Review of Current Strategies for Delivering Alzheimer’s Disease Drugs across the Blood-Brain Barrier

 ,
,
Abstract
:1. Introduction
2. Pathophysiology of Alzheimer’s Disease
2.1. Amyloid Hypothesis
2.2. Tau Protein
2.3. Others
3. Blood-Brain Barrier
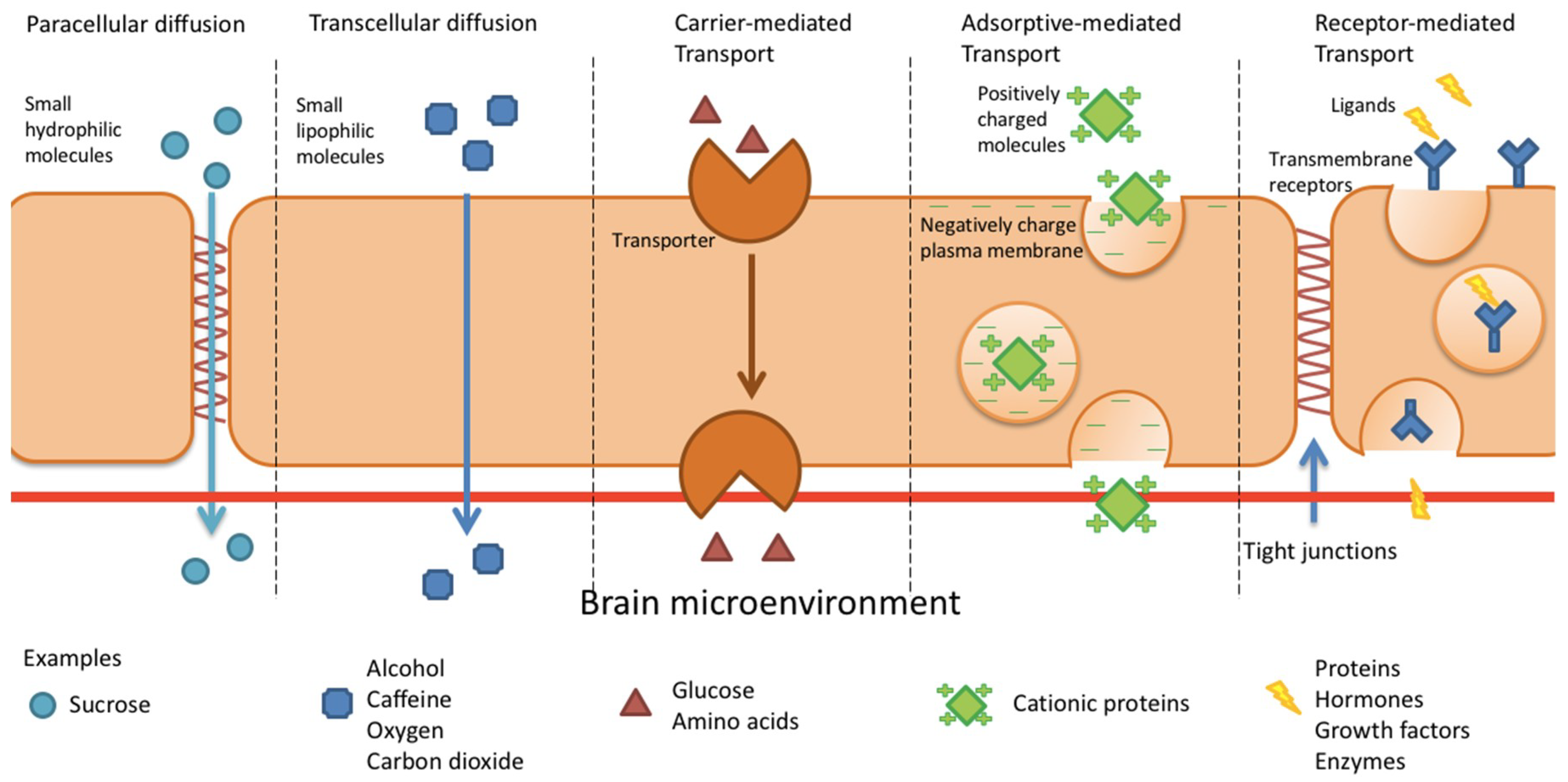
4. BBB Permeation Mechanisms
4.1. Paracellular and Transcellular Diffusion
4.2. Carrier-Mediated Transport
4.3. Adsorptive-Mediated Transcytosis
4.3.1. Syn-B Vectors
4.3.2. TAT-Derived Peptides
4.4. Receptor-Mediated Transcytosis
4.4.1. Transferrin Receptor
4.4.2. Insulin Receptor
4.4.3. Low Density Lipoprotein Receptor
5. Role of the Blood-Brain Barrier in Drug Delivery
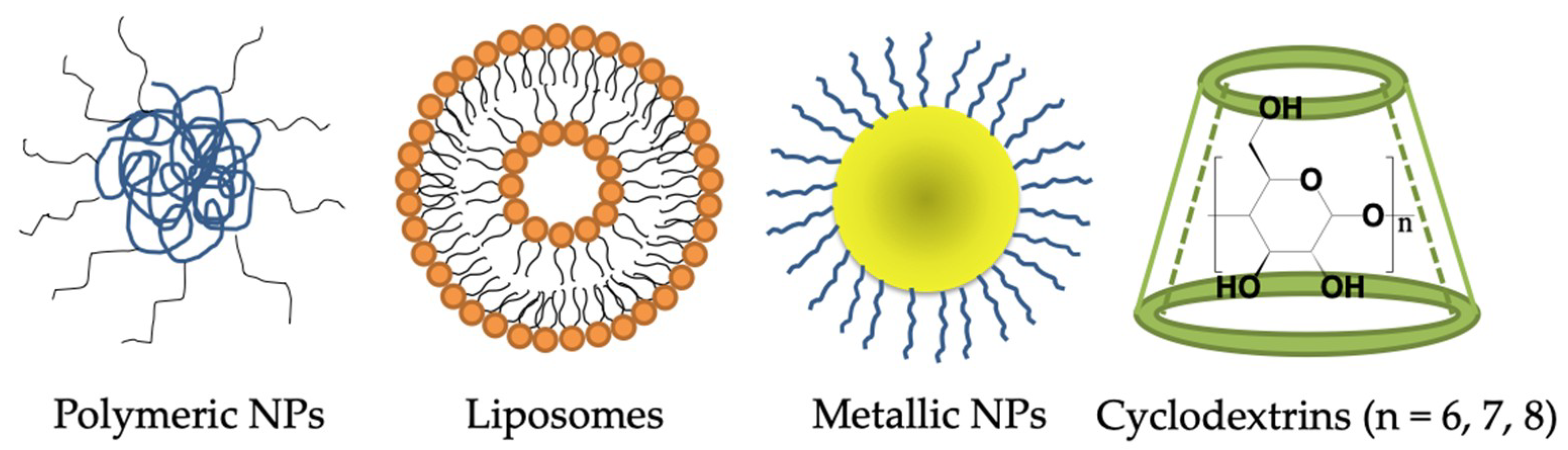
6. Nanotechnology-Based Drug Delivery Systems for Penetrating the BBB to Treat AD
6.1. Polymeric Nanoparticles
6.2. Lipid-Based Nanocarriers—Liposome
6.3. Metal-Based Nanoparticles
6.4. Others—Cyclodextrins
7. Discussion and Future Perspective
8. Conclusions
Funding
Conflicts of Interest
References
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological Alterations in Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s Association. 2018 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2018, 14, 367–429. [Google Scholar] [CrossRef]
- Zhang, M.; Schmitt-Ulms, G.; Sato, C.; Xi, Z.; Zhang, Y.; Zhou, Y.; George-Hyslop, P.S.; Rogaeva, E. Drug Repositioning for Alzheimer’s Disease Based on Systematic “omics” Data Mining. PLoS ONE 2016, 11, e0168812. [Google Scholar] [CrossRef] [PubMed]
- Lleó, A. Current Therapeutic Options for Alzheimer’s Disease. Curr. Genom. 2007, 8, 550–558. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.L.; Morstorf, T.; Zhong, K. Alzheimer’s disease drug-development pipeline: Few candidates, frequent failures. Alzheimer’s Res. Ther. 2014, 6, 37. [Google Scholar] [CrossRef]
- Swanson, C.J.; Zhang, Y.; Dhadda, S.; Wang, J.; Kaplow, J.; Lai, R.Y.; Lannfelt, L.; Kramer, L.D.; Luthman, J. Treatment of early AD subjects with BAN2401, an anti-Aβ protofibril monoclonal antibody, significantly clears amyloid plaque and reduces clinical decline. Alzheimer’s Dement. 2018, 14, P1668. [Google Scholar] [CrossRef]
- Redzic, Z. Molecular biology of the blood-brain and the blood-cerebrospinal fluid barriers: Similarities and differences. Fluids Barriers CNS 2011, 8, 3. [Google Scholar] [CrossRef]
- Pardridge, W.M. The Blood-Brain Barrier: Bottleneck in Brain Drug Development. NeuroRx 2005, 2, 3–14. [Google Scholar] [CrossRef]
- Banks, W.A. Characteristics of compounds that cross the blood-brain barrier. BMC Neurol. 2009, 9, S3. [Google Scholar] [CrossRef] [Green Version]
- Sercombe, L.; Veerati, T.; Moheimani, F.; Wu, S.Y.; Sood, A.K.; Hua, S. Advances and Challenges of Liposome Assisted Drug Delivery. Front. Pharmacol. 2015, 6, 286. [Google Scholar] [CrossRef]
- Tiwari, G.; Tiwari, R.; Sriwastawa, B.; Bhati, L.; Pandey, S.; Pandey, P.; Bannerjee, S.K. Drug delivery systems: An updated review. Int. J. Pharm. Investig. 2012, 2, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Allen, T.M.; Cullis, P.R. Drug delivery systems: Entering the mainstream. Science 2004, 303, 1818–1822. [Google Scholar] [CrossRef] [PubMed]
- Beason-Held, L.L.; Goh, J.O.; An, Y.; Kraut, M.A.; O’Brien, R.J.; Ferrucci, L.; Resnick, S.M. Changes in Brain Function Occur Years before the Onset of Cognitive Impairment. J. Neurosci. 2013, 33, 18008–18014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bateman, R.J.; Xiong, C.; Benzinger, T.L.; Fagan, A.M.; Goate, A.; Fox, N.C.; Marcus, D.S.; Cairns, N.J.; Xie, X.; Blazey, T.M.; et al. Clinical and Biomarker Changes in Dominantly Inherited Alzheimer’s Disease. N. Engl. J. Med. 2012, 367, 795–804. [Google Scholar] [CrossRef] [PubMed]
- A Elman, J.; Oh, H.; Madison, C.M.; Baker, S.L.; Vogel, J.W.; Marks, S.M.; Crowley, S.; O’Neil, J.P.; Jagust, W.J. Neural compensation in older people with brain amyloid-β deposition. Nat. Neurosci. 2014, 17, 1316–1318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Chen, W.-D.; Wang, Y.-D. β-Amyloid: The key peptide in the pathogenesis of Alzheimer’s disease. Front. Pharmacol. 2015, 6, 221. [Google Scholar] [CrossRef] [PubMed]
- Louise, C.S. Alzheimer’s amyloid fibrils: Structure and assembly. BBA—Mol. Basis Dis. 2000, 1502, 16–30. [Google Scholar] [CrossRef]
- Singh, S.K.; Srivastav, S.; Yadav, A.K.; Srikrishna, S.; Perry, G. Overview of Alzheimer’s Disease and Some Therapeutic Approaches Targeting Aβ by Using Several Synthetic and Herbal Compounds. Oxidative Med. Cell. Longev. 2016, 2016, 1–22. [Google Scholar] [CrossRef]
- Wang, F.; Zhou, X.-L.; Yang, Q.-G.; Xu, W.-H.; Wang, F.; Chen, Y.-P.; Chen, G.-H. A Peptide That Binds Specifically to the β-Amyloid of Alzheimer’s Disease: Selection and Assessment of Anti-β-Amyloid Neurotoxic Effects. PLoS ONE 2011, 6, e27649. [Google Scholar] [CrossRef]
- Reddy, P.H.; Beal, M.F. Amyloid beta, mitochondrial dysfunction and synaptic damage: Implications for cognitive decline in aging and Alzheimer’s disease. Trends Mol. Med. 2008, 14, 45–53. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Swomley, A.M.; Sultana, R. Amyloid β-Peptide (1–42)-Induced Oxidative Stress in Alzheimer Disease: Importance in Disease Pathogenesis and Progression. Antioxid. Redox Signal. 2013, 19, 823–835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masahir, K.; Isao, O.; Shoko, Y.; Midori, K.; Yutaka, S. Membrane Incorporation, Channel Formation, and Disruption of Calcium Homeostasis by Alzheimer’s β-Amyloid Protein. Int. J. Alzheimer’s Dis. 2011, 2011. [Google Scholar] [CrossRef]
- Demuro, A.; Parker, I.; Stutzmann, G.E. Calcium signaling and amyloid toxicity in Alzheimer disease. J. Biol. Chem. 2010, 285, 12463–12468. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Hussain, M.D.; Yan, L.J. Microglia, neuroinflammation, and beta-amyloid protein in Alzheimer’s disease. Int. J. Neurosci. 2014, 124, 307–321. [Google Scholar] [CrossRef] [PubMed]
- De Paula, V.d.J.R.; Guimarães, F.M.; Diniz, B.S.; Forlenza, O.V. Neurobiological pathways to Alzheimer’s disease: Amyloid-beta, TAU protein or both? Dement. Neuropsychol. 2009, 3, 188–194. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.H.; Bastianetto, S.; Mennicken, F.; Ma, W.; Kar, S. Amyloid beta peptide induces tau phosphorylation and loss of cholinergic neurons in rat primary septal cultures. Neuroscience 2002, 115, 201–211. [Google Scholar] [CrossRef]
- Nisbet, R.M.; Polanco, J.-C.; Ittner, L.M.; Götz, J. Tau aggregation and its interplay with amyloid-β. Acta Neuropathol. 2015, 129, 207–220. [Google Scholar] [CrossRef]
- Gong, C.-X.; Iqbal, K. Hyperphosphorylation of Microtubule-Associated Protein Tau: A Promising Therapeutic Target for Alzheimer Disease. Curr. Med. Chem. 2008, 15, 2321–2328. [Google Scholar] [CrossRef]
- Gendron, T.F.; Petrucelli, L. The role of tau in neurodegeneration. Mol. Neurodegener. 2009, 4, 13. [Google Scholar] [CrossRef] [Green Version]
- Lambert, J.C.; Ibrahim-Verbaas, C.A.; Harold, D.; Naj, A.C.; Sims, R.; Bellenguez, C.; Jun, G.; DeStefano, A.L.; Bis, J.C.; Beecham, G.W.; et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 2013, 45, 1452–1458. [Google Scholar] [CrossRef]
- Liu, C.-C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein E and Alzheimer disease: Risk, mechanisms, and therapy. Nat. Rev. Neurol. 2013, 9, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Nishitsuji, K.; Hosono, T.; Nakamura, T.; Bu, G.; Michikawa, M. Apolipoprotein E regulates the integrity of tight junctions in an isoform-dependent manner in an in vitro blood-brain barrier model. J. Biol. Chem. 2011, 286, 17536–17542. [Google Scholar] [CrossRef] [PubMed]
- Lamartinière, Y.; Boucau, M.C.; Dehouck, L.; Krohn, M.; Pahnke, J.; Candela, P.; Gosselet, F.; Fenart, L. ABCA7 downregulation modifies cellular cholesterol homeostasis and decreases amyloid-β peptide efflux in an in vitro model of the blood-brain barrier. J. Alzheimer’s Dis. 2018, 64, 1195–1211. [Google Scholar] [CrossRef]
- Abbott, N.J.; Patabendige, A.A.; Dolman, D.E.; Yusof, S.R.; Begley, D.J. Structure and function of the blood-brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, E.M.; Blazquez, J.L.; Guerra, M. The design of barriers in the hypothalamus allows the median eminence and the arcuate nucleus to enjoy private milieus: The former opens to the portal blood and the latter to the cerebrospinal fluid. Peptides 2010, 31, 757–776. [Google Scholar] [CrossRef] [PubMed]
- Zlokovic, B.V. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron 2008, 57, 178–201. [Google Scholar] [CrossRef] [PubMed]
- Bailey, T.L.; Rivara, C.B.; Rocher, A.B.; Hof, P.R. The nature and effects of cortical microvascular pathology in aging and Alzheimer’s disease. Neurol. Res. 2004, 26, 573–588. [Google Scholar] [CrossRef]
- Montagne, A.; Nation, D.A.; Pa, J.; Sweeney, M.D.; Toga, A.W.; Zlokovic, B.V. Brain imaging of neurovascular dysfunction in Alzheimer’s disease. Acta Neuropathol. 2016, 131, 687–707. [Google Scholar] [CrossRef] [Green Version]
- Wong, A.D.; Ye, M.; Levy, A.F.; Rothstein, J.D.; Bergles, D.E.; Searson, P.C. The blood-brain barrier: An engineering perspective. Front. Neuroeng. 2013, 6, 7. [Google Scholar] [CrossRef]
- Serlin, Y.; Shelef, I.; Knyazer, B.; Friedman, A. Anatomy and Physiology of the Blood-Brain Barrier. Semin. Cell Dev. Biol. 2015, 38, 2–6. [Google Scholar] [CrossRef]
- Volterra, A.; Meldolesi, J. Astrocytes, from brain glue to communication elements: The revolution continues. Nat. Rev. Neurosci. 2005, 6, 626–640. [Google Scholar] [CrossRef] [PubMed]
- Peppiatt, C.M.; Howarth, C.; Mobbs, P.; Attwell, D. Bidirectional control of CNS capillary diameter by pericytes. Nature 2006, 443, 700–704. [Google Scholar] [CrossRef] [Green Version]
- Hawkins, B.T.; Davis, T.P. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol. Rev. 2005, 57, 173–185. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, L. Modern methods for delivery of drugs across the blood-brain barrier. Adv. Drug Deliv. Rev. 2012, 64, 640–665. [Google Scholar] [CrossRef]
- Fischer, H.; Gottschlich, R.; Seelig, A. Blood-Brain Barrier Permeation: Molecular Parameters Governing Passive Diffusion. J. Membr. Biol. 1998, 165, 201–211. [Google Scholar] [CrossRef] [Green Version]
- Rishton, G.M.; LaBonte, K.; Williams, A.J.; Kassam, K.; Kolovanov, E. Computational approaches to the prediction of blood-brain barrier permeability: A comparative analysis of central nervous system drugs versus secretase inhibitors for Alzheimer’s disease. Curr. Opin. Drug Discov. Dev. 2006, 9, 303–313. [Google Scholar]
- Kelder, J.; Grootenhuis, P.D.J.; Bayada, D.M. Polar molecular surface as a dominating determinant for oral absorption and brain penetration of drugs. Pharm. Res. 1999, 16, 1514–1519. [Google Scholar] [CrossRef]
- Alam, M.I.; Beg, S.; Samad, A.; Baboota, S.; Kohli, K.; Ali, J.; Ahuja, A.; Akbar, M. Strategy for effective brain drug delivery. Eur. J. Pharm. Sci. 2010, 40, 385–403. [Google Scholar] [CrossRef]
- Fong, C.W. Permeability of the Blood-Brain Barrier: Molecular Mechanism of Transport of Drugs and Physiologically Important Compounds. J. Membr. Biol. 2015, 248, 651–669. [Google Scholar] [CrossRef] [PubMed]
- Simpson, I.A.; Vannucci, S.J.; DeJoseph, M.R.; Hawkins, R.A. Glucose transporter asymmetries in the bovine blood-brain barrier. J. Biol. Chem. 2001, 276, 12725–12729. [Google Scholar] [CrossRef] [PubMed]
- Winkler, E.A.; Nishida, Y.; Sagare, A.P.; Rege, S.V.; Bell, R.D.; Perlmutter, D.; Sengillo, J.D.; Hillman, S.; Kong, P.; Nelson, A.R.; et al. GLUT1 reductions exacerbate Alzheimer’s disease vasculo-neuronal dysfunction and degeneration. Nat. Neurosci. 2015, 18, 521–530. [Google Scholar] [CrossRef]
- Pardridge, W.M. Recent developments in peptide drug delivery to the brain. Pharmacol. Toxicol. 1992, 71, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Bickel, U.; Yoshikawa, T.; Pardridge, W.M. Delivery of peptides and proteins through the blood-brain barrier. Adv. Drug Deliv. Rev. 2001, 46, 247–279. [Google Scholar] [CrossRef]
- Kumagai, A.K.; Eisenberg, J.B.; Pardridge, W.M. Absorptive-mediated endocytosis of cationized albumin and a beta-endorphin-cationized albumin chimeric peptide by isolated brain capillaries. Model system of blood-brain barrier transport. J. Biol. Chem. 1987, 262, 15214–15219. [Google Scholar] [PubMed]
- Kokryakov, V.N.; Harwig, S.S.; Panyutich, E.A.; Shevchenko, A.A.; Aleshina, G.M.; Shamova, O.V.; Korneva, H.A.; Lehrer, R.I. Protegrins: Leukocyte antimicrobial peptides that combine features of corticostatic defensins and tachyplesins. FEBS Lett. 1993, 327, 231–236. [Google Scholar] [CrossRef]
- Drin, G.; Rousselle, C.; Scherrmann, J.M.; Rees, A.R.; Temsamani, J. Peptide delivery to the brain via adsorptive-mediated endocytosis: Advances with SynB vectors. AAPS Pharmsci. 2002, 4, 61–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rousselle, C.; Smirnova, M.; Clair, P.; Lefauconnier, J.M.; Chavanieu, A.; Calas, B.; Scherrmann, J.M.; Temsamani, J. Enhanced delivery of doxorubicin into the brain via a peptide-vector-mediated strategy: Saturation kinetics and specificity. J. Pharmacol. Exp. Ther. 2001, 296, 124–131. [Google Scholar]
- Rousselle, C.; Clair, P.; Temsamani, J.; Scherrmann, J.M. Improved brain delivery of benzylpenicillin with a peptide-vector-mediated strategy. J. Drug Target. 2002, 10, 309–315. [Google Scholar] [CrossRef]
- Vivès, E.; Brodin, P.; Lebleu, B. A truncated HIV-1 Tat protein basic domain rapidly translocates through the plasma membrane and accumulates in the cell nucleus. J. Biol. Chem. 1997, 272, 16010–16017. [Google Scholar] [CrossRef]
- Wender, P.A.; Mitchell, D.J.; Pattabiraman, K.; Pelkey, E.T.; Steinman, L.; Rothbard, J.B. The design, synthesis, and evaluation of molecules that enable or enhance cellular uptake: Peptoid molecular transporters. Proc. Natl. Acad. Sci. USA 2000, 97, 13003–13008. [Google Scholar] [CrossRef] [Green Version]
- Santra, S.; Yang, H.; Stanley, J.T.; Holloway, P.H.; Moudgil, B.M. Rapid and effective labeling of brain tissue using TAT-conjugated CdS:Mn/ZnS quantum dots. Chem. Commun. 2005, 25, 3144–3146. [Google Scholar] [CrossRef]
- Di Fede, G.; Catania, M.; Maderna, E.; Morbin, M.; Moda, F.; Colombo, L.; Rossi, A.; Cagnotto, A.; Virgilio, T.; Palamara, L.; et al. Tackling amyloidogenesis in Alzheimer’s disease with A2V variants of Amyloid-β. Sci. Rep. 2016, 6, 20949. [Google Scholar] [CrossRef] [Green Version]
- Giunta, B.; Hou, H.; Zhu, Y.; Rrapo, E.; Tian, J. HIV-1 Tat contributes to Alzheimer’s disease-like pathology in PSAPP mice. Int. J. Clin. Exp. Pathol. 2009, 2, 433–443. [Google Scholar] [PubMed]
- Hategan, A.; A Bianchet, M.; Steiner, J.; Karnaukhova, E.; Masliah, E.; Fields, A.; Lee, M.-H.; Dickens, A.M.; Haughey, N.; Dimitriadis, E.K.; et al. HIV-Tat protein and amyloid β peptide form multifibrillar structures that cause neurotoxicity. Nat. Struct. Mol. Biol. 2017, 24, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Luo, X.; Liu, X.; Liu, D.; Wang, X.; Guo, Z.; Zhu, L.; Tian, Q.; Yang, X.; Wang, J.-Z. Intraperitoneal Administration of a Novel TAT-BDNF Peptide Ameliorates Cognitive Impairments via Modulating Multiple Pathways in Two Alzheimer’s Rodent Models. Sci. Rep. 2015, 5, 15032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Y.-S.; Chen, Z.-T.; Liao, T.-Y.; Lin, C.; Shen, H.C.-H.; Wang, Y.-H.; Chang, C.-W.; Liu, R.-S.; Chen, R.P.-Y.; Tu, P.-H.; et al. An intranasally delivered peptide drug ameliorates cognitive decline in Alzheimer transgenic mice. Embo Mol. Med. 2017, 9, 703–715. [Google Scholar] [CrossRef]
- Van Rooy, I.; Cakir-Tascioglu, S.; Hennink, W.E.; Storm, G.; Schiffelers, R.M.; Mastrobattista, E. In Vivo Methods to Study Uptake of Nanoparticles into the Brain. Pharm. Res. 2011, 28, 456–471. [Google Scholar] [CrossRef]
- Lajoie, J.M.; Shusta, E.V. Targeting receptor-mediated transport for delivery of biologics across the blood-brain barrier. Annu. Rev. Pharmacol. Toxicol. 2015, 55, 613–631. [Google Scholar] [CrossRef]
- Moos, T.; Morgan, E.H. Transferrin and transferrin receptor function in brain barrier systems. Cell. Mol. Neurobiol. 2000, 20, 77–95. [Google Scholar] [CrossRef]
- Kalaria, R.N.; Sromek, S.M.; Grahovac, I.; Harik, S.I. Transferrin receptors of rat and human brain and cerebral microvessels and their status in Alzheimer’s disease. Brain Res. 1992, 585, 87–93. [Google Scholar] [CrossRef]
- Zhang, Y.; Pardridge, W.M. Rapid transferrin efflux from brain to blood across the blood-brain barrier. J. Neurochem. 2001, 76, 1597–1600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Boer, A.G.; Gaillard, P.J. Drug targeting to the brain. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 323–355. [Google Scholar] [CrossRef] [PubMed]
- Jefferies, W.A.; Brandon, M.R.; Hunt, S.V.; Williams, A.F.; Gatter, K.C.; Mason, D.Y. Transferrin receptor on endothelium of brain capillaries. Nature 1984, 312, 162–163. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Buciak, J.; Yang, J.; Pardridge, W.M. Vector-mediated delivery of 125I-labeled beta-amyloid peptide A beta 1-40 through the blood-brain barrier and binding to Alzheimer disease amyloid of the A beta 1-40/vector complex. Proc. Natl. Acad. Sci. USA 1995, 92, 10227–10231. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M.; Wu, D.; Sakane, T. Combined use of carboxyl-directed protein pegylation and vector-mediated blood-brain barrier drug delivery system optimizes brain uptake of brain-derived neurotrophic factor following intravenous administration. Pharm. Res. 1998, 15, 576–582. [Google Scholar] [CrossRef] [PubMed]
- Boado, R.J.; Tsukamoto, H.; Pardridge, W.M. Drug delivery of antisense molecules to the brain for treatment of Alzheimer’s disease and cerebral AIDS. J. Pharm. Sci. 1998, 87, 1308–1315. [Google Scholar] [CrossRef] [PubMed]
- Paterson, J.; Webster, C.I. Exploiting transferrin receptor for delivering drugs across the blood-brain barrier. Drug Discov. Today Technol. 2016, 20, 49–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.J.; Engelhardt, B.; Lesley, J.; Bickel, U.; Pardridge, W.M. Targeting rat anti-mouse transferrin receptor monoclonal antibodies through blood-brain barrier in mouse. J. Pharmacol. Exp. Ther. 2000, 292, 1048–1052. [Google Scholar]
- Pardridge, W.M.; Eisenberg, J.; Yang, J. Human blood-brain barrier insulin receptor. J. Neurochem. 1985, 44, 1771–1778. [Google Scholar] [CrossRef]
- Frölich, L.; Blum-Degen, D.; Bernstein, H.G.; Engelsberger, S. Brain insulin and insulin receptors in aging and sporadic Alzheimer’s disease. J. Neural Transm. (Vienna) 1998, 105, 423–438. [Google Scholar] [CrossRef]
- Qiu, W.Q.; Walsh, D.M.; Ye, Z.; Vekrellis, K.; Zhang, J.; Podlisny, M.B.; Rosner, M.R.; Safavi, A.; Hersh, L.B.; Selkoe, D.J. Insulin-degrading enzyme regulates extracellular levels of amyloid beta-protein by degradation. J. Biol. Chem. 1998, 273, 32730–32738. [Google Scholar] [CrossRef] [PubMed]
- Shiiki, T.; Ohtsuki, S.; Kurihara, A.; Naganuma, H.; Nishimura, K.; Tachikawa, M.; Hosoya, K.; Terasaki, T. Brain insulin impairs amyloid-beta(1-40) clearance from the brain. J. Neurosci. 2004, 24, 9632–9637. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Helmerhorst, E.; Taddei, K.; Plewright, B. Alzheimer’s beta-amyloid peptides compete for insulin binding to the insulin receptor. J. Neurosci. 2002, 22, RC221. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M.; Kang, Y.S.; Buciak, J.L.; Yang, J. Human insulin receptor monoclonal antibody undergoes high affinity binding to human brain capillaries in vitro and rapid transcytosis through the blood-brain barrier in vivo in the primate. Pharm. Res. 1995, 12, 807–816. [Google Scholar] [CrossRef] [PubMed]
- Coloma, M.J.; Lee, H.J.; Kurihara, A.; Landaw, E.M.; Boado, R.J.; Morrison, S.L.; Pardridge, W.M. Transport across the primate blood-brain barrier of a genetically engineered chimeric monoclonal antibody to the human insulin receptor. Pharm. Res. 2000, 17, 266–274. [Google Scholar] [CrossRef] [PubMed]
- Hwang, W.Y.; Foote, J. Immunogenicity of engineered antibodies. Methods 2005, 36, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Boado, R.J.; Zhang, Y.F.; Zhang, Y.; Pardridge, W.M. Humanization of anti-human insulin receptor antibody for drug targeting across the human blood-brain barrier. Biotechnol. Bioeng. 2007, 96, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Boado, R.J.; Zhang, Y.; Zhang, Y.; Xia, C.F.; Pardridge, W.M. Fusion antibody for Alzheimer’s disease with bidirectional transport across the blood-brain barrier and abeta fibril disaggregation. Bioconjug. Chem. 2007, 18, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Boado, R.J.; Lu, J.Z.; Hui, E.K.-W.; Pardridge, W.M. IgG-Single Chain Fv Fusion Protein Therapeutic for Alzheimer’s Disease: Expression in CHO Cells and Pharmacokinetics and Brain Delivery in the Rhesus Monkey. Biotechnol. Bioeng. 2010, 105, 627–635. [Google Scholar] [CrossRef]
- Ohshima-Hosoyama, S.; Simmons, H.A.; Goecks, N.; Joers, V.; Swanson, C.R.; Bondarenko, V.; Velotta, R.; Brunner, K.; Wood, L.D.; Hruban, R.H.; et al. A monoclonal antibody-GDNF fusion protein is not neuroprotective and is associated with proliferative pancreatic lesions in parkinsonian monkeys. PLoS ONE 2012, 7, e39036. [Google Scholar] [CrossRef]
- Holtzman, D.M.; Herz, J.; Bu, G. Apolipoprotein E and Apolipoprotein E Receptors: Normal Biology and Roles in Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006312. [Google Scholar] [CrossRef] [PubMed]
- Bu, G.; Maksymovitch, E.A.; Nerbonne, J.M.; Schwartz, A.L. Expression and function of the low density lipoprotein receptor-related protein (LRP) in mammalian central neurons. J. Biol. Chem. 1994, 269, 18521–18528. [Google Scholar] [PubMed]
- Gaillard, P.J.; Visser, C.C.; de Boer, A.G. Targeted delivery across the blood-brain barrier. Expert Opin. Drug Deliv. 2005, 2, 299–309. [Google Scholar] [CrossRef]
- Harris-White, M.E.; Frautschy, S.A. Low density lipoprotein receptor-related proteins (LRPs), Alzheimer’s and cognition. Curr. Drug Targets CNS Neurol. Disord. 2005, 4, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Hughes, S.R.; Khorkova, O.; Goyal, S.; Knaeblein, J.; Heroux, J.; Riedel, N.G.; Sahasrabudhe, S. Alpha2-macroglobulin associates with beta-amyloid peptide and prevents fibril formation. Proc. Natl. Acad. Sci. USA 1998, 95, 3275–3280. [Google Scholar] [CrossRef]
- Narita, M.; Holtzman, D.M.; Schwartz, A.L.; Bu, G. Alpha2-macroglobulin complexes with and mediates the endocytosis of beta-amyloid peptide via cell surface low-density lipoprotein receptor-related protein. J. Neurochem. 1997, 69, 1904–1911. [Google Scholar] [CrossRef] [PubMed]
- Blacker, D.; Wilcox, M.A.; Laird, N.M.; Rodes, L.; Horvath, S.M. Alpha-2 macroglobulin is genetically associated with Alzheimer disease. Nat. Genet. 1998, 19, 357–360. [Google Scholar] [CrossRef]
- Actor, J.K.; Hwang, S.-A.; Kruzel, M.L. Lactoferrin as a Natural Immune Modulator. Curr. Pharm. Des. 2009, 15, 1956–1973. [Google Scholar] [CrossRef]
- Chen, H.; Tang, L.; Qin, Y.; Yin, Y.; Tang, J.; Tang, W.; Sun, X.; Zhang, Z.; Liu, J.; He, Q. Lactoferrin-modified procationic liposomes as a novel drug carrier for brain delivery. Eur. J. Pharm. Sci. 2010, 40, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.Q.; Ke, W.L.; Qu, Y.H.; Zhu, J.H.; Pei, Y.Y.; Jiang, C. Characterization of lactoferrin receptor in brain endothelial capillary cells and mouse brain. J. Biomed. Sci. 2007, 14, 121–128. [Google Scholar] [CrossRef]
- Kawamata, T.; Tooyama, I.; Yamada, T.; Walker, D.G.; McGeer, P.L. Lactotransferrin immunocytochemistry in Alzheimer and normal human brain. Am. J. Pathol. 1993, 142, 1574–1585. [Google Scholar]
- Kennard, M.L.; Feldman, H.; Yamada, T.; Jefferies, W.A. Serum levels of the iron binding protein p97 are elevated in Alzheimer’s disease. Nat. Med. 1996, 2, 1230–1235. [Google Scholar] [CrossRef] [PubMed]
- Demeule, M.; Poirier, J.; Jodoin, J.; Bertrand, Y.; Desrosiers, R.R. High transcytosis of melanotransferrin (P97) across the blood-brain barrier. J. Neurochem. 2002, 83, 924–933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richardson, D.R.; Morgan, E.H. Development of a potential protein vector (NeuroTrans) to deliver drugs across the blood–brain barrier. Int. Congr. Ser. 2004, 1277, 171–184. [Google Scholar] [CrossRef]
- Pan, W.; Kastin, A.J.; Zankel, T.C.; van Kerkhof, P.; Terasaki, T.; Bu, G. Efficient transfer of receptor-associated protein (RAP) across the blood-brain barrier. J. Cell Sci. 2004, 117, 5071–5078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, M.; Ku, T.; Chong, K.; Yoo, M.J.; Choi, C. Minimally invasive molecular delivery into the brain using optical modulation of vascular permeability. Proc. Natl. Acad. Sci. USA 2011, 108, 9256–9261. [Google Scholar] [CrossRef] [Green Version]
- Raliya, R.; Saha, D.; Chadha, T.S.; Raman, B.; Biswas, P. Non-invasive aerosol delivery and transport of gold nanoparticles to the brain. Sci. Rep. 2017, 7, 44718. [Google Scholar] [CrossRef] [Green Version]
- Hanson, L.R.; Fine, J.M.; Svitak, A.L.; Faltesek, K.A. Intranasal Administration of CNS Therapeutics to Awake Mice. J. Vis. Exp. 2013, 4440. [Google Scholar] [CrossRef]
- Etame, A.B.; Diaz, R.J.; Smith, C.A.; Mainprize, T.G.; Hynynen, K.; Rutka, J.T. Focused ultrasound disruption of the blood-brain barrier: A new frontier for therapeutic delivery in molecular neurooncology. Neurosurg. Focus 2012, 32, E3. [Google Scholar] [CrossRef]
- Lipsman, N.; Meng, Y.; Bethune, A.J.; Huang, Y.; Lam, B.; Masellis, M.; Herrmann, N.; Heyn, C.; Aubert, I.; Boutet, A.; et al. Blood-brain barrier opeining in Alzheimer’s disease using MR-guided focused ultrasound. Nat. Commun. 2018, 9, 2336. [Google Scholar] [CrossRef]
- Gosselet, F.; Saint-Pol, J.; Candela, P.; Fenart, L. Amyloid-β peptides, Alzheimer’s disease and the blood-brain barrier. Curr. Alzheimer Res. 2013, 10, 1015–1033. [Google Scholar] [CrossRef] [PubMed]
- Suri, S.S.; Fenniri, H.; Singh, B. Nanotechnology-based drug delivery systems. J. Occup. Med. Toxicol. 2007, 2, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, I.; Saeed, K.; Khan, I. Nanoparticles: Properties, applications and toxicities. Arab. J. Chem. 2017. [Google Scholar] [CrossRef]
- Makadia, H.K.; Siegel, S.J. Poly Lactic-co-Glycolic Acid (PLGA) as Biodegradable Controlled Drug Delivery Carrier. Polymers 2011, 3, 1377–1397. [Google Scholar] [CrossRef] [PubMed]
- Sathya, S.; Shanmuganathan, B.; Saranya, S.; Vaidevi, S.; Ruckmani, K.; Pandima, D.K. Phytol-loaded PLGA nanoparticle as a modulator of Alzheimer’s toxic Aβ peptide aggregation and fibrillation associated with impaired neuronal cell function. Artif. Cellsnanomed. Biotechnol. 2017, 25, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Fornaguera, C.; Feiner-Gracia, N.; Calderó, G.; García-Celma, M.J.; Solans, C. Galantamine-loaded PLGA nanoparticles, from nano-emulsion templating, as novel advanced drug delivery systems to treat neurodegenerative diseases. Nanoscale 2015, 7, 12076–12084. [Google Scholar] [CrossRef] [PubMed]
- Mathew, A.; Fukuda, T.; Nagaoka, Y.; Hasumura, T.; Morimoto, H.; Yoshida, Y.; Maekawa, T.; Venugopal, K.; Kumar, D.S. Curcumin Loaded-PLGA Nanoparticles Conjugated with Tet-1 Peptide for Potential Use in Alzheimer’s Disease. PLoS ONE 2012, 7, e32616. [Google Scholar] [CrossRef] [PubMed]
- Anand, P.; Nair, H.B.; Sung, B.; Kunnumakkara, A.B.; Yadav, V.R.; Tekmal, R.R.; Aggarwal, B.B. Design of Curcumin Loaded PLGA Nanoparticles Formulation with Enhanced Cellular Uptake, and Increased Bioactivity in vitro and Superior Bioavailability in vivo. Biochem. Pharmacol. 2010, 79, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, S.K.; Agarwal, S.; Seth, B.; Yadav, A.; Nair, S.; Bhatnagar, P. Curcumin-loaded nanoparticles potently induce adult neurogenesis and reverse cognitive deficits in Alzheimer’s disease model via canonical Wnt/β-catenin pathway. ACS Nano 2014, 8, 76–103. [Google Scholar] [CrossRef] [PubMed]
- Huang, N.; Lu, S.; Liu, X.-G.; Zhu, J.; Wang, Y.-J.; Liu, R.-T. PLGA nanoparticles modified with a BBB-penetrating peptide co-delivering Aβ generation inhibitor and curcumin attenuate memory deficits and neuropathology in Alzheimer’s disease mice. Oncotarget 2017, 8, 81001–81013. [Google Scholar] [CrossRef]
- Sánchez-López, E.; Ettcheto, M.; Egea, M.A.; Espina, M.; Calpena, A.C. New potential strategies for Alzheimer’s disease prevention: Pegylated biodegradable dexibuprofen nanospheres administration to APPswe/PS1dE9. Nanomedicine 2017, 13, 1171–1182. [Google Scholar] [CrossRef] [PubMed]
- Carradori, D.; Balducci, C.; Re, F.; Brambilla, D. Antibody-functionalized polymer nanoparticle leading to memory recovery in Alzheimer’s disease-like transgenic mouse model. Nanomedicine 2018, 14, 609–618. [Google Scholar] [CrossRef]
- Akbarzadeh, A.; Rezaei-Sadabady, R.; Davaran, S. Liposome: Classification, preparation, and applications. Nanoscale Res. Lett. 2013, 8, 102. [Google Scholar] [CrossRef] [PubMed]
- Milla, P.; Dosio, F.; Cattel, L. PEGylation of proteins and liposomes: A powerful and flexible strategy to improve the drug delivery. Curr. Drug Metab. 2012, 13, 105–119. [Google Scholar] [CrossRef] [PubMed]
- Mutlu, N.B.; Değim, Z.; Yilmaz, S.; Eiz, D.; Nacar, A. New perspective for the treatment of Alzheimer diseases: Liposomal rivastigmine formulations. Drug Dev. Ind. Pharm. 2011, 37, 775–789. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Shao, X.; Zhang, C.; Tan, Y. Intranasal H102 Peptide-Loaded Liposomes for Brain Delivery to Treat Alzheimer’s Disease. Pharm. Res. 2015, 32, 3837–3849. [Google Scholar] [CrossRef]
- Chen, Z.L.; Huang, M.; Wang, X.R. Transferrin-modified liposome promotes α-mangostin to penetrate the blood-brain barrier. Nanomedicine 2016, 12, 421–430. [Google Scholar] [CrossRef]
- Kuo, Y.C.; Wang, C.T. Protection of SK-N-MC cells against β-amyloid peptide-induced degeneration using neuron growth factor-loaded liposomes with surface lactoferrin. Biomaterials 2014, 35, 5954–5964. [Google Scholar] [CrossRef]
- Kuo, Y.; Tsao, C. Neuroprotection against apoptosis of SK-N-MC cells using RMP-7- and lactoferrin-grafted liposomes carrying quercetin. Int. J. Nanomed. 2017, 12, 2857–2869. [Google Scholar] [CrossRef]
- Markoutsa, E.; Papadia, K.; Giannou, A.D. Mono and dually decorated nanoliposomes for brain targeting, in vitro and in vivo studies. Pharm. Res. 2014, 31, 1275–1289. [Google Scholar] [CrossRef]
- Balducci, C.; Mancini, S.; Minniti, S.; La Vitola, P.; Zotti, M.; Sancini, G.; Mauri, M.; Cagnotto, A.; Colombo, L.; Fiordaliso, F.; et al. Multifunctional Liposomes Reduce Brain β-Amyloid Burden and Ameliorate Memory Impairment in Alzheimer’s Disease Mouse Models. J. Neurosci. 2014, 34, 14022–14031. [Google Scholar] [CrossRef] [PubMed]
- Mourtas, S.; Lazar, A.N.; Markoutsa, E. Multifunctional nanoliposomes with curcumin-lipid derivative and brain targeting functionality with potential applications for Alzheimer disease. Eur. J. Med. Chem. 2014, 80, 175–183. [Google Scholar] [CrossRef]
- Shankar, P.D.; Shobana, S.; Karuppusamy, I. A review on the biosynthesis of metallic nanoparticles (gold and silver) using bio-components of microalgae: Formation mechanism and applications. Enzym. Microb. Technol. 2016, 95, 28–44. [Google Scholar] [CrossRef] [PubMed]
- Yuna, K.; Ji-Hyun, P.; Hyojin, L.; Jwa-Min, N. How Do the Size, Charge and Shape of Nanoparticles Affect Amyloid β Aggregation on Brain Lipid Bilayer? Sci. Rep. 2016, 6, 19548. [Google Scholar] [CrossRef]
- Praça, C.; Rai, A.; Santos, T.; Cristovão, A.C.; Pinho, S.L.; Cecchelli, R.; Dehouck, M.P.; Bernardino, L.; Ferreira, L.S. A nanoformulation for the preferential accumulation in adult neurogenic niches. J. Control Release 2018, 284, 57–72. [Google Scholar] [CrossRef] [PubMed]
- Nan, G.; Hanjun, S.; Kai, D.; Jinsong, R.; Xiaogang, Q. Gold-Nanoparticle-Based Multifunctional Amyloid-b Inhibitor against Alzheimer’s Disease. Chemistry 2015, 21, 829–835. [Google Scholar] [CrossRef]
- Martin Del Valle, E.M. Cyclodextrins and their uses: A review. Process Biochem. 2004, 39, 1033–1046. [Google Scholar] [CrossRef]
- Coisne, C.; Tilloy, S.; Monflier, E.; Wils, D.; Fenart, L.; Gosselet, F. Cyclodextrins as Emerging Therapeutic Tools in the Treatment of Cholesterol-Associated Vascular and Neurodegenerative Diseases. Molecules 2016, 21, 1748. [Google Scholar] [CrossRef]
- Yao, J.; Ho, D.; Calingasan, N.Y.; Pipalia, N.H.; Lin, M.T.; Bea, M.F. Neuroprotection by cyclodextrin in cell and mouse models of Alzheimer disease. J. Exp. Med. 2012, 209, 2501–2513. [Google Scholar] [CrossRef] [Green Version]
- Vecsernyés, M.; Fenyvesi, F.; Bácskay, I.; Deli, M.A.; Szente, L.; Fenyvesi, É. Cyclodextrins, blood-brain barrier, and treatment of neurological diseases. Arch. Med. Res. 2014, 45, 711–729. [Google Scholar] [CrossRef]
- Tilloy, S.; Monnaert, V.; Fenart, L. Methylated beta-cyclodextrin as P-gp modulators for deliverance of doxorubicin across an in vitro model of blood-brain barrier. Bioorg. Med. Chem. Lett. 2006, 16, 2154–2157. [Google Scholar] [CrossRef] [PubMed]
- Gil, E.S.; Wu, L.; Xu, L.; Lowe, T.L. β-cyclodextrin-poly(β-amino ester) nanoparticles for sustained drug delivery across the blood-brain barrier. Biomacromolecules 2012, 13, 3533–3541. [Google Scholar] [CrossRef] [PubMed]
- Helms, H.C.C.; Abbott, N.J.; Burek, M.; Cecchelli, R.; Couraud, P.-O.; A Deli, M.; Förster, C.; Galla, H.J.; A Romero, I.; Shusta, E.V.; et al. In vitro models of the blood-brain barrier: An overview of commonly used brain endothelial cell culture models and guidelines for their use. J. Cereb. Blood Flow Metab. 2016, 36, 862–890. [Google Scholar] [CrossRef] [PubMed]
- Shawahna, R.; Decleves, X.; Scherrmann, J.M. Hurdles with using in vitro models to predict human blood-brain barrier drug permeability: A special focus on transporters and metabolizing enzymes. Curr. Drug Metab. 2013, 14, 120–136. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, Q.; Wang, X. Drug-in-cyclodextrin-in-liposomes: A novel drug delivery system for flurbiprofen. Int. J. Pharm. 2015, 492, 40–45. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Component | Features in AD | Ref. |
|---|---|---|
| Capillaries | Total length is shorter. | [36] |
| GLUT1 | Downregulate, result in reduction of Aβ clearance | [51] |
| Transferrin receptor | Number of receptors in hippocampus are less than normal. | [70] |
| Insulin receptor | Brain insulin receptor density decreases with aging. | [80] |
| Lactoferrin | Expression is upregulated. | [101] |
| Melanotransferrin | [102] |
| Targeting Vectors | Pathway | Features | Ref. |
|---|---|---|---|
| Syn-B vectors | Adsorptive-mediated transcytosis | Cross the BBB without compromising the BBB integrity | [55,56,57] |
| TAT-derived peptides | Penetrating ability is related to the arginine residues in TAT. Non-specific transduction, induce Aβ deposition, tau phosphorylation and subsequent neuronal death in AD development | [60,61,62,63,64,65] | |
| Polyarginines | Highly hydrophilic and cationic nature is responsible for charge repulsion which makes endocytosis possible. | [66] | |
| Transferrin | Receptor-mediated transcytosis (Transferrin receptor) | Compete with endogenous transferrin in the blood, affect cellular uptake of iron by the brain | [71,72] |
| OX 26 | Bind at extracellular domain without affecting transferrin binding, brain targeting effect is species-specific. | [73,74,75,76,78] | |
| MAb 8D3 | |||
| RI7217 | |||
| Insulin | Receptor-mediated transcytosis (Insulin receptor) | Affect the clearance of Aβ and result in higher level of extracellular Aβ, short serum half-life, disturb insulin metabolism | [53,80,81,82,83] |
| MAb83-14 | Species-specific. Only transport across the BBB in Old-World primates | [84] | |
| HIRMAb | Can be evaluated in animal model and humans | [85,86,87] | |
| IgG | Recognize both human transferrin and insulin receptors with ability to penetrate the BBB | [89] | |
| ApoE | Receptor-mediated transcytosis (Low density lipoprotein receptor) | APOE4 allele is a genetic risk factor for late-onset AD, ApoE can regulate the integrity of tight junctions | [31,32] |
| Lactoferrin | Expression is greatly upregulated in both neurons and glia in AD. | [98,99,100,101] | |
| Melanotransferrin | Stronger BBB-penetrating ability than lactotransferrin in bovine, not change integrity of the brain capillary endothelial cell monolayer | [102,103] |
| Encapsulated Agents | Carrier Composition | Targeting Vectors | Elevated Model | Therapeutic Effects | Ref. |
|---|---|---|---|---|---|
| Phytol | PLGA, PVA | -- | Neuro2a cells, without in vivo data | Sustained Release, show anti-amyloid activity, show neuron protective effect | [115] |
| Galantamine | Polysorbate 80, PLGA | -- | HeLa cells, SH-SY5Y cells, without in vivo data | Sustained release, show AchE inhibition ability | [116] |
| Curcumin | PLGA, PVA | Tet-1 peptide | GI-1 glioma cells | Show anti-amyloid activity, show neuronal targeting effect | [117] |
| Curcumin | PLGA-PEG-5000 | -- | Mice | Increase drug serum level, longer half-life | [118] |
| Curcumin | PLGA, PVA | -- | Neural stem cells, neurospheres, rats | Internalized by cells in vitro, cross the BBB in vivo, improve memory and cognitive ability, inhibit Aβ-induced neurodegeneration | [119] |
| Curcumin | PLGA-PEG-3400 | CRT peptide and S1 inhibitor | bEnd3 cells, SH-SY5Y cells, BV2 cells, mice | Nontoxic to neuron cells, decrease Aβ burden, gliosis and inflammation in vivo, improve spatial memory and recognition | [120] |
| Dexibuprofen | PLGA-PEG, PVA | -- | PC12 cells, bEnd3 cells, glial cells, APPswe/PS1dE9 mice | Nontoxic to cells in vitro, increase the BBB permeation coefficient, reduce memory impairment | [121] |
| -- | P(HDCA-co-RCA-co-MePEGCA), MePEGCA-co-Bio-PEGCA-co-HDCA | Anti-Aβ1–42 MAb | Tg2576 mice | Reduce triton-soluble Aβ peptides and oligomers levels in the brain | [122] |
| Encapsulated Agents | Carrier Composition | Targeting Vectors | Elevated Model | Therapeutic Effects | Ref. |
|---|---|---|---|---|---|
| Rivastigmine | Cholesterol, DPPC, Methyl cellulose, dimethyl-β-CD, sodium taurocholate | -- | Balb-C type mice | Increase amount of drug delivered into the brain | [125] |
| H102 peptide | EPC, DSPE-PEG2000 and cholesterol | -- | SD rat | Enhance peptide stability, increase amount of peptide delivered into the brain, improve spatial memory impairment, increase activities of ChAT and IDE | [126] |
| α-Mangostin | DSPC, cholesterol, DSPE-PEG2000, DSPE-PEG2000-COOH | Transferrin | bEnd3 cells, astrocytes, SD rat | Penetrate in vitro BBB model without destroying the structure of liposomes, improve bioavailability of drug and increase amount of drug in brain | [127] |
| -- | CHETA, DDAB, DOPE, PC | Lactoferrin | BCE cells, astrocytes, Kunming Mice | Enhance the uptake of Lf-procationic liposomes | [99] |
| NGF | DPPC, DSPE-PEG2000, DSPE-PEG2000-CA | Lactoferrin | HBME cells, human astrocytes, SK-N-MC cells, without in vivo data | Accelerate drug delivery across the BBB model, prevent Aβ-induced neurotoxicity | [128] |
| Quercetin | DPPC, cardiolipin, DSPE-PEG2000-CA, SPC, stearylamine, cholesterol | Lactoferrin, RMP-7 | HBME cells, SK-N-MC cells, human astrocytes, without in vivo data | Slightly enhance paracellular drug delivery, protect neurodegeneration caused by Aβ-induced neurotoxicity | [129] |
| -- | DSPC, DSPE-PEG2000, DSPE-PEG-Mal, DSPE-PEG2000-biotin | OX 26/RI7217/ApoE3/OX26 + ApoE3/RI7217 + ApoE3 | hCMEC/D3 cells, FVB Mice | Cellular uptake of dual functionalized-liposomes was nearly twice as compared to mono-functionalized liposomes. In vivo results did not comply with in vitro results as ApoE peptide was inactivated by serum proteins. | [130] |
| -- | Sphingomyelin and cholesterol | Phosphatidic acid, mApoE | APP/presenilin 1 mice | Decrease total Aβ fibrils and oligomers in brain, slow neurodegeneration | [131] |
| -- | DSPC, DSPE-PEG2000 and cholesterol | Lipid-PEG-curcumin derivative, OX26, RI1227 | Brain from AD patient | Able to bind amyloid deposits | [132] |
| Encapsulated Agents | Carrier Composition | Targeting Vectors | Elevated Model | Therapeutic Effects | Ref. |
|---|---|---|---|---|---|
| -- | AuNPs | -- | -- | Large AuNPs induce amorphous aggregates on the brain lipid bilayer. Smaller AuNPs induce protofibrillar Aβ structures. | [134] |
| -- | Amine-modified AuNPs, Citrate-modified AuNPs | -- | -- | Positively charged AuNPs attached to Aβ more tightly. | [134] |
| -- | AuNRs, AuNCs | -- | -- | Aβ was preferentially bound to the long axis of AuNRs and fewer fibrils were formed. All the facets of AuNCs interacted with Aβ to produce the fibril networks. | [134] |
| -- | AuNPs, AuNRs | Transferrin | CD34+-derived ECs with bovine pericytes, C57BL/6 inbred strain mice | Cross the BBB both in vitro and vivo. With the use of NIR, irradiation, formulations could preferentially accumulate in the neurogenic niches. | [135] |
| -- | AuNPs | POMD, LPFFD peptide | PC12 cells, S4880202 normal mice | Synergistic effects in inhibiting Aβ activity and Aβ-induced cytotoxicity in vitro, penetrate the BBB in vivo | [136] |
| HP-β-CD | HP-β-CD | -- | SwN2a cells, Tg19959 mice | Reduce the levels of Aβ42 and membrane cholesterol in vitro, improve spatial learning and memory, reduce Aβ plaque deposition and tau immunoreactive dystrophic neurites in vivo | [139] |
| Doxorubicin | Rame-β-CD or crysme- β-CD | -- | BCE cells | Increase the transport of doxorubicin, modulate P-gp activity | [141] |
| Doxorubicin | β-CD, poly(β-amino ester) | -- | BME cells | High permeability across the in vitro BBB models | [142] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wong, K.H.; Riaz, M.K.; Xie, Y.; Zhang, X.; Liu, Q.; Chen, H.; Bian, Z.; Chen, X.; Lu, A.; Yang, Z. Review of Current Strategies for Delivering Alzheimer’s Disease Drugs across the Blood-Brain Barrier. Int. J. Mol. Sci. 2019, 20, 381. https://doi.org/10.3390/ijms20020381
Wong KH, Riaz MK, Xie Y, Zhang X, Liu Q, Chen H, Bian Z, Chen X, Lu A, Yang Z. Review of Current Strategies for Delivering Alzheimer’s Disease Drugs across the Blood-Brain Barrier. International Journal of Molecular Sciences. 2019; 20(2):381. https://doi.org/10.3390/ijms20020381
Chicago/Turabian StyleWong, Ka Hong, Muhammad Kashif Riaz, Yuning Xie, Xue Zhang, Qiang Liu, Huoji Chen, Zhaoxiang Bian, Xiaoyu Chen, Aiping Lu, and Zhijun Yang. 2019. "Review of Current Strategies for Delivering Alzheimer’s Disease Drugs across the Blood-Brain Barrier" International Journal of Molecular Sciences 20, no. 2: 381. https://doi.org/10.3390/ijms20020381
APA StyleWong, K. H., Riaz, M. K., Xie, Y., Zhang, X., Liu, Q., Chen, H., Bian, Z., Chen, X., Lu, A., & Yang, Z. (2019). Review of Current Strategies for Delivering Alzheimer’s Disease Drugs across the Blood-Brain Barrier. International Journal of Molecular Sciences, 20(2), 381. https://doi.org/10.3390/ijms20020381