Tocotrienols Modulate a Life or Death Decision in Cancers


Abstract
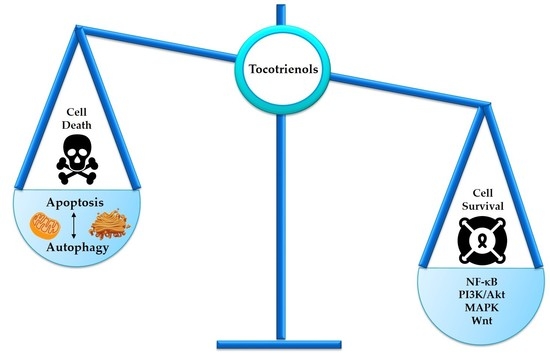
:
1. Introduction
2. Programmed Cell Death in Cancer
3. Tocotrienols Act as a Potent Apoptosis Inducer
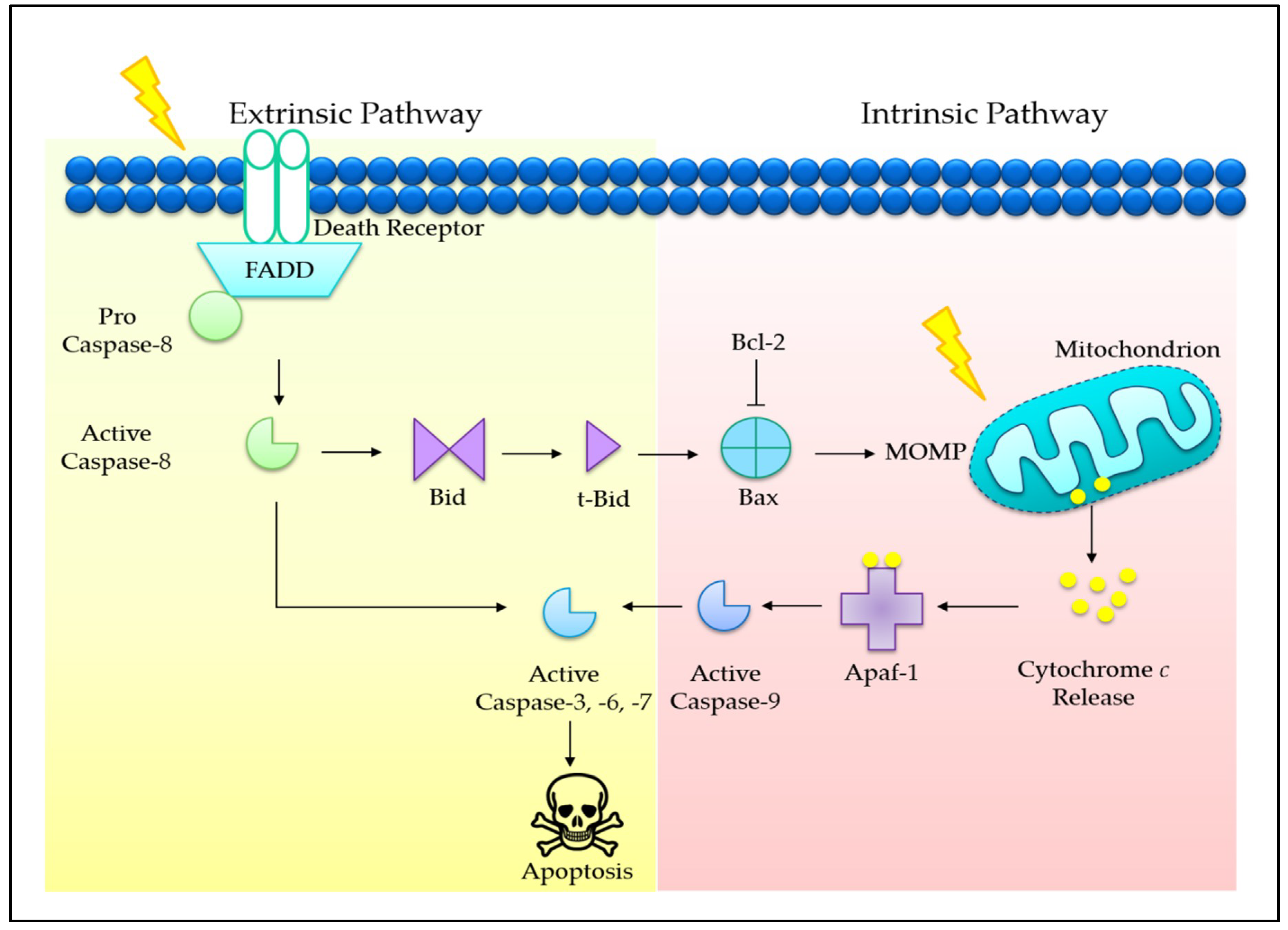
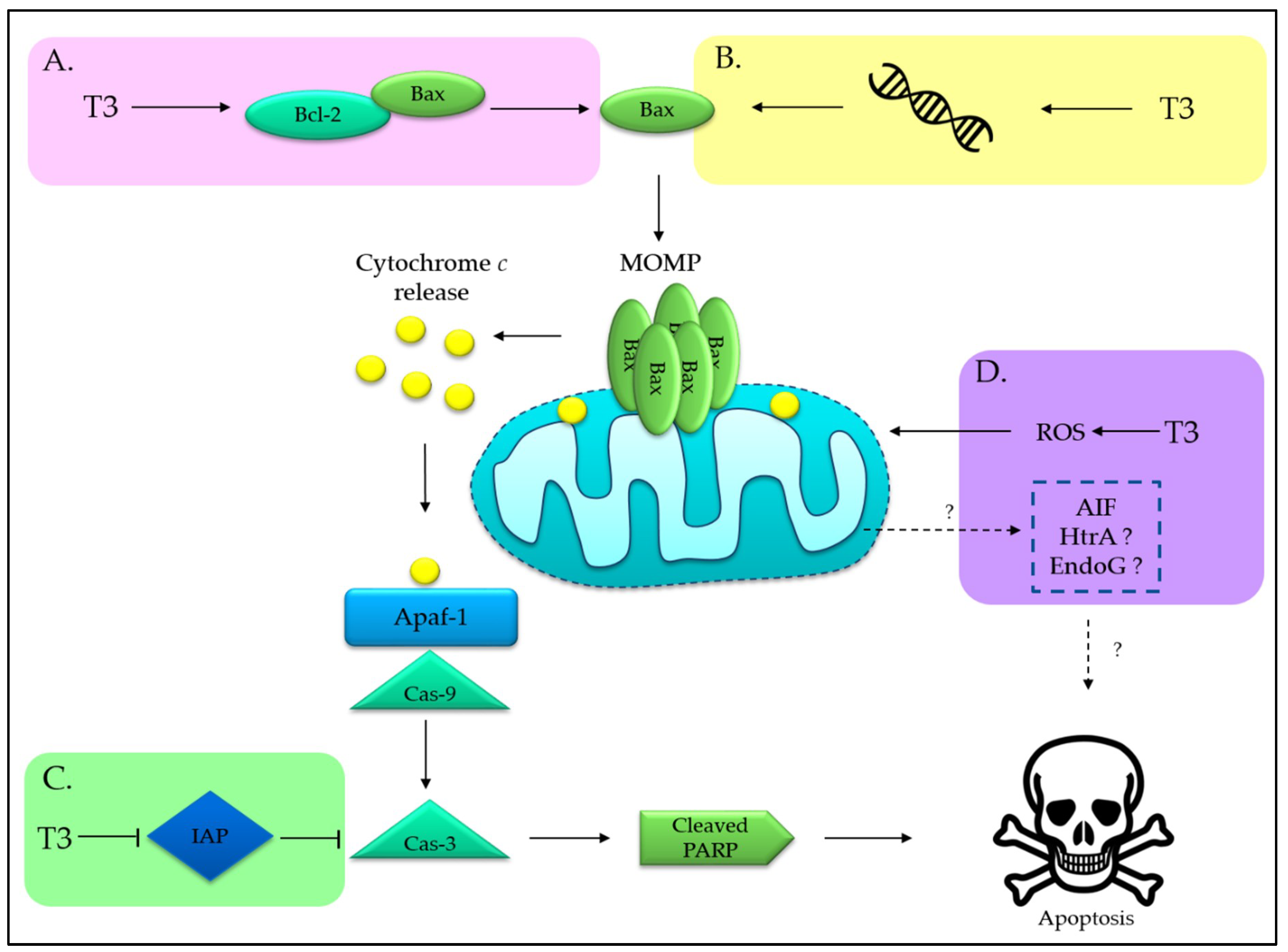
3.1. Tocotrienols Induce Mitochondria-Mediated Apoptosis
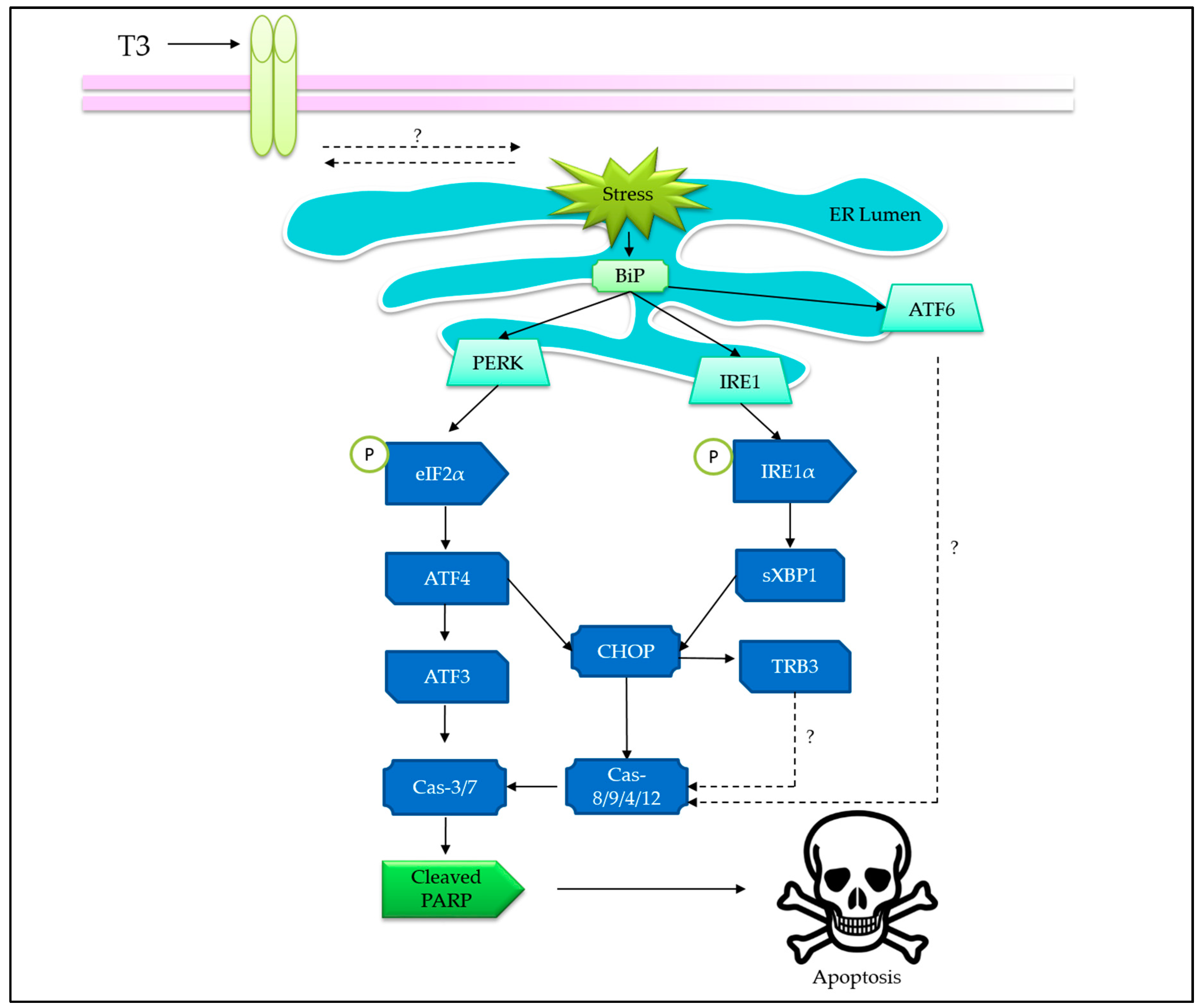
3.2. Tocotrienols Induce Endoplasmic Reticulum Stress
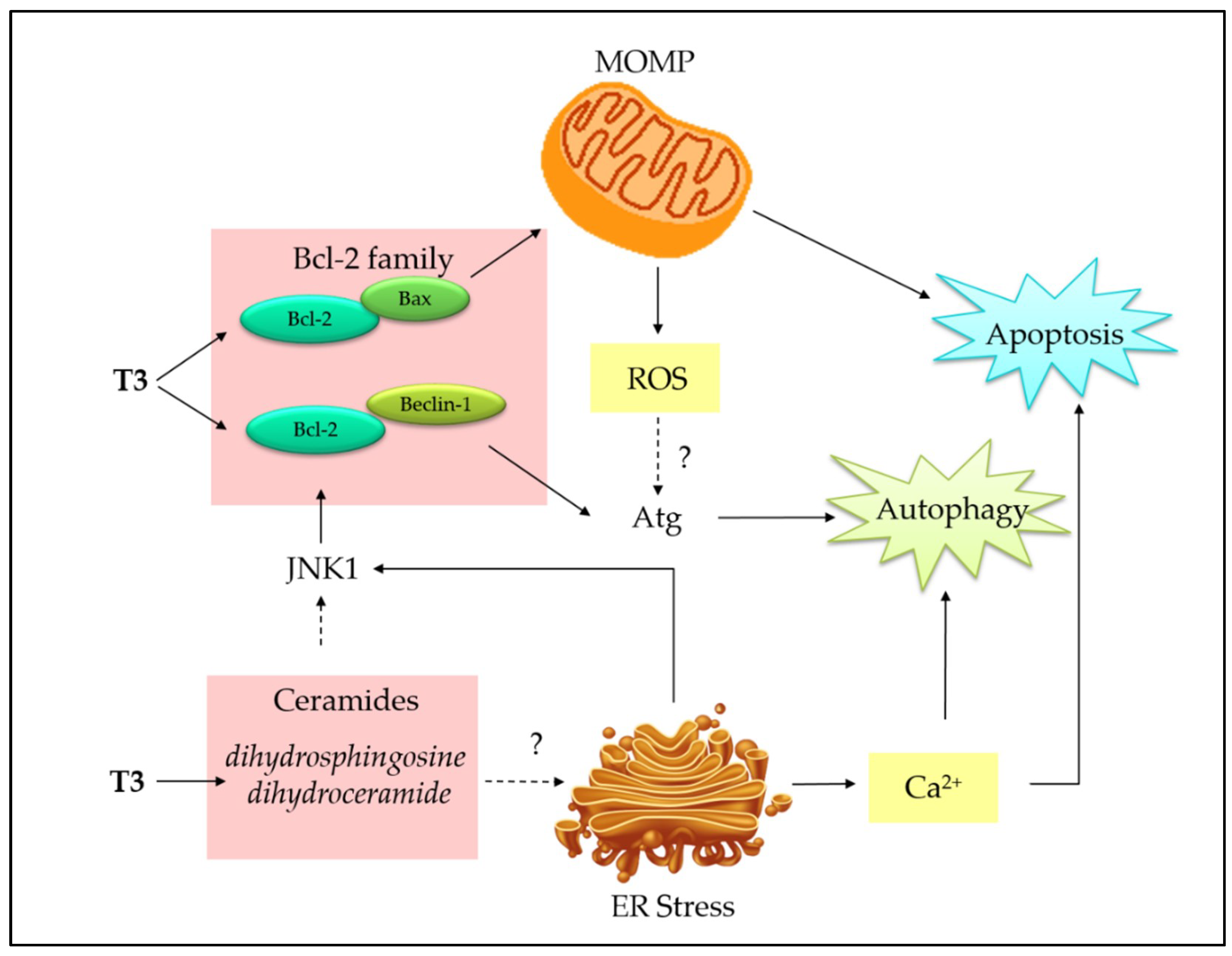
3.3. Co-Involvement of Endoplasmic Reticulum Stress and Mitochondria-Mediated Apoptosis
3.4. Co-Involvement of Extrinsic and Intrinsic Pathways
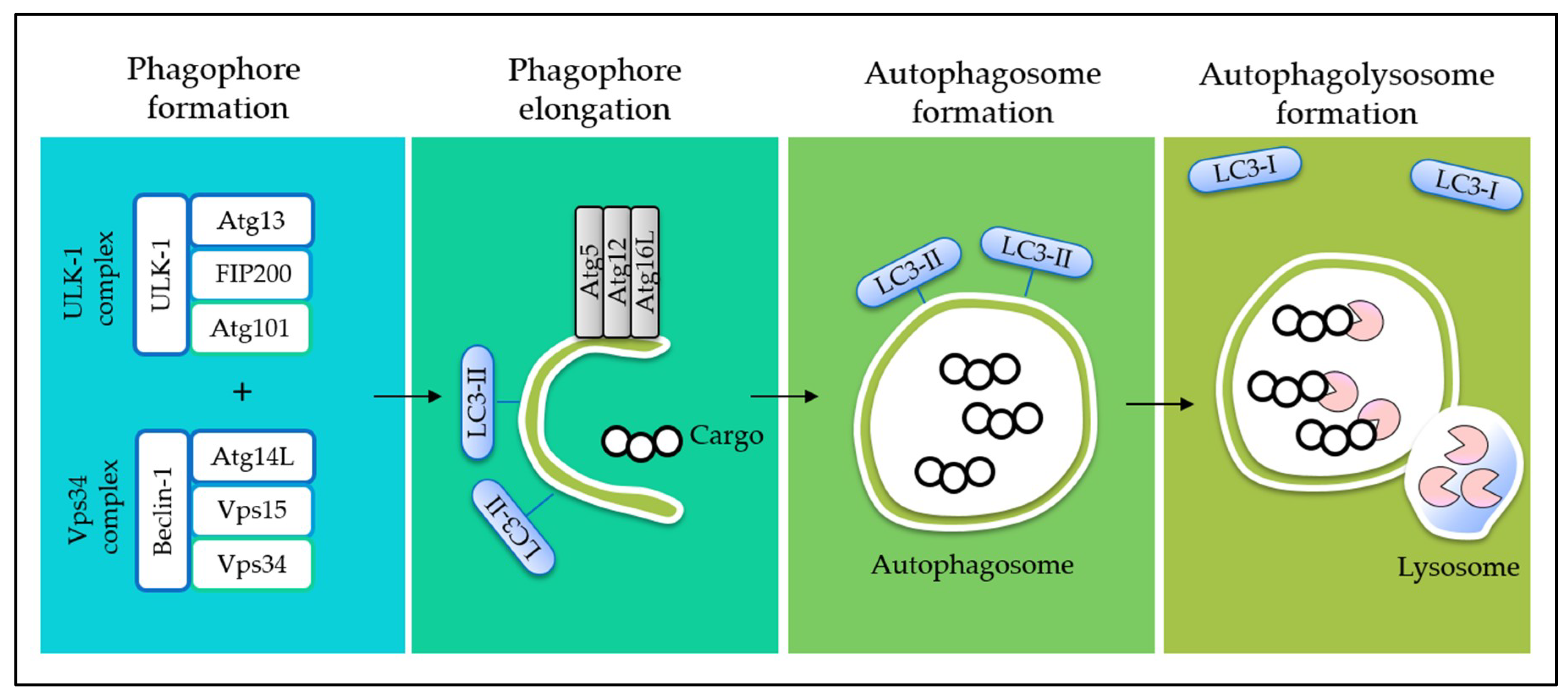
4. Interplay between Autophagy and Apoptosis
5. Tocotrienols Target Prosurvival Signaling Pathways
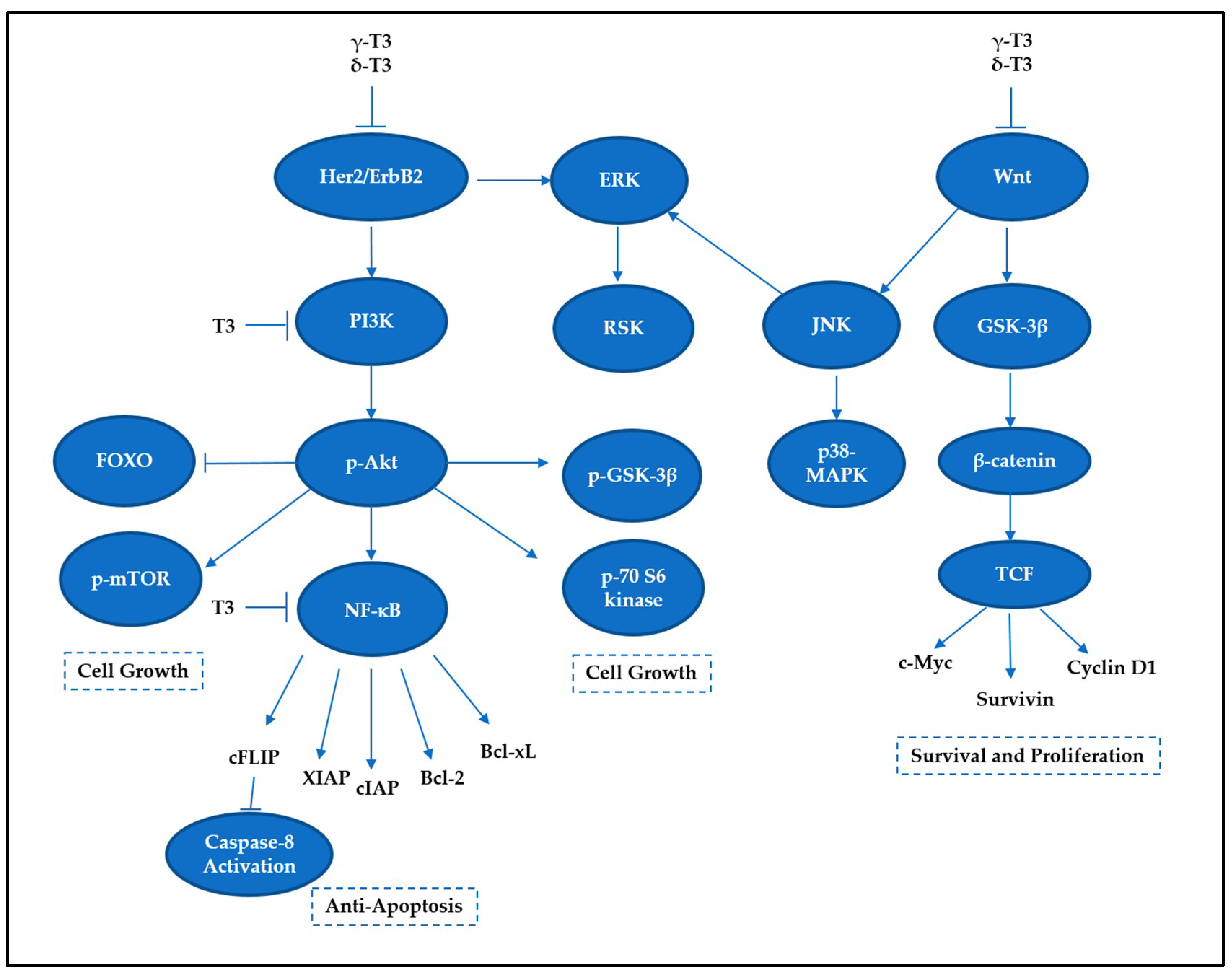
5.1. NF-κB Family
5.2. PI3K/Akt Signaling
5.3. MAP Kinase Signaling
5.4. Wnt Signaling
6. Current and Future Perspectives of Tocotrienols
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 3-MA | 3-Methyladenine |
| ABCG2 | ATP-binding cassette subfamily G member 2 |
| AIF | Apoptosis-inducing factor |
| Akt | PKB or protein kinase B |
| ANS | 8-Anilino-1-naphthalenesulfonic acid ammonium salt |
| Apaf-1 | Apoptotic protease activating factor 1 |
| APC | Adenomatous polyposis coli |
| ATF4 | Activating transcription factor 4 |
| ATF6 | Activating transcription factor 6 |
| Atg | Autophagy-related protein |
| Bf1-1/A1 | Bcl-2-related protein A1 |
| BH3 | Bcl-2 homology 3 |
| BiP | GRP78, glucose-regulated protein |
| CDK | Cyclin-dependent kinase |
| CHOP | CCAAT-enhancer-binding protein homologous protein |
| cIAP | Cellular inhibitor of apoptosis |
| COX-2 | Cyclooxygenase-2 |
| CXCR4 | C-X-C motif chemokine receptor 4 |
| CYP450 | Cytochrome P450 enzyme |
| DR | Death receptor |
| EGR1 | Early growth response protein 1 |
| eIF2-α | Eukaryotic initiation factor 2 alpha |
| ELAM-2 | Endothelial cell adhesion molecule-2 |
| EndoG | Endonuclease G |
| ER | Endoplasmic reticulum |
| ERK1/2 | Extracellular signal-regulated protein kinases 1 and 2 |
| ERO1α | Endoplasmic reticulum oxidation 1 |
| ETK1 | Epithelial and endothelial tyrosine kinase |
| FDFT1 | Farnesyl-diphosphate farnesyltransferase 1 |
| GSK-β | Glycogen synthase kinase 3 beta |
| Hes-1 | Hairy and enhancer of split-1 |
| hTERT | Human telomerase reverse transcriptase |
| HtrA | High temperature requirement A |
| IAP | Inhibitor of apoptosis protein |
| ICAM-1 | Intercellular adhesion molecules-1 |
| Id-1 | Inhibitor of differentiation/DNA binding |
| IκB | Inhibitor of kappa B |
| IKKα/β | IκB kinase alpha/beta |
| IL-6 | Interleukin 6 |
| IRE-1 | Inositol-requiring enzyme 1 |
| JNK | c-Jun N-terminal kinase |
| LAMP-1 | Lysosomal-associated membrane protein 1 |
| LC3 | Microtubule-associated protein 1A/1B-light chain 3 |
| LRP | Low-density lipoprotein receptor-related protein |
| MAPK | Mitogen-activated protein kinase |
| MC1R | Melanocortin 1 receptor |
| Mcl-1 | Myeloid cell leukemia 1 |
| MDR1 | Multidrug resistance protein-1 |
| MEK | Mitogen-activated protein kinase |
| MIC-1 | Macrophage inhibitory cytokine 1 |
| MITF | Melanogenesis associated transcription factor |
| MMP | Matrix metalloproteinases |
| MOMP MPTP | Mitochondrial outer membrane permeabilization Mitochondrial permeability transition pore |
| mTOR | Mammalian target of rapamycin |
| NF-κB | Nuclear factor kappa B |
| Oct-4 | Octamer-binding transcription factor 4 |
| p27Kip1 | Cyclin-dependent kinase inhibitor 1B |
| PARP | Poly(ADP-ribose) polymerase |
| PCD | Programmed cell death |
| PCNA | Proliferating cell nuclear antigen |
| PDK1 | 3-phosphoinositide-dependent protein kinase-1 |
| PERK | Protein kinase R (PKR)-like endoplasmic reticulum kinase |
| PI3K | Phosphoinositide 3-kinase |
| PXR | Pregame-X-receptor |
| ROS | Reactive oxygen species |
| RSK | Ribosomal protein S6 kinase |
| SAPK/JNK | Stress-activated protein kinase/c-Jun NH2-terminal kinase |
| SHP-1 | Src homology region 2 domain-containing phosphatase-1 |
| Sox-2 | Sex determining region Y-box 2 |
| SXR | Steroid and xenobiotic receptor |
| TGF-β1 | Transforming growth factor beta 1 |
| TRA1 | Transcription-associated protein 1 |
| TRB3 | Tribbles-related Protein 3 |
| TYRP | Tyrosinase-related proteins |
| UGT1A1 | Glucuronosyltransferase 1A1 |
| ULK | Unc-51-like kinase |
| uPA | Urokinase-type plasminogen activator |
| UPR | Unfolded protein response |
| US | United States |
| VCAM-1 | Vascular cell adhesion molecule-1 |
| VEGF | Vascular endothelial growth factor |
| Vps | Vacuolar protein sorting |
| Wnt | Wingless/integrase |
| XBP | X-box binding protein |
| XIAP | X-linked inhibitor of apoptosis protein |
References
- Bray, F.; Ferlay, J.; Soerjamataram, I.; Siegel, R.L.; Torrer, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- National Cancer Institute. Cancer Statistics-National Cancer Institute. Available online: https://www.cancer.gov/about-cancer/understanding/statistics (accessed on 18 December 2018).
- Mai, C.W.; Chung, F.F.L.; Leong, C.O. Targeting legumain as a novel therapeutic strategy in cancers. Curr. Drug Targets 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- Mai, C.W.; Kang, Y.B.; Pichika, M.R. Should a Toll-like receptor 4 (TLR-4) agonist or antagonist be designed to treat cancer? TLR-4: Its expression and effects in the ten most common cancers. Onco-Targets Ther. 2013, 6, 1573–1587. [Google Scholar] [PubMed]
- Chung, F.F.L.; Mai, C.W.; Ng, P.Y.; Leong, C.O. Cytochrome P450 2W1 (CYP2W1) in colorectal cancers. Curr. Cancer Drug Targets 2016, 16, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Mai, C.W.; Kang, Y.B.; Nadarajah, V.D.; Hamzah, A.S.; Pichika, M.R. Drug-like dietary vanilloids induce anticancer activity through proliferation inhibition and regulation of bcl-related apoptotic proteins. Phyther. Res. 2018, 32, 1108–1118. [Google Scholar] [CrossRef] [PubMed]
- Soo, H.C.; Chung, F.F.L.; Lim, K.H.; Yap, V.A.; Bradshaw, T.D.; Hii, L.W.; Tan, S.H.; See, S.J.; Tan, Y.F.; Leong, C.O.; et al. Cudraflavone C induces tumor-specific apoptosis in colorectal cancer cells through inhibition of the phosphoinositide 3-Kinase (PI3K)-AKT Pathway. PLoS ONE 2017, 12, e0170551. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.K.; Hemaroopini, S.; Gan, S.Y.; Loo, S.M.; Low, J.R.; Jong, V.Y.M.; Soo, H.C.; Leong, C.O.; Mai, C.W.; Chee, C.F. In vitro cytotoxic activity of isolated compounds from Malaysian Calophyllum species. Med. Chem. Res. 2016, 25, 1686–1694. [Google Scholar] [CrossRef]
- Abubakar, I.; Loh, H.S. A review on ethnobotany, pharmacology and phytochemistry of Tabernaemontana corymbosa. J. Pharm. Pharmacol. 2016, 68, 423–432. [Google Scholar] [CrossRef] [Green Version]
- Rasol, N.E.; Ahmad, F.B.; Lim, X.Y.; Chung, F.F.L.; Leong, C.O.; Mai, C.W.; Bihud, N.V.; Zaki, H.M.; Ismail, N.H. Cytotoxic lactam and naphthoquinone alkaloids from roots of Goniothalamus lanceolatus Miq. Phytochem. Lett. 2018, 24, 51–55. [Google Scholar] [CrossRef]
- Yap, V.A.; Qazzaz, M.E.; Raja, V.J.; Bradshaw, T.D.; Loh, H.S.; Sim, K.S.; Yong, K.T.; Low, Y.Y.; Lim, K.H. Fistulopsines A and B antiproliferative septicine-type alkaloids from Ficus fistulosa. Phytochem. Lett. 2016, 15, 136–141. [Google Scholar] [CrossRef]
- Chung, F.F.; Tan, P.F.; Raja, V.J.; Tan, B.S.; Lim, K.H.; Kam, T.S.; Hii, L.W.; Tan, S.H.; See, S.J.; Tan, Y.F.; et al. Jerantinine A induces tumor-specific cell death through modulation of splicing factor 3b subunit 1 (SF3B1). Sci. Rep. 2017, 7. [Google Scholar] [CrossRef]
- De Silva, L.; Chuah, L.H.; Meganathan, P.; Fu, J.Y. Tocotrienol and cancer metastasis. Biofactors 2016, 42, 149–162. [Google Scholar] [CrossRef] [PubMed]
- Miyazawa, T.; Shibata, A.; Nakagawa, K.; Tsuzuki, T. Anti-angiogenic function of tocotrienol. Asia Pac. J. Clin. Nutr. 2008, 17, 253–256. [Google Scholar] [PubMed]
- Sailo, B.L.; Banik, K.; Padmavathi, G.; Javadi, M.; Bordoloi, D.; Kunnumakkara, A.B. Tocotrienols: The promising analogues of vitamin E for cancer therapeutics. Pharmacol. Res. 2018, 130, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Kabir, M.I.; Adnan, M.; Rahman, M.M. Natural sources of tocotrienols: A note on absorption. J. Silico Vitr. Pharmacol. 2017, 3, 1–5. [Google Scholar] [CrossRef]
- Chang, P.N.; Yap, W.N.; Lee, D.T.; Ling, M.T.; Wong, Y.C.; Yap, Y.L. Evidence of gamma-tocotrienol as an apoptosis-inducing, invasion-suppressing, and chemotherapy drug-sensitizing agent in human melanoma cells. Nutr. Cancer 2009, 61, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Hussein, D.; Mo, H. δ-Tocotrienol-mediated suppression of the proliferation of human PANC-1, MIA PaCa-2, and BxPC-3 pancreatic carcinoma cells. Pancreas 2009, 38, e124–e136. [Google Scholar] [CrossRef] [PubMed]
- Yap, W.N.; Chang, P.N.; Han, H.Y.; Lee, D.T.; Ling, M.T.; Wong, Y.C.; Yap, Y.L. Gamma-tocotrienol suppresses prostate cancer cell proliferation and invasion through multiple-signalling pathways. Br. J. Cancer 2008, 99, 1832–1841. [Google Scholar] [CrossRef]
- Abubakar, I.B.; Lim, K.H.; Kam, T.S.; Loh, H.S. Enhancement of apoptotic activities on brain cancer cells via the combination of γ-tocotrienol and jerantinine A. Phytomedicine 2017, 30, 74–84. [Google Scholar] [CrossRef]
- Agarwal, M.K.; Agarwal, M.L.; Athar, M.; Gupta, S. Tocotrienol-rich fraction of palm oil activates p53, modulates Bax/Bcl2 ratio and induces apoptosis independent of cell cycle association. Cell Cycle 2004, 13, 205–211. [Google Scholar] [CrossRef]
- Srivastava, J.K.; Gupta, S. Tocotrienol-rich fraction of palm oil induces cell cycle arrest and apoptosis selectively in human prostate cancer cells. Biochem. Biophys. Res. Commun. 2006, 346, 447–453. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.S.; Zhang, S.J.; Li, Q.; Liu, Y.H.; He, N.; Zhang, J.; Zhou, P.H.; Li, M.; Guan, T.; Liu, J.R. Tocotrienol-rich fraction (TRF) suppresses the growth of human colon cancer xenografts in Balb/C nude mice by the Wnt pathway. PLoS ONE 2015, 10, e0122175. [Google Scholar] [CrossRef] [PubMed]
- Zugazagoitia, J.; Guedes, C.; Ponce, S.; Ferrer, I.; Molina-Pinelo, S.; Paz-Ares, L. Current challenges in cancer treatment. Clin. Ther. 2016, 38, 1551–1566. [Google Scholar] [CrossRef]
- De Palma, M.; Hanahan, D. The biology of personalized cancer medicine: Facing individual complexities underlying hallmark capabilities. Mol. Oncol. 2012, 6, 111–127. [Google Scholar] [CrossRef]
- Labi, V.; Erlacher, M. How cell death shapes cancer. Cell Death Dis. 2015, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meganathan, P.; Fu, J.Y. Biological properties of tocotrienols: Evidence in human dtudies. Int. J. Mol. Sci. 2016, 17, 1682. [Google Scholar] [CrossRef] [PubMed]
- Chin, K.Y.; Pang, K.L.; Soelaiman, I.N. Tocotrienol and its role in chronic diseases. In Anti-Inflammatory Nutraceuticals and Chronic Diseases; Gupta, S., Prasad, S., Aggarwal, B., Eds.; Springer International Publishing: Cham, Switzerland, 2016; Volume 928, pp. 97–130. [Google Scholar]
- Ahsan, H.; Ahad, A.; Iqbal, J.; Siddiqui, W.A. Pharmacological potential of tocotrienols: A review. Nutr. Metab. 2014, 11. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, Y.; Steller, H. Programmed cell death in animal development and disease. Cell 2011, 147, 742–758. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S. Regulation of cell death in cancer-possible implications for immunotherapy. Front. Oncol. 2013, 3. [Google Scholar] [CrossRef]
- Kantari, C.; Walczak, H. Caspase-8 and Bid: Caught in the act between death receptors and mitochondria. Biochim. Biophys. Acta Mol. Cell Res. 2011, 1813, 558–563. [Google Scholar] [CrossRef] [Green Version]
- Wong, R.S.Y. Apoptosis in cancer: From pathogenesis to treatment. J. Exp. Clin. Cancer Res. 2011, 30. [Google Scholar] [CrossRef]
- Kalkavan, H.; Green, D.R. MOMP, cell suicide as a BCL-2 family business. Cell Death Differ. 2018, 25, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Harris, M.H.; Thompson, C.B. The role of the Bcl-2 family in the regulation of outer mitochondrial membrane permeability. Cell Death Differ. 2000, 7, 1182–1191. [Google Scholar] [CrossRef] [Green Version]
- Brentnall, M.; Rodriguez-Menocal, L.; De Guevara, R.L.; Cepero, E.; Boise, L.H. Caspase-9, caspase-3 and caspase-7 have distinct roles during intrinsic apoptosis. BMC Cell Biol. 2013, 14, 32. [Google Scholar] [CrossRef] [PubMed]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Nesaretnam, K.S.; Kanga, R.; Abdul-Razak, G.; Veerasenan, S.D.; Gomez, P.A. Effectiveness of tocotrienol-rich fraction combined with tamoxifen in the management of women with early breast cancer: A pilot clinical trial. Breast Cancer Res. 2010, 12. [Google Scholar] [CrossRef]
- Parajuli, P.; Tiwari, R.V.; Sylvester, P.W. Anti-proliferative effects of gamma-tocotrienol are associated with suppression of c-Myc expression in mammary tumour cells. Cell Prolif. 2015, 48, 421–435. [Google Scholar] [CrossRef]
- Lim, S.W.; Loh, H.S.; Ting, K.N.; Bradshaw, T.D.; Zeenathul, N.A. Cytotoxicity and apoptotic activities of alpha-, gamma- and delta-tocotrienol isomers on human cancer cells. BMC Complement. Altern. Med. 2014, 14, 469. [Google Scholar] [CrossRef]
- Abubakar, I.B.; Lim, K.H.; Kam, T.S.; Loh, H.S. Synergistic cytotoxic effects of combined δ-tocotrienol and jerantinine B on human brain and colon cancers. J. Ethnopharmacol. 2016, 184, 107–118. [Google Scholar] [CrossRef]
- Xu, W.; Du, M.; Zhao, Y.; Wang, Q.; Sun, W.; Chen, B. γ-Tocotrienol inhibits cell viability through suppression of β-catenin/Tcf signaling in human colon carcinoma HT-29 cells. J. Nutr. Biochem. 2012, 23, 800–807. [Google Scholar] [CrossRef]
- Abubakar, I.B.; Lim, K.H.; Kam, T.S.; Loh, H.S. Jerantinine B enhances the mitochondria-mediated apoptosis by p53 Activation in human glioblastoma cells via a combination with δ-tocotrienol. J. Biol. Act. Prod. Nat. 2018, 8, 21–27. [Google Scholar] [CrossRef]
- Burdeos, G.C.; Ito, J.; Eitsuka, T.; Nakagawa, K.; Kimura, F.; Miyazawa, T. δ and γ tocotrienols suppress human hepatocellular carcinoma cell proliferation via regulation of Ras-Raf-MEK-ERK pathway-associated upstream signaling. Food Funct. 2016, 7, 4170–4174. [Google Scholar] [CrossRef]
- Sakai, M.; Okabe, M.; Tachibana, H.; Yamada, K. Apoptosis induction by gamma-tocotrienol in human hepatoma Hep3B cells. J. Nutr. Biochem. 2006, 17, 672–676. [Google Scholar] [CrossRef]
- Xu, W.; Mi, Y.; He, P.; He, S.; Niu, L. γ-Tocotrienol inhibits proliferation and induces apoptosis via the mitochondrial pathway in human cancer HeLa cells. Molecules 2017, 22, 1299. [Google Scholar]
- Yamasaki, M.; Nishimura, M.; Sakakibara, Y.; Suiko, M.; Morishita, K.; Nishiyama, K. Delta-tocotrienol induces apoptotic cell death via depletion of intracellular squalene in ED40515 cells. Food Funct. 2014, 5, 2842–2849. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, N.V.; Guntipalli, P.K.; Mo, H. δ-Tocotrienol-mediated cell cycle arrest and apoptosis in human melanoma cells. Anticancer Res. 2010, 30, 4937–4944. [Google Scholar]
- Fulda, S.; Galluzzi, L.; Kroemer, G. Targeting mitochondria for cancer therapy. Nat. Rev. Drug Discov. 2010, 9, 447–464. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Loo, G. Disruption of mitochondria during tocotrienol-induced apoptosis in MDA-MB-231 human breast cancer cells. Biochem. Pharmacol. 2004, 67, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Inoue, A.; Takitani, K.; Koh, M.; Kawakami, C.; Kuno, T.; Tamai, H. Induction of apoptosis by γ-tocotrienol in human cancer cell lines and leukemic blasts from patients: Dependency on Bid, cytochrome c, and caspase pathway. Nutr. Cancer 2011, 63, 763–770. [Google Scholar] [CrossRef]
- Abubakar, I.B.; Lim, K.H.; Loh, H.S. Alkaloid extracts of Ficus species and palm oil-derived tocotrienols synergistically inhibit proliferation of human cancer cells. Nat. Prod. Res. 2014, 29, 1–4. [Google Scholar]
- Rickmann, M.; Vaquero, E.C.; Malagelada, J.R.; Molero, X. Tocotrienols induce apoptosis and autophagy in rat pancreatic stellate cells through the mitochondrial death pathway. Gastroenterology 2007, 132, 2518–2532. [Google Scholar] [CrossRef] [PubMed]
- Komar, H.M.; Serpa, G.; Kerscher, C.; Schwoegl, E.; Mace, T.A.; Jin, M.; Yang, M.C.; Chen, C.S.; Bloomston, M.; Ostrowski, M.C.; et al. Inhibition of Jak/STAT signaling reduces the activation of pancreatic stellate cells in vitro and limits caerulein-induced chronic pancreatitis in vivo. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, P.; Li, F.; Manu, K.A.; Shanmugam, M.K.; Loo, S.Y.; Kumar, A.P.; Sethi, G. γ-Tocotrienol is a novel inhibitor of constitutive and inducible STAT3 signalling pathway in human hepatocellular carcinoma: Potential role as an antiproliferative, pro-apoptotic and chemosensitizing agent. Br. J. Pharmacol. 2011, 163, 283–298. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.L.; Liu, J.R.; Liu, H.K.; Qi, G.Y.; Sun, X.R.; Sun, W.G.; Chen, B.Q. Inhibition of proliferation and induction of apoptosis by gamma-tocotrienol in human colon carcinoma HT-29 cells. Nutrition 2009, 25, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.K.; Then, S.M.; Mazlan, M.; Raja Abdul Rahman, R.N.; Jamal, R.; Wan Ngah, W.Z. Gamma-tocotrienol acts as a BH3 mimetic to induce apoptosis in neuroblastoma SH-SY5Y cells. J. Nutr. Biochem. 2016, 31, 28–37. [Google Scholar] [CrossRef]
- Wang, C.; Husain, K.; Zhang, A.; Centeno, B.A.; Chen, D.T.; Tong, Z.; Sebti, S.M.; Malafa, M.P. EGR-1/Bax pathway plays a role in vitamin E δ-tocotrienol-induced apoptosis in pancreatic cancer cells. J. Nutr. Biochem. 2015, 26, 797–807. [Google Scholar] [CrossRef] [Green Version]
- Prasad, S.; Gupta, S.C.; Tyagi, A.K.; Aggarwal, B.B. γ-Tocotrienol suppresses growth and sensitises human colorectal tumours to capecitabine in a nude mouse xenograft model by down-regulating multiple molecules. Br. J. Cancer 2016, 115, 814–824. [Google Scholar] [CrossRef] [Green Version]
- Kannappan, R.; Ravindran, J.; Prasad, S.; Sung, B.; Yadav, V.R.; Reuter, S.; Chaturvedi, M.M.; Aggarwal, B.B. Gamma-tocotrienol promotes TRAIL-induced apoptosis through reactive oxygen species/extracellular signal-regulated kinase/p53-mediated upregulation of death receptors. Mol. Cancer Ther. 2010, 9, 2196–2207. [Google Scholar] [CrossRef] [PubMed]
- Wilankar, C.; Khan, N.M.; Checker, R.; Sharma, D.; Patwardhan, R.; Gota, V.; Sandur, S.K.; Devasagayam, T.P. γ-Tocotrienol induces apoptosis in human T cell lymphoma through activation of both intrinsic and extrinsic pathways. Curr. Pharm. Des. 2011, 17, 2176–2189. [Google Scholar] [CrossRef]
- Senft, D.; Ronai, Z.A. UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem. Sci. 2015, 40, 141–148. [Google Scholar] [CrossRef] [Green Version]
- Avril, T.; Vauléon, E.; Chevet, E. Endoplasmic reticulum stress signaling and chemotherapy resistance in solid cancers. Oncogenesis 2017, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sano, R.; Reed, J.C. ER stress-induced cell death mechanisms. Biochim. Biophys. Acta Mol. Cell Res. 2013, 1833, 3460–3470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winter, E.; Chiaradia, D.C.; Silva, A.H.; Nunes, R.J.; Yunes, R.A.; Creczynski-Pasa, T.B. Involvement of extrinsic and intrinsic apoptotic pathways together with endoplasmic reticulum stress in cell death induced by naphthylchalcones in a leukemic cell line: Advantages of multi-target action. Toxicol. Vitro 2014, 28, 769–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krajarng, A.; Imoto, M.; Tashiro, E.; Fujimaki, T.; Shinjo, S.; Watanapokasin, R. Apoptosis induction associated with the ER stress response through up-regulation of JNK in HeLa cells by gambogic acid. BMC Complement. Altern. Med. 2015, 15, 26. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, X.; Li, W.; Miao, H.; Huang, S.; Zhou, Y.; Sun, Y.; Li, Z.; Guo, Q.; Zhao, L. Activation of endoplasmic reticulum stress and the extrinsic apoptotic pathway in human lung cancer cells by the new synthetic flavonoid, LZ-205. Oncotarget 2016, 7, 87257–87270. [Google Scholar] [CrossRef] [Green Version]
- Wali, V.B.; Bachawal, S.V.; Sylvester, P.W. Endoplasmic reticulum stress mediates gamma-tocotrienol-induced apoptosis in mammary tumor cells. Apoptosis 2009, 14, 1366–1377. [Google Scholar] [CrossRef] [PubMed]
- Patacsil, D.; Tran, A.T.; Cho, Y.S.; Suy, S.; Saenz, F.; Malyukova, I.; Ressom, H.; Collins, S.P.; Clarke, R.; Kumar, D. Gamma-tocotrienol induced apoptosis is associated with unfolded protein response in human breast cancer cells. J. Nutr. Biochem. 2012, 23, 93–100. [Google Scholar] [CrossRef] [Green Version]
- Comitato, R.; Guantario, R.; Leoni, G.; Nesaretnam, K.; Ronci, M.B.; Canali, R.; Virgili, F. Tocotrienols induce endoplasmic reticulum stress and apoptosis in cervical cancer cells. Genes Nutr. 2016, 11. [Google Scholar] [CrossRef]
- Montagnani, M.M.; Marzagalli, M.; Moretti, R.M.; Beretta, G.; Casati, L.; Comitato, R.; Gravina, G.L.; Festuccia, C.; Limonta, P. Vitamin E δ-tocotrienol triggers endoplasmic reticulum stress-mediated apoptosis in human melanoma cells. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef]
- Dadey, D.Y.; Kapoor, V.; Khudanyan, A.; Urano, F.; Kim, A.H.; Thotala, D.; Hallahan, D.E. The ATF6 pathway of the ER stress response contributes to enhanced viability in glioblastoma. Oncotarget 2016, 7, 2080–2092. [Google Scholar] [CrossRef]
- Park, S.K.; Sanders, B.G.; Kline, K. Tocotrienols induce apoptosis in breast cancer cell lines via an endoplasmic reticulum stress-dependent increase in extrinsic death receptor signaling. Breast Cancer Res. Treat. 2010, 124, 361–375. [Google Scholar] [CrossRef] [PubMed]
- Marchi, S.; Patergnani, S.; Pinton, P. The endoplasmic reticulum–mitochondria connection: One touch, multiple functions. Biochim. Biophys. Acta Bioenerg. 2014, 1837, 461–469. [Google Scholar] [CrossRef] [Green Version]
- Iurlaro, R.; Muñoz-Pinedo, C. Cell death induced by endoplasmic reticulum stress. FEBS J. 2016, 283, 2640–2652. [Google Scholar] [CrossRef] [PubMed]
- Bustos, G.; Cruz, P.; Lovy, A.; Cárdenas, C. Endoplasmic reticulum–mitochondria calcium communication and the regulation of mitochondrial metabolism in cancer: A novel potential target. Front. Oncol. 2017, 7. [Google Scholar] [CrossRef]
- Rodriguez, D.; Rojas-Rivera, D.; Hetz, C. Integrating stress signals at the endoplasmic reticulum: The BCL-2 protein family rheostat. Biochim. Biophys. Acta Mol. Cell Res. 2011, 1813, 564–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galehdar, Z.; Swan, P.; Fuerth, B.; Callaghan, S.M.; Park, D.S.; Cregan, S.P. Neuronal apoptosis induced by endoplasmic reticulum stress is regulated by ATF4-CHOP-mediated induction of the Bcl-2 homology 3-only member PUMA. J. Neurosci. 2010, 30, 16938–16948. [Google Scholar] [CrossRef] [PubMed]
- Pike, L.R.G.; Phadwal, K.; Simon, A.K.; Harris, A.L. ATF4 orchestrates a program of BH3-only protein expression in severe hypoxia. Mol. Biol. Rep. 2012, 39, 10811–10822. [Google Scholar] [CrossRef]
- Ng, K.L.; Ammu, K.R.; Selvaduray, K.R. Apoptosis gene network regulated by delta-tocotrienol in K562 chronic myeloid leukemia cells. J. Oil Palm Res. 2016, 29, 251–261. [Google Scholar]
- Fulda, S.; Debatin, K.M. Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene 2006, 25, 4798–4811. [Google Scholar] [CrossRef] [Green Version]
- Rudner, J.; Jendrossek, V.; Lauber, K.; Daniel, P.T.; Wesselborg, S.; Belka, C. Type I and type II reactions in TRAIL-induced apoptosis—Results from dose–response studies. Oncogene 2005, 24, 130–140. [Google Scholar] [CrossRef]
- Tran, A.T.; Ramalinga, M.; Kedir, H.; Clarke, R.; Kumar, D. Autophagy inhibitor 3-methyladenine potentiates apoptosis induced by dietary tocotrienols in breast cancer cells. Eur. J. Nutr. 2015, 54, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Gump, J.M.; Thorburn, A. Autophagy and apoptosis: What is the connection? Trends Cell Biol. 2011, 21, 387–392. [Google Scholar] [CrossRef]
- Vaquero, E.C.; Rickmann, M.; Molero, X. Tocotrienols balancing the mitochondrial crosstalk between apoptosis and autophagy tocotrienols induce apoptosis and autophagy in rat pancreatic stellate cells through the mitochondrial death pathway. Autophagy 2007, 3, 652–654. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, R.V.; Parajuli, P.; Sylvester, P.W. Synergistic anticancer effects of combined γ-tocotrienol and oridonin treatment is associated with the induction of autophagy. Mol. Cell. Biochem. 2015, 408, 123–137. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Rao, X.; Kim, C.Y.; Freiser, H.; Zhang, Q.; Jiang, Z.; Li, G. Gamma-tocotrienol induces apoptosis and autophagy in prostate cancer cells by increasing intracellular dihydrosphingosine and dihydroceramide. Int. J. Cancer 2012, 130, 685–693. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, R.V.; Parajuli, P.; Sylvester, P.W. γ-Tocotrienol-induced autophagy in malignant mammary cancer cells. Exp. Biol. Med. 2013, 239, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, R.V.; Parajuli, P.; Sylvester, P.W. γ-Tocotrienol-induced endoplasmic reticulum stress and autophagy act concurrently to promote breast cancer cell death. Biochem. Cell Biol. 2015, 93, 306–320. [Google Scholar] [CrossRef] [PubMed]
- De Palma, C.; Perrotta, C. Clinical lipidology ceramide as a target of chemotherapy: Its role in apoptosis and autophagy. Clin. Lipidol. 2017, 7, 111–119. [Google Scholar] [CrossRef]
- Pinton, P.; Giorgi, C.; Siviero, R.; Zecchini, E.; Rizzuto, R. Calcium and apoptosis: ER-mitochondria Ca2+ transfer in the control of apoptosis. Oncogene 2008, 27, 6407–6418. [Google Scholar] [CrossRef] [Green Version]
- Kwang, S.A.; Sethi, G.; Krishnan, K.; Aggarwal, B.B. γ-tocotrienol inhibits nuclear factor-κB signaling pathway through inhibition of receptor-interacting protein and TAK1 leading to suppression of antiapoptotic gene products and potentiation of apoptosis. J. Biol. Chem. 2007, 282, 809–820. [Google Scholar]
- Aggarwal, B.; Prasad, S.; Sung, B.; Krishnan, S.; Guha, S. Prevention and treatment of colorectal cancer by natural agents from mother nature. Curr. Colorectal Cancer Rep. 2013, 9, 37–56. [Google Scholar] [CrossRef] [PubMed]
- Campbell, S.E.; Whaley, S.G.; Phillips, R.; Aggarwal, B.B.; Stimmel, J.B.; Leesnitzer, L.; Blanchard, S.G.; Stone, W.L.; Muenyi, C.; Krishnan, K. Gamma tocotrienol and prostate cancer: The regulation of two independent pathways to potentiate cell growth inhibition and apoptosis. J. Oil Palm Res. 2008, 33–43. [Google Scholar]
- Shah, S.J.; Sylvester, P.W. Gamma-tocotrienol inhibits neoplastic mammary epithelial cell proliferation by decreasing Akt and nuclear factor kappaB activity. Exp. Biol. Med. 2005, 230, 235–241. [Google Scholar] [CrossRef]
- Rajasinghe, L.D.; Gupta, S.V. Tocotrienol-rich mixture inhibits cell proliferation and induces apoptosis via down-regulation of the Notch-1/NF-κB pathways in NSCLC cells. Nutr. Diet. Suppl. 2017, 9, 103–114. [Google Scholar] [CrossRef]
- Kani, K.; Momota, Y.; Harada, M.; Yamamura, Y.; Aota, K.; Yamanoi, T.; Takano, H.; Motegi, K.; Azuma, M. γ-tocotrienol enhances the chemosensitivity of human oral cancer cells to docetaxel through the downregulation of the expression of NF-κB-regulated anti-apoptotic gene products. Int. J. Oncol. 2013, 42, 75–82. [Google Scholar] [CrossRef]
- Kunnumakkara, A.B.; Sung, B.; Ravindran, J.; Diagaradjane, P.; Deorukhkar, A.; Dey, S.; Koca, C.; Yadav, V.R.; Tong, Z.; Gelovani, J.G.; et al. Gamma-tocotrienol inhibits pancreatic tumors and sensitizes them to gemcitabine treatment by modulating the inflammatory microenvironment. Cancer Res. 2010, 70, 8695–8705. [Google Scholar] [CrossRef] [PubMed]
- Husain, K.; Francois, R.A.; Yamauchi, T.; Perez, M.; Sebti, S.M.; Malafa, M.P. Vitamin E δ-tocotrienol augments the antitumor activity of gemcitabine and suppresses constitutive NF-kB activation in pancreatic cancer. Mol. Cancer Ther. 2011, 10, 2363–2372. [Google Scholar] [CrossRef]
- Manu, K.A.; Shanmugam, M.K.; Ramachandran, L.; Li, F.; Fong, C.W.; Kumar, A.P.; Tan, P.; Sethi, G. First evidence that gamma-tocotrienol inhibits the growth of human gastric cancer and chemosensitizes it to capecitabine in a xenograft mouse model through the modulation of NF-kappaB pathway. Clin Cancer Res. 2012, 18, 2220–2229. [Google Scholar] [CrossRef]
- Song, G.; Ouyang, G.; Bao, S. The activation of Akt/PKB signaling pathway and cell survival. J. Cell. Mol. Med. 2005, 9, 59–71. [Google Scholar] [CrossRef] [Green Version]
- Samant, G.V.; Sylvester, P.W. Gamma-tocotrienol inhibits ErbB3-dependent PI3K/Akt mitogenic signalling in neoplastic mammary epithelial cells. Cell Prolif. 2006, 39, 563–574. [Google Scholar] [CrossRef]
- Sylvester, P.W.; Shah, S. Intracellular mechanisms mediating tocotrienol-induced apoptosis in neoplastic mammary epithelial cells. Asia Pac. J. Clin. Nutr. 2005, 14, 366–373. [Google Scholar]
- Sylvester, P.W.; Ayoub, N.M. Tocotrienols target PI3K/Akt signaling in anti-breast cancer therapy. Anticancer Agents Med. Chem. 2013, 13, 1039–1047. [Google Scholar] [CrossRef] [PubMed]
- Shin-Kang, S.; Ramsauer, V.P.; Lightner, J.; Chakraborty, K.; Stone, W.; Campbell, S.; Reddy, S.A.; Krishnan, K. Tocotrienols inhibit AKT and ERK activation and suppress pancreatic cancer cell proliferation by suppressing the ErbB2 pathway. Free Radic. Biol. Med. 2011, 51, 1164–1174. [Google Scholar] [CrossRef]
- Sui, X.; Kong, N.; Ye, L.; Han, W.; Zhou, J.; Zhang, Q.; He, C.; Pan, H. JNK MAPK pathways control the balance of apoptosis and autophagy in response to chemotherapeutic agents. Cancer Lett. 2014, 344, 174–179. [Google Scholar] [CrossRef]
- Wada, T.; Penninger, J.M. Mitogen-activated protein kinases in apoptosis regulation. Oncogene 2004, 23, 2838–2849. [Google Scholar] [CrossRef] [PubMed]
- Koul, H.K.; Pal, M.; Koul, S. Role of p38 MAP kinase signal transduction in solid tumors. Genes Cancer 2013, 4, 342–359. [Google Scholar] [CrossRef]
- Mebratu, Y.; Tesfaigzi, Y. How ERK1/2 activation controls cell proliferation and cell death: Is subcellular localization the answer? Cell Cycle 2009, 8, 1168–1175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehud, W.; Rony, S. The dynamic subcellular localization of ERK: Mechanisms of translocation and role in various organelles. Curr. Opin. Cell Biol. 2016, 39, 15–20. [Google Scholar]
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt signaling in cancer. Oncogene 2017, 36, 1461–1473. [Google Scholar] [CrossRef]
- Gehrke, I.; Gandhirajan, R.K.; Kreuzer, K.A. Targeting the WNT/β-catenin/TCF/LEF1 axis in solid and haematological cancers: Multiplicity of therapeutic options. Eur. J. Cancer 2009, 45, 2759–2767. [Google Scholar] [CrossRef]
- Li, H.; Mo, J.; Jia, G.; Liu, C.; Luan, Z.; Guan, Y. Activation of Wnt signaling inhibits the pro-apoptotic role of Notch in gastric cancer cells. Mol. Med. Rep. 2013, 7, 1751–1756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.; He, N.; Zhang, J.; Li, D.; Liu, Y.; Zhang, J. Experimental study on delta-tocotrienol inhibits the Wnt pathway in the colon cancer cell SW620. Wei Sheng Yan Jiu 2012, 41, 900–904. [Google Scholar]
- Ahmed, R.A.; Alawin, O.A.; Sylvester, P.W. γ-Tocotrienol reversal of epithelial-to-mesenchymal transition in human breast cancer cells is associated with inhibition of canonical Wnt signalling. Cell Prolif. 2016, 49, 460–470. [Google Scholar] [CrossRef] [PubMed]
- Sen, C.K.; Khanna, S.; Roy, S. Tocotrienols: Vitamin E beyond tocopherols. Life Sci. 2006, 78, 2088–2098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, J.Y.; Che, H.L.; Tan, D.M.Y.; Teng, K.T. Bioavailability of tocotrienols: Evidence in human studies. Nutr. Metab. 2014, 11. [Google Scholar] [CrossRef] [PubMed]
- Abuasal, B.S.; Lucas, C.; Peyton, B.; Alayoubi, A.; Nazzal, S.; Sylvester, P.W.; Kaddoumi, A. Enhancement of intestinal permeability utilizing solid lipid nanoparticles increases γ-tocotrienol oral bioavailability. Lipids 2012, 47, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Abu-Fayyad, A.; Lucas, C.; Peyton, B.; Alayoubi, A.; Nazzal, S.; Sylvester, P.W.; Kaddoumi, A. PEGylated γ-tocotrienol isomer of vitamin E: Synthesis, characterization, in vitro cytotoxicity, and oral bioavailability. Eur. J. Pharm. Biopharm. 2015, 96, 185–195. [Google Scholar] [CrossRef]
- Tan, D.M.Y.; Fu, J.Y.; Wong, F.S.; Er, H.M.; Chen, Y.S.; Nesaretnam, K. Tumor regression and modulation of gene expression via tumor-targeted tocotrienol niosomes. Nanomedicine 2017, 12, 2487–2502. [Google Scholar] [CrossRef]
- Pham, J.; Nayel, A.; Hoang, C.; Elbayoumi, T. Enhanced effectiveness of tocotrienol-based nano-emulsified system for topical delivery against skin carcinomas. Drug Deliv. 2016, 23, 1514–1524. [Google Scholar] [CrossRef]
- Viola, V.; Pilolli, F.; Piroddi, M.; Pierpaoli, E.; Orlando, F.; Provinciali, M.; Betti, M.; Mazzini, F.; Galli, F. Why tocotrienols work better: Insights into the in vitro anti-cancer mechanism of vitamin E. Genes Nutr. 2012, 7, 29–41. [Google Scholar] [CrossRef]
- Zhou, C.; Tabb, M.M.; Sadatrafiei, A.; Grün, F.; Blumberg, B. Tocotrienols activate the steroid and xenobiotic receptor, SXR, and selectively regulate expression of its target genes. Drug Metab. Dispos. 2004, 32, 1075–1082. [Google Scholar] [CrossRef] [PubMed]
- Brigelius-Flohé, R. Induction of drug metabolizing enzymes by vitamin E. J. Plant Physiol. 2005, 162, 797–802. [Google Scholar] [CrossRef]
- Chen, D.H.; Zhang, X.S. Targeted therapy: Resistance and re-sensitization. Chin. J. Cancer 2015, 34, 496–501. [Google Scholar] [CrossRef] [PubMed]
- Melisi, D.; Piro, G.; Tamburrino, A.; Carbone, C.; Tortora, G. Rationale and clinical use of multitargeting anticancer agents. Curr. Opin. Pharmacol. 2013, 13, 536–542. [Google Scholar] [CrossRef]
- Reddy, A.S.; Zhang, S. Polypharmacology: Drug discovery for the future. Expert Rev. Clin. Pharmacol. 2013, 6, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Eitsuka, T.; Tatewaki, N.; Nishida, H.; Nakagawa, K.; Miyazawa, T. Synergistic anticancer effect of tocotrienol combined with chemotherapeutic agents or dietary components: A review. Int. J. Mol. Sci. 2016, 17, 1605. [Google Scholar] [CrossRef]
- Springett, G.M.; Husain, K.; Neuger, A.; Centeno, B.; Chen, D.; Hutchinson, T.Z.; Lush, R.M.; Sebti, S.; Malafa, M.P. A Phase I safety, pharmacokinetic, and pharmacodynamic presurgical trial of vitamin E delta-tocotrienol in patients with pancreatic ductal neoplasia. EBioMedicine 2015, 2, 1987–1995. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.S.; Li, D.M.; He, N.; Liu, Y.H.; Wang, C.H.; Jiang, S.Q.; Chen, B.Q.; Liu, J.R. γ-Tocotrienol induces paraptosis-like cell death in human colon carcinoma SW620 Cells. PLoS ONE 2013, 8, e57779. [Google Scholar] [CrossRef]
- Zhang, J.S.; Li, D.M.; He, N.; Liu, Y.H.; Wang, C.H.; Jiang, S.Q.; Chen, B.Q.; Liu, J.R. A paraptosis-like cell death induced by δ-tocotrienol in human colon carcinoma SW620 cells is associated with the suppression of the Wnt signaling pathway. Toxicology 2011, 285, 8–17. [Google Scholar] [CrossRef]
- El-Khattouti, A.; Selimovic, D.; Haikel, Y.; Hassan, M. Crosstalk between apoptosis and autophagy: Molecular mechanisms and therapeutic strategies in cancer. J. Cell Death 2013, 6, 37–55. [Google Scholar] [CrossRef]
- Ye, C.; Zhao, W.; Li, M.; Zhuang, J.; Yan, X.; Lu, Q.; Chang, C.; Huang, X.; Zhou, J.; Xie, B.; et al. δ-tocotrienol induces human bladder cancer cell growth arrest, apoptosis and chemosensitization through inhibition of STAT3 pathway. PLoS ONE 2015, 10, e0122712. [Google Scholar] [CrossRef]
- Lim, S.W.; Loh, H.S.; Ting, K.N.; Bradshaw, T.D.; Zeenathul, N.A. Antiproliferation and induction of caspase-8-dependent mitochondria-mediated apoptosis by β-tocotrienol in human lung and brain cancer cell lines. Biomed. Pharmacother. 2014, 68, 1105–1115. [Google Scholar] [CrossRef] [PubMed]
- Loganathan, R.; Selvaduray, K.R.; Nesaretnam, K.; Radhakrishnan, A.K. Tocotrienols promote apoptosis in human breast cancer cells by inducing poly(ADP-ribose) polymerase cleavage and inhibiting nuclear factor kappa-B activity. Cell Prolif. 2013, 46, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Ju, H.; Shen, C.; Tong, Z. miR-429 mediates δ-tocotrienol-induced apoptosis in triple-negative breast cancer cells by targeting XIAP. Int. J. Clin. Exp. Med. 2015, 8, 15648–15656. [Google Scholar] [PubMed]
- Comitato, R.; Leoni, G.; Canali, R.; Ambra, R.; Nesaretnam, K.; Virgili, F. Tocotrienols activity in MCF-7 breast cancer cells: Involvement of ERβ signal transduction. Mol. Nutr. Food Res. 2010, 54, 669–678. [Google Scholar] [CrossRef] [PubMed]
- Viola, V.; Ciffolilli, S.; Legnaioli, S.; Piroddi, M.; Betti, M.; Mazzini, F.; Pierpaoli, E.; Provinciali, M.; Galli, F. Mitochondrial-dependent anticancer activity of δ-tocotrienol and its synthetic derivatives in HER-2/neu overexpressing breast adenocarcinoma cells. BioFactors 2013, 39, 485–493. [Google Scholar] [CrossRef] [PubMed]
- Shun, M.C.; Yu, W.; Gapor, A.; Parsons, R.; Atkinson, J.; Sanders, B.G.; Kline, K. Pro-apoptotic mechanisms of action of a novel vitamin E analog (α-TEA) and a naturally occurring form of vitamin E (δ-Tocotrienol) in MDA-MB-435 human breast cancer cells. Nutr. Cancer 2004, 48, 95–105. [Google Scholar] [CrossRef]
- Wu, S.J.; Ng, L.T. Tocotrienols inhibited growth and induced apoptosis in human HeLa cells through the cell cycle signaling pathway. Integr. Cancer Ther. 2010, 9, 66–72. [Google Scholar] [CrossRef]
- Hasani, N.A.; Yusoff, P.A.; Bak, K.; Abdul-Gapor, M.T.; Wan-Ngah, W.Z. The possible mechanism of action of palm oil γ-tocotrienol and α-tocopherol on the cervical carcinoma CaSki cell apoptosis. Biomed. Res. 2008, 19, 194–200. [Google Scholar]
- Hasani, N.A.; Bak, K.; Wan Ngah, W.Z. The anti proliferative effect of palm oil γ-tocotrienol involves alterations in MEK-2 and ERK-2 protein expressions in CaSki cells. Asian Biomed. 2011, 5, 601–609. [Google Scholar]
- Yang, Z.; Hang, X.; Jin, H.; Koo, P.T.; Tsang, D.J.; Yang, C.S. Synergistic actions of atorvastatin with γ-tocotrienol and celecoxib against human colon cancer HT29 and HCT116 cells. Int. J. Cancer 2010, 126, 852–863. [Google Scholar] [CrossRef] [PubMed]
- Shibata, A.; Nakagawa, K.; Sookwong, P.; Tsuduki, T.; Asai, A.; Miyazawa, T. α-Tocopherol attenuates the cytotoxic effect of δ-tocotrienol in human colorectal adenocarcinoma cells. Biochem. Biophys. Res. Commun. 2010, 397, 214–219. [Google Scholar] [CrossRef]
- Eitsuka, T.; Tatewaki, N.; Nishida, H.; Nakagawa, K.; Miyazawa, T. A combination of d-tocotrienol and ferulic acid synergistically inhibits telomerase activity in DLD-1 human colorectal adenocarcinoma cells. J. Nutr. Sci. Vitaminol. 2016, 62, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Eitsuka, T.; Nakagawa, K.; Miyazawa, T. Down-regulation of telomerase activity in DLD-1 human colorectal adenocarcinoma cells by tocotrienol. Biochem. Biophys. Res. Commun. 2006, 348, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Wang, Q.; Chen, B.; Liu, J.; Liu, H.; Xu, W. γ-Tocotrienol-induced apoptosis in human gastric cancer SGC-7901 cells is associated with a suppression in mitogen-activated protein kinase signalling. Br. J. Nutr. 2008, 99, 1247–1254. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Xu, W.; Liu, H.; Liu, J.; Wang, Q.; Zhou, J.; Dong, F.; Chen, B. γ-Tocotrienol induces mitochondria-mediated apoptosis in human gastric adenocarcinoma SGC-7901 cells. J. Nutr. Biochem. 2009, 20, 276–284. [Google Scholar] [CrossRef]
- Rajasinghe, L.D.; Pindiprolu, R.H.; Gupta, S.V. Delta-tocotrienol inhibits non-small-cell lung cancer cell invasion via the inhibition of NF-κB, uPA activator, and MMP-9. Onco-Targets Ther. 2018, 11, 4301–4314. [Google Scholar] [CrossRef]
- Ji, X.; Wang, Z.; Sarkar, F.H.; Gupta, S.V. Delta-tocotrienol augments cisplatin-induced suppression of non-small cell lung cancer cells via inhibition of the Notch-1 pathway. Anticancer Res. 2012, 32, 2647–2656. [Google Scholar]
- Husain, K.; Centeno, B.A.; Coppola, D.; Trevino, J.; Sebti, S.M.; Malafa, M.P. δ-Tocotrienol, a natural form of vitamin E, inhibits pancreatic cancer stem-like cells and prevents pancreatic cancer metastasis. Oncotarget 2017, 8, 31554–31567. [Google Scholar] [CrossRef]
- Hodul, P.J.; Dong, Y.; Husain, K.; Pimiento, J.M.; Chen, J.; Zhang, A.; Francois, R.; Pledger, W.J.; Coppola, D.; Sebti, S.M.; et al. Vitamin E δ-tocotrienol induces p27Kip1-dependent cell-cycle arrest in pancreatic cancer cells via an E2F-1-dependent mechanism. PLoS ONE 2013, 8, e52526. [Google Scholar] [CrossRef]
- Eitsuka, T.; Tatewaki, N.; Nishida, H.; Nakagawa, K.; Miyazawa, T. Synergistic inhibition of cancer cell proliferation with a combination of delta-tocotrienol and ferulic acid. Biochem. Biophys. Res. Commun. 2014, 453, 606–611. [Google Scholar] [CrossRef]
- Luk, S.U.; Yap, W.N.; Chiu, Y.T.; Lee, D.T.; Ma, S.; Lee, T.K.; Vasireddy, R.S.; Wong, Y.C.; Ching, Y.P.; Nelson, C.; et al. Gamma-tocotrienol as an effective agent in targeting prostate cancer stem cell-like population. Int. J. Cancer 2011, 128, 2182–2191. [Google Scholar] [CrossRef] [PubMed]
- Marzagalli, M.; Moretti, R.M.; Messi, E.; Marelli, M.M.; Fontana, F.; Anastasia, A.; Bani, M.R.; Beretta, G.; Limonta, P. Targeting melanoma stem cells with the Vitamin E derivative δ-tocotrienol. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Ng, L.T.; Lin, L.T.; Chen, C.L.; Chen, H.W.; Wu, S.J.; Lin, C.C. Anti-melanogenic effects of δ-tocotrienol are associated with tyrosinase-related proteins and MAPK signaling pathway in B16 melanoma cells. Phytomedicine 2014, 21, 978–983. [Google Scholar] [CrossRef] [PubMed]
- Wada, S.; Naito, Y.; Matsushita, Y.; Nouchi, M.; Kawai, M.; Minami, E.; Aoi, W.; Ikeda, S.; Higashi, A.; Yoshikawa, T. δ-Tocotrienol suppresses tumorigenesis by inducing apoptosis and blocking the COX-2/PGE2 pathway that stimulates tumor–stromal interactions in colon cancer. J. Funct. Foods 2017, 35, 428–435. [Google Scholar] [CrossRef]
- Husain, K.; Centeno, B.A.; Chen, D.T.; Fulp, W.J.; Perez, M.; Lee, G.Z.; Luetteke, N.; Hingorani, S.R.; Sebti, S.M.; Malafa, M.P. Prolonged survival and delayed progression of pancreatic intraepithelial neoplasia in LSL-Kras; Pdx-1-Cre mice by vitamin E δ-tocotrienol. Carcinogenesis 2013, 34, 858–863. [Google Scholar] [CrossRef] [PubMed]
- Yap, W.N.; Zaiden, N.; Luk, S.Y.; Lee, D.T.; Ling, M.T.; Wong, Y.C.; Yap, Y.L. In vivo evidence of gamma-tocotrienol as a chemosensitizer in the treatment of hormone-refractory prostate cancer. Pharmacology 2010, 85, 248–258. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer Type | Cell Line(s) | Molecular Target(s) | Reference(s) |
|---|---|---|---|
| Bladder | T24 5637 J82 UMUC-3 | ↑ p21, p27, Bax, caspase-3, cleaved PARP, SHP-1 ↓ cyclin D1, Bcl-2, Bcl-xL, Mcl-1, ETK phosphorylation, STAT3 | [133] |
| Brain | U87MG | ↑ caspase-8, Bid, cytochrome c, Bax | [41,134] |
| Breast | MDA-MB-231 | ↑ caspase-8, caspase-9, caspase-7, caspase-3, cleaved PARP, DR5, DR4, p-JNK, p-c-Jun, p-p38, BiP, ATF3, ATF4, p-PERK, p-IRE1α, p-eIF2α, CHOP, LC3-II/I, Beclin-1, Bax ↓ NF-κB, cyclin D1, cyclin D3, CDK4, Bcl-2, PI3K, p-AKT, p-mTOR, XIAP | [69,73,88,135,136] |
| MCF-7 | ↑ caspase-8, caspase-9, caspase-7, caspase-3, Bax, cleaved PARP, ATF3, BiP, CHOP, p-PERK, p-IRE1α, p-EIF2α, ATF4, LC3-II/I, Beclin-1, DR5, p-JNK, p-c-Jun, p-p38, MIC-1, EGR-1, cathepsin D ↓ cyclin D1, cyclin D3, CDK4, NF-κB, Bcl-2, PI3K, p-AKT, p-mTOR | [69,70,73,88,136,137] | |
| +SA | ↑ LC3-II/I, Beclin-1, Bax, cleaved PARP, cleaved caspase-3 ↓ Bcl-2, PI3K, p-AKT, p-mTOR | [88] | |
| SKBR3 | ↓ p-ERK1/2 | [138] | |
| MDA-MB-435 | ↑ cleaved PARP, p-JNK-1, JNK1, p-c-Jun, c-Jun, TGFβRII, TGFβRI | [139] | |
| Cervix | HeLa | ↑ Bax, cytochrome c, caspase-12, caspase-9, caspase-8, caspase-3, IL-6, XBP-1, p-IRE-1α, BiP, CHOP, XBP-1, cleaved PARP ↓ PCNA, cyclin D3, p16, CDK6 | [46,47,140,141] |
| CaSki | ↑ p53, Bax, caspase-3 ↓ MEK-2, ERK | [141,142] | |
| Colon | HT29 | ↑ p21, Bax, caspase-9, caspase-3 ↓ Bcl-2, NF-κB p65, β-catenin, cyclin D1, c-Myc, survivin | [42,56,143,144] |
| SW620 | ↓ Wnt-1, β-catenin, cyclin D1, c-Jun, MMP-7 | [130,131] | |
| HCT116 | ↑ p21 ↓ cIAP-1, cIAP-2, survivin, cyclin D1, c-Myc, MMP-9, VEGF, ICAM-1, CXCR4, NF-κB | [59,144] | |
| DLD-1 | ↑ p21, p27, caspase-7, caspase-9 ↓ hTERT | [144,145,146] | |
| Gastric | SGC-7901 | ↑ Bax, caspase-3, caspase-9, cleaved PARP ↓ Bcl-2, c-Myc, p-ERK1/2, Raf-1 | [147,148] |
| SNU-5 | ↓ NF-κB | [100] | |
| SNU-16 | ↑ cleaved PARP ↓ cyclin D1, Bcl-2, MMP-9, CXCR4, VEGF, NF-κB | [100] | |
| Leukemia | ED40515 | ↑ caspase-3, caspase-6, caspase-7, caspase-9, PARP, Bcl-2, Bcl-xL, XIAP ↓ FDFT1, NF-κB | [47] |
| HL-60 | ↑ cleaved Bid, cytochrome c release, caspase-8, caspase-9, caspase-3 | [51] | |
| NB-4 | ↑ cleaved Bid, cytochrome c release, caspase-8, caspase-9, caspase-3 | [51] | |
| Lung | A549 | ↑ caspase-3, caspase-8, Bid, cytochrome c, Bax, cleaved PARP ↓ Notch-1, Hes-1, Bcl-2, NF-κB, uPA, survivin, Bcl-XL, MMP-9 | [41,134,149,150] |
| H520 | ↑ PARP, caspase-3 ↓ Notch-1, Hes-1, Bcl-2, NF-κB, survivin, Bcl-XL | [96,149] | |
| H1299 | ↓ Notch-1, Hes-1, uPA, MMP-9 | [149] | |
| Pancreas | MIA PaCa-2 | ↑ E-cadherin, EGR-1, Bax, p27Kip1 ↓ NF-κB, Bcl-2, cIAP-1, survivin, cyclin D1, c-Myc, COX-2, VEGF, MMP-9, ICAM-1, CXCR4, N-cadherin, vimentin, p-MEK, p-AKT, p-GSK-β | [58,98,151,152] |
| L3.6pl | ↑ E-cadherin ↓ N-cadherin, vimentin, VEGF, MMP-9 | [151] | |
| BXPC3 | ↑ p27Kip1 ↓ p-MEK, p-AKT, p-ERK | [152] | |
| SW1990 | ↑ p27Kip1 ↓ p-MEK, p-AKT, p-ERK | [152] | |
| PANC-1 | ↑ p21 | [153] | |
| Pancreatic cancer stem cell | ↑ cleaved PARP ↓ Nanog, Sox-2, Oct-4, Notch-1, p-AKT, pERK | [151] | |
| Prostate | PC-3 | ↑ caspase-9, cytochrome c, cleaved PARP, LC3-II ↓ p-Akt, β-catenin, Id-1, Bcl-2 | [19,87] |
| PC-3 (stem cell-like) | ↑ caspase-3, cleaved PARP ↓ Id-1 | [154] | |
| LNCaP | ↑ caspase-9, caspase-8, caspase-7, caspase-3, cytochrome c, cleaved PARP, LC3-II ↓ Id-1, p-Akt | [19,87] | |
| Skin | G361 | ↑ cleaved PARP, caspase-7, caspase-9, caspase-3, E-cadherin, β-catenin, γ-catenin ↓ Snail, vimentin, α-SME, Twist | [17] |
| C32 | ↑ cleaved PARP, caspase-7, caspase-9, caspase-3, IκB, p-ATF2, p-c-Jun, p-SAPK/JNK ↓ PI3K p85, p-IKKα/β, IκBα/β, NF-κB p65, EGFR, Id-1, Id-3 | [17] | |
| A375 (stem cell-like) | ↓ ABCG2 | [155] | |
| BLM | ↑ caspase-3, caspase-4, cleaved PARP, Bax, BiP, PERK, p-eIF2α, IRE1α, ATF4, CHOP ↓ Bcl-2 | [71] | |
| A375 | ↑ caspase-3, caspase-4, cleaved PARP, BiP, PERK, p-eIF2α, IRE1α, ATF4, CHOP, ERO1α ↓ Bcl-2, CDK4, Ras, caspase-3 | [48,71] | |
| A2058 | ↓ CDK4, Ras, caspase-3 | [48] | |
| B16 | ↑ p-ERK ↓ Tyrosinase, MC1R, MITF, TYRP-1, TYRP-2, p-p38 | [156] |
| Cancer Type | Tocotrienol(s) | Anticancer Effect(s)/Molecular Target(s) | Reference |
|---|---|---|---|
| Colon | TRF or δ-T3-enriched diet |
| [157] |
| Colon | TRF |
| [23] |
| Gastric | γ-T3 |
| [100] |
| Pancreas | γ-T3 |
| [98] |
| Pancreas | δ-T3 |
| [151] |
| Pancreas | δ-T3 |
| [152] |
| Pancreas | δ-T3 |
| [158] |
| Prostate | γ-T3 |
| [154] |
| Prostate | γ-T3 |
| [87] |
| Prostate | γ-T3 |
| [159] |
| Skin | δ-T3 |
| [155] |
| Skin | δ-T3 |
| [71] |
| Cancer Type | Target Application(s) of Tocotrienols | Drugs Involved | Phase: Status | ClinicalTrials.gov Identifier |
|---|---|---|---|---|
| Breast | Adjunct cancer treatment | TRF and Tamoxifen | Pilot trial: Completed in 2010 | NCT01157026 |
| Breast | Health supplement | Gamma-Delta Tocotrienols and TRF | 1: Completed in 2013 | NCT01571921 |
| Breast | Neoadjuvant treatment | Epirubicin, Cyclophosphamide, Docetaxel, Paclitaxel, Trastuzumab, Pertuzumab and Tocotrienols | 2: Ongoing | NCT02909751 |
| Colon | Adjunct cancer treatment | Irinotecan, Oxaliplatin, Calcium Folinate, 5-Fluorouracil and Tocotrienols | 2: Ongoing | NCT02705300 |
| Lung | Adjunct cancer treatment | Cisplatin, Vinorelbine, Carboplatin and Tocotrienols | 3: Ongoing | NCT02644252 |
| Ovary | Adjunct cancer treatment | Bevacizumab and Tocotrienols | 2: Ongoing | NCT02399592 |
| Ovary | Cancer treatment | Cabazitaxel and/or Tocotrienols | 2: Ongoing | NCT02560337 |
| Pancreas | Cancer treatment | δ-T3 | 1: Completed in 2016 | NCT00985777 |
| Pancreas | Health supplement | δ-T3 | 1: Completed in 2016 | NCT01450046 |
| Pancreas | Health supplement | δ-T3 | 1: Completed in 2016 | NCT01446952 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tham, S.-Y.; Loh, H.-S.; Mai, C.-W.; Fu, J.-Y. Tocotrienols Modulate a Life or Death Decision in Cancers. Int. J. Mol. Sci. 2019, 20, 372. https://doi.org/10.3390/ijms20020372
Tham S-Y, Loh H-S, Mai C-W, Fu J-Y. Tocotrienols Modulate a Life or Death Decision in Cancers. International Journal of Molecular Sciences. 2019; 20(2):372. https://doi.org/10.3390/ijms20020372
Chicago/Turabian StyleTham, Shiau-Ying, Hwei-San Loh, Chun-Wai Mai, and Ju-Yen Fu. 2019. "Tocotrienols Modulate a Life or Death Decision in Cancers" International Journal of Molecular Sciences 20, no. 2: 372. https://doi.org/10.3390/ijms20020372
APA StyleTham, S. -Y., Loh, H. -S., Mai, C. -W., & Fu, J. -Y. (2019). Tocotrienols Modulate a Life or Death Decision in Cancers. International Journal of Molecular Sciences, 20(2), 372. https://doi.org/10.3390/ijms20020372