J.; Jannink, J.L.; Akdemir, D.; Poland, J.; Heslot, N.; Sorrells, M.E. Training set optimization under population structure in genomic selection. Theor. Appl. Genet. 2015, 128, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Rutkoski, J.E.; Poland, J.A.; Singh, R.P.; Huerta-Espino, J.; Bhavani, S.; Barbier, H.; Rouse, M.N.; Jannink, J.-L.; Sorrells, M.E. Genomic selection for quantitative adult plant stem rust resistance in wheat. Plant Genome 2014, 7. [Google Scholar] [CrossRef]
- McElroy, M.S.; Navarro, A.J.R.; Mustiga, G.; Stack, C.; Gezan, S.; Pena, G.; Sarabia, W.; Saquicela, D.; Sotomayor, I.; Douglas, G.M.; et al. Prediction of cacao (Theobroma cacao) resistance to Moniliophthora spp. diseases via genome-wide association analysis and genomic selection. Front. Plant Sci. 2018, 9, 343. [Google Scholar] [CrossRef]
- Enciso-Rodriguez, F.; Douches, D.; Lopez-Cruz, M.; Coombs, J.; de Los Campos, G. Genomic selection for late blight and common scab resistance in tetraploid potato (Solanum tuberosum). G3 Genes Genomes Genet. 2018, 8, 2471–2481. [Google Scholar] [CrossRef]
- Daetwyler, H.D.; Pong-Wong, R.; Villanueva, B.; Woolliams, J.A. The impact of genetic architecture on genome-wide evaluation methods. Genetics 2010, 185, 1021–1031. [Google Scholar] [CrossRef]
- Jannink, J.L.; Lorenz, A.J.; Iwata, H. Genomic selection in plant breeding: From theory to practice. Brief Funct. Genom. 2010, 9, 166–177. [Google Scholar] [CrossRef]
- Thavamanikumar, S.; Dolferus, R.; Thumma, B.R. Comparison of genomic selection models to predict flowering time and spike grain number in two hexaploid wheat doubled haploid populations. G3 Genes Genomes Genet. 2015, 5, 1991–1998. [Google Scholar] [CrossRef]
- VanRaden, P.M.; Van Tassell, C.P.; Wiggans, G.R.; Sonstegard, T.S.; Schnabel, R.D.; Taylor, J.F.; Schenkel, F.S. Invited review: Reliability of genomic predictions for North American Holstein bulls. J. Dairy Sci. 2009, 92, 16–24. [Google Scholar] [CrossRef]
- Clark, S.A.; Hickey, J.M.; van der Werf, J.H. Different models of genetic variation and their effect on genomic evaluation. Genet. Sel. Evol. 2011, 43, 18. [Google Scholar] [CrossRef] [Green Version]
- Rutkoski, J.; Singh, R.P.; Huerta-Espino, J.; Bhavani, S.; Poland, J.; Jannink, J.L.; Sorrells, M.E. Genetic gain from phenotypic and genomic selection for quantitative resistance to stem rust of wheat. Plant Genome 2015, 8. [Google Scholar] [CrossRef]
- Gonzalez-Camacho, J.M.; Ornella, L.; Perez-Rodriguez, P.; Gianola, D.; Dreisigacker, S.; Crossa, J. Applications of machine learning methods to genomic selection in breeding wheat for rust resistance. Plant Genome 2018, 11. [Google Scholar] [CrossRef]
- Liabeuf, D.; Sim, S.C.; Francis, D.M. Comparison of marker-based genomic estimated breeding values and phenotypic evaluation for selection of bacterial spot resistance in tomato. Phytopathology 2018, 108, 392–401. [Google Scholar] [CrossRef]
- Ornella, L.; Singh, S.; Perez, P.; Burgueño, J.; Singh, R.; Tapia, E.; Bhavani, S.; Dreisigacker, S.; Braun, H.-J.; Mathews, K.; et al. Genomic prediction of genetic values for resistance to wheat rusts. Plant Genome 2012, 5. [Google Scholar] [CrossRef]
- Endelman, J.B. Ridge regression and other kernels for genomic selection with R package rrBLUP. Plant Genome 2011, 4, 250–255. [Google Scholar] [CrossRef]
- Piepho, H.P. Ridge regression and extensions for genomewide selection in maize. Crop Sci. 2009, 49, 1165–1176. [Google Scholar] [CrossRef]
- Bassi, F.M.; Bentley, A.R.; Charmet, G.; Ortiz, R.; Crossa, J. Breeding schemes for the implementation of genomic selection in wheat (Triticum spp.). Plant Sci. 2016, 242, 23–36. [Google Scholar] [CrossRef]
- Lin, C.S.; Poushinsky, G. A modified augmented design (type 2) for rectangular plots. Can. J. Plant Sci. 1985, 65, 743–749. [Google Scholar] [CrossRef]
- You, F.M.; Duguid, S.D.; Thambugala, D.; Cloutier, S. Statistical analysis and field evaluation of the type 2 modified augmented design (MAD) in phenotyping of flax (Linum usitatissimum) germplasms in multiple environments. Aust. J. Crop Sci. 2013, 7, 1789–1800. [Google Scholar]
- Browning, S.R.; Browning, B.L. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am. J. Hum. Genet. 2007, 81, 1084–1097. [Google Scholar] [CrossRef] [PubMed]
- Price, A.L.; Patterson, N.J.; Plenge, R.M.; Weinblatt, M.E.; Shadick, N.A.; Reich, D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006, 38, 904–909. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Pressoir, G.; Briggs, W.H.; Vroh Bi, I.; Yamasaki, M.; Doebley, J.F.; McMullen, M.D.; Gaut, B.S.; Nielsen, D.M.; Holland, J.B.; et al. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat. Genet. 2006, 38, 203–208. [Google Scholar] [CrossRef]
- Zhou, X.; Stephens, M. Genome-wide efficient mixed-model analysis for association studies. Nat. Genet. 2012, 44, 821–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Huang, M.; Fan, B.; Buckler, E.S.; Zhang, Z. Iterative usage of fixed and random effect models for powerful and efficient genome-wide association studies. PLoS Genet. 2016, 12, e1005767. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.B.; Feng, J.Y.; Ren, W.L.; Huang, B.; Zhou, L.; Wen, Y.J.; Zhang, J.; Dunwell, J.M.; Xu, S.; Zhang, Y.M. Improving power and accuracy of genome-wide association studies via a multi-locus mixed linear model methodology. Sci. Rep. 2016, 6, 19444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, Y.J.; Zhang, H.; Ni, Y.L.; Huang, B.; Zhang, J.; Feng, J.Y.; Wang, S.B.; Dunwell, J.M.; Zhang, Y.M.; Wu, R. Methodological implementation of mixed linear models in multi-locus genome-wide association studies. Brief. Bioinform. 2017, 19, 700–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamba, C.L.; Ni, Y.L.; Zhang, Y.M. Iterative sure independence screening EM-Bayesian LASSO algorithm for multi-locus genome-wide association studies. PLoS Comput. Biol. 2017, 13, e1005357. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Feng, J.Y.; Ni, Y.L.; Wen, Y.J.; Niu, Y.; Tamba, C.L.; Yue, C.; Song, Q.; Zhang, Y.M. pLARmEB: Integration of least angle regression with empirical Bayes for multilocus genome-wide association studies. Heredity 2017, 118, 517–524. [Google Scholar] [CrossRef]
- Ren, W.L.; Wen, Y.J.; Dunwell, J.M.; Zhang, Y.M. pKWmEB: Integration of Kruskal-Wallis test with empirical Bayes under polygenic background control for multi-locus genome-wide association study. Heredity 2017, 120, 208–218. [Google Scholar] [CrossRef]
- mrMLM. Available online: https://cran.r-project.org/web/packages/mrMLM/index.html (accessed on 25 August 2018).
- De los Campos, G.; Naya, H.; Gianola, D.; Crossa, J.; Legarra, A.; Manfredi, E.; Weigel, K.; Cotes, J.M. Predicting quantitative traits with regression models for dense molecular markers and pedigree. Genetics 2009, 182, 375–385. [Google Scholar] [CrossRef] [PubMed]
- De los Campos, G.; Hickey, J.M.; Pong-Wong, R.; Daetwyler, H.D.; Calus, M.P. Whole-genome regression and prediction methods applied to plant and animal breeding. Genetics 2013, 193, 327–345. [Google Scholar] [CrossRef] [PubMed]
- de Los Campos, G.; Perez, P.; Vazquez, A.I.; Crossa, J. Genome-enabled prediction using the BLR (Bayesian Linear Regression) R-package. Methods Mol. Biol. 2013, 1019, 299–320. [Google Scholar] [PubMed]
- Yang, J.; Benyamin, B.; McEvoy, B.P.; Gordon, S.; Henders, A.K.; Nyholt, D.R.; Madden, P.A.; Heath, A.C.; Martin, N.G.; Montgomery, G.W.; et al. Common SNPs explain a large proportion of the heritability for human height. Nat. Genet. 2010, 42, 565–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. GCTA: A tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2011, 88, 76–82. [Google Scholar] [CrossRef]
- You, F.M.; Xiao, J.; Li, P.; Yao, Z.; Jia, G.; He, L.; Kumar, S.; Soto-Cerda, B.; Duguid, S.D.; Booker, H.M.; et al. Genome-wide association study and selection signatures detect genomic regions associated with seed yield and oil quality in flax. Int. J. Mol. Sci. 2018, 19, 2303. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data Set | s | Range | CV (%) |
|---|---|---|---|
| PS-2012 | 5.57 ± 1.86 | 1.00–9.00 | 32.76 |
| PS-2013 | 5.69 ± 1.91 | 2.00–9.00 | 33.20 |
| PS-2014 | 6.86 ± 2.07 | 1.00–9.00 | 29.41 |
| PS-2015 | 6.11 ± 1.55 | 1.00–9.00 | 25.44 |
| PS-2016 | 6.72 ± 1.37 | 2.00–9.00 | 20.39 |
| PS-mean | 6.22 ± 1.32 | 1.80–9.00 | 21.27 |
| PS Dataset | Marker Set | |||
|---|---|---|---|---|
| SNP-500QTL | SNP-134QTL | SNP-67QTL | SNP-52347 | |
| PS-mean | 0.72 ± 0.04 | 0.27 ± 0.05 | 0.29 ± 0.05 | 0.54 ± 0.07 |
| PS-2012 | 0.64 ± 0.06 | 0.18 ± 0.05 | 0.16 ± 0.04 | 0.43 ± 0.08 |
| PS-2013 | 0.63 ± 0.06 | 0.12 ± 0.04 | 0.12 ± 0.04 | 0.38 ± 0.08 |
| PS-2014 | 0.65 ± 0.06 | 0.23 ± 0.05 | 0.20 ± 0.05 | 0.45 ± 0.08 |
| PS-2015 | 0.56 ± 0.06 | 0.20 ± 0.05 | 0.17 ± 0.04 | 0.44 ± 0.09 |
| PS-2016 | 0.53 ± 0.06 | 0.18 ± 0.05 | 0.18 ± 0.05 | 0.38 ± 0.07 |
| Marker Set | PS Dataset | s) 1 | s) 1 |
|---|---|---|---|
| SNP-500QTL | PS-mean | 0.92 ± 0.02a | 1.84 ± 0.04a |
| PS-2012 | 0.84 ± 0.03b | 1.68 ± 0.06b | |
| PS-2013 | 0.81 ± 0.04c | 1.62 ± 0.07c | |
| PS-2014 | 0.82 ± 0.04c | 1.63 ± 0.07c | |
| PS-2015 | 0.76 ± 0.05d | 1.52 ± 0.09d | |
| PS-2016 | 0.76 ± 0.05d | 1.52 ± 0.11d | |
| SNP-134QTL | PS-mean | 0.75 ± 0.06e | 1.49 ± 0.11e |
| PS-2012 | 0.68 ± 0.06f | 1.36 ± 0.11f | |
| PS-2013 | 0.60 ± 0.07ij | 1.19 ± 0.14ij | |
| PS-2014 | 0.60 ± 0.07i | 1.21 ± 0.14i | |
| PS-2015 | 0.47 ± 0.09o | 0.94 ± 0.18o | |
| PS-2016 | 0.56 ± 0.09l | 1.12 ± 0.17l | |
| SNP-67QTL | PS-mean | 0.76 ± 0.05d | 1.53 ± 0.1d |
| PS-2012 | 0.67 ± 0.06g | 1.35 ± 0.11g | |
| PS-2013 | 0.60 ± 0.07ij | 1.20 ± 0.14ij | |
| PS-2014 | 0.60 ± 0.07ij | 1.20 ± 0.14ij | |
| PS-2015 | 0.50 ± 0.09n | 1.00 ± 0.17n | |
| PS-2016 | 0.59 ± 0.08k | 1.17 ± 0.17k | |
| SNP-52347 | PS-mean | 0.67 ± 0.07g | 1.33 ± 0.14g |
| PS-2012 | 0.63 ± 0.06h | 1.27 ± 0.12h | |
| PS-2013 | 0.59 ± 0.07jk | 1.19 ± 0.14jk | |
| PS-2014 | 0.53 ± 0.08m | 1.06 ± 0.17m | |
| PS-2015 | 0.38 ± 0.09q | 0.77 ± 0.17q | |
| PS-2016 | 0.46 ± 0.09p | 0.93 ± 0.18p |
| PS Dataset for Prediction | r | RE |
|---|---|---|
| PS-mean | 0.98 | 1.96 |
| PS-2012 | 0.73 | 1.46 |
| PS-2013 | 0.71 | 1.42 |
| PS-2014 | 0.81 | 1.62 |
| PS-2015 | 0.71 | 1.43 |
| PS-2016 | 0.77 | 1.55 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, L.; Xiao, J.; Rashid, K.Y.; Jia, G.; Li, P.; Yao, Z.; Wang, X.; Cloutier, S.; You, F.M. Evaluation of Genomic Prediction for Pasmo Resistance in Flax. Int. J. Mol. Sci. 2019, 20, 359. https://doi.org/10.3390/ijms20020359
He L, Xiao J, Rashid KY, Jia G, Li P, Yao Z, Wang X, Cloutier S, You FM. Evaluation of Genomic Prediction for Pasmo Resistance in Flax. International Journal of Molecular Sciences. 2019; 20(2):359. https://doi.org/10.3390/ijms20020359
Chicago/Turabian StyleHe, Liqiang, Jin Xiao, Khalid Y. Rashid, Gaofeng Jia, Pingchuan Li, Zhen Yao, Xiue Wang, Sylvie Cloutier, and Frank M. You. 2019. "Evaluation of Genomic Prediction for Pasmo Resistance in Flax" International Journal of Molecular Sciences 20, no. 2: 359. https://doi.org/10.3390/ijms20020359
APA StyleHe, L., Xiao, J., Rashid, K. Y., Jia, G., Li, P., Yao, Z., Wang, X., Cloutier, S., & You, F. M. (2019). Evaluation of Genomic Prediction for Pasmo Resistance in Flax. International Journal of Molecular Sciences, 20(2), 359. https://doi.org/10.3390/ijms20020359