The Role of O-GlcNAcylation for Protection against Ischemia-Reperfusion Injury

 ,
, {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
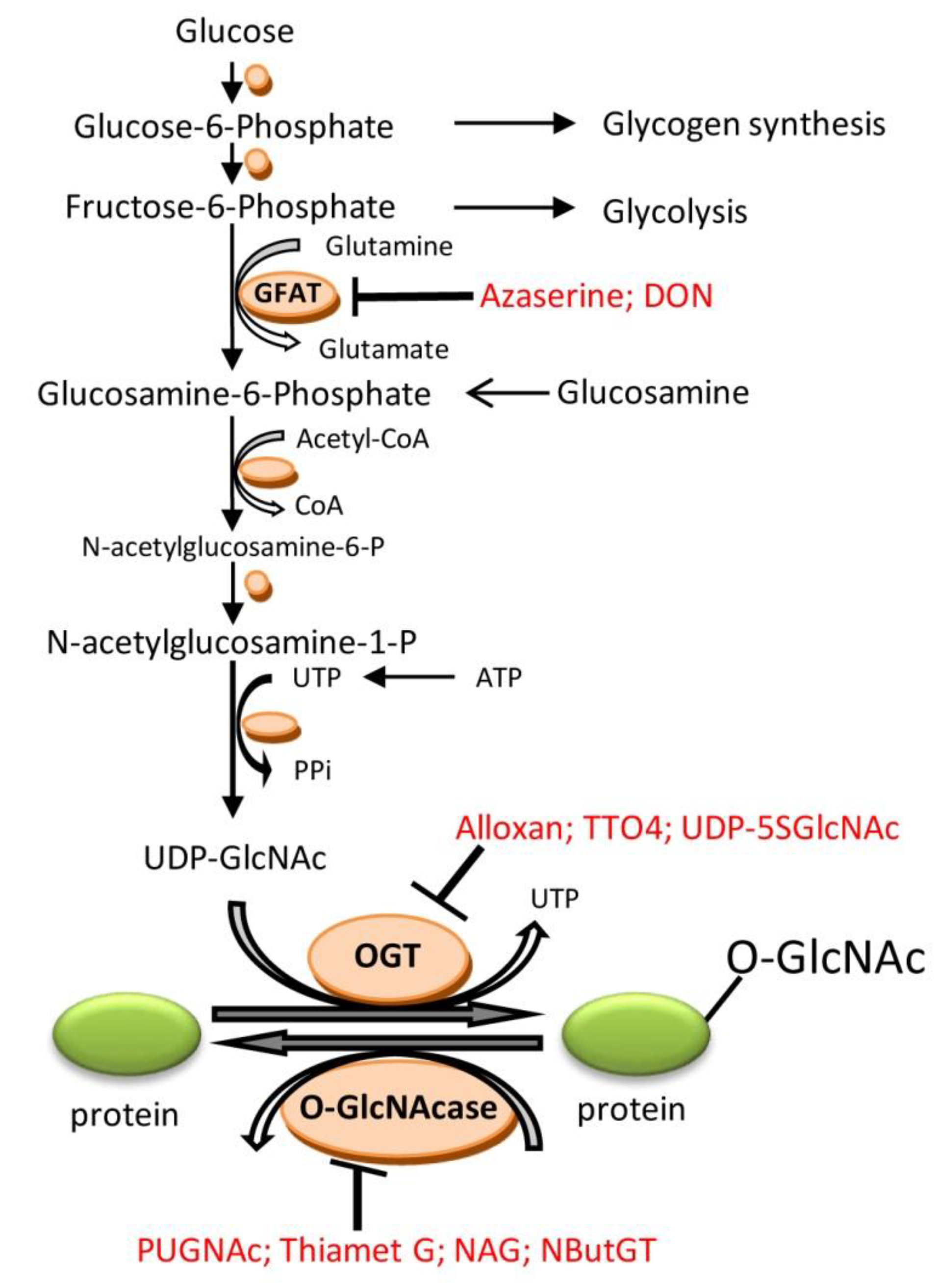
2. O-Linked β-N-Acetylglucosamine (O-GlcNAc)
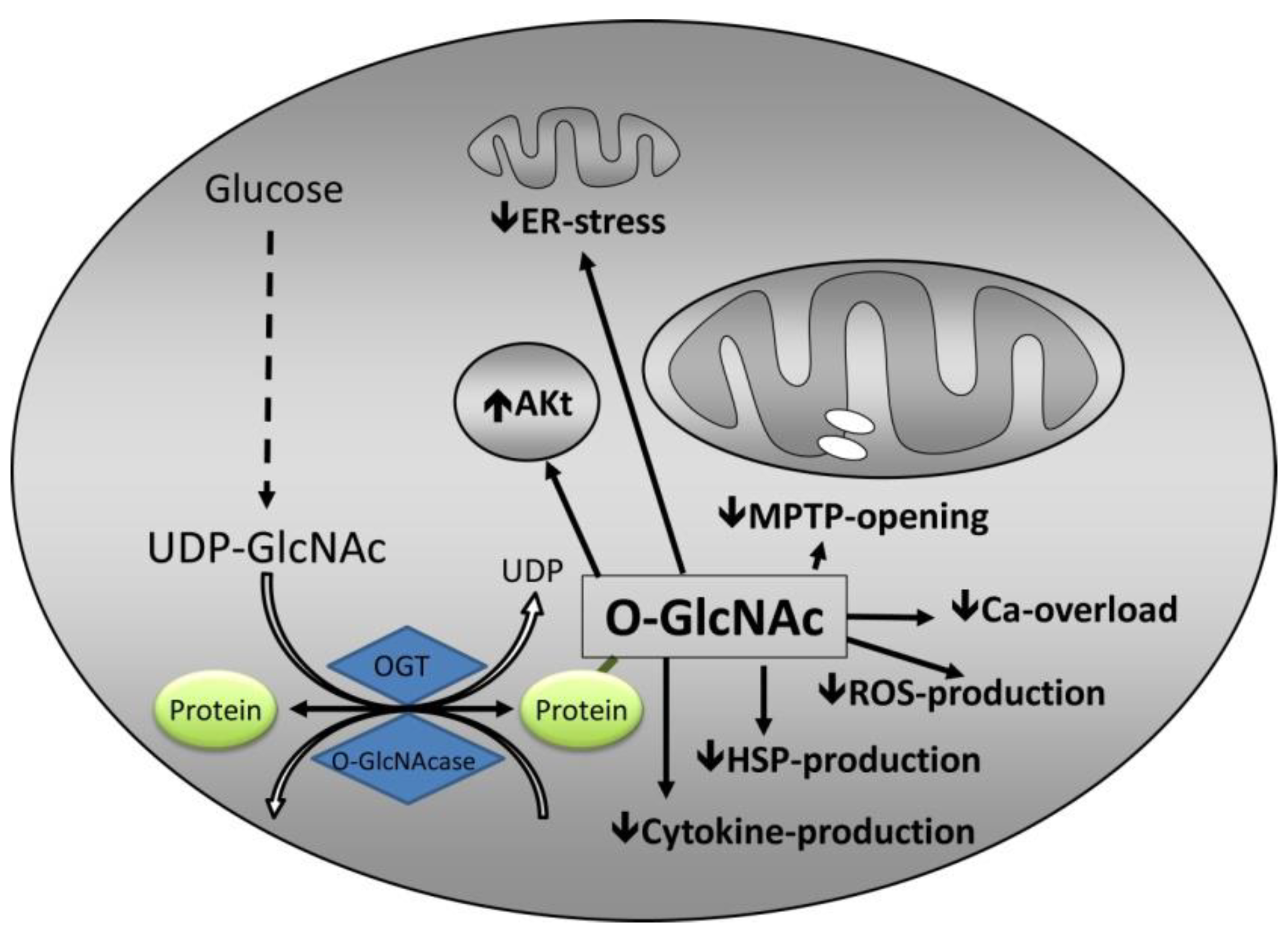
3. Mechanisms by Which O-GlcNAc Confers Protection
3.1. Calcium Overload
3.2. mPTP Opening
3.3. Endoplasmic Reticulum Stress
3.4. Inflammation
3.5. Heat Shock Proteins
3.6. Interaction with Established Cardioprotective Pathways
3.7. Impact of O-GlcNAcylation on Cardiac Function
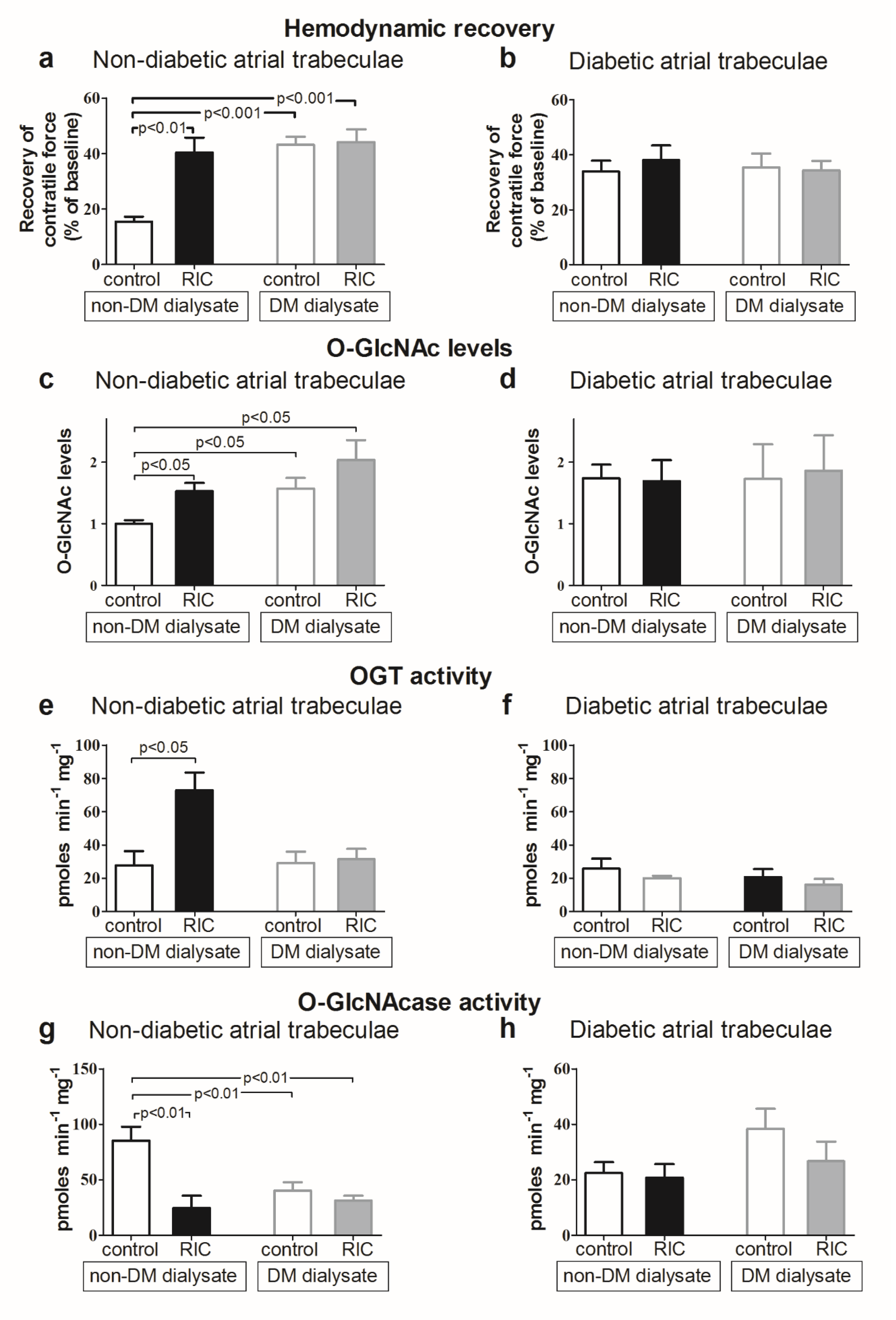
3.8. Impact of Diabetes Mellitus on O-GlcNAcylation and Cardioprotective Efficacy
3.9. Pharmacological Modulation of O-GlcNAcylation and Role for Cardioprotection
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ANT | Adenine nucleotide translocase |
| CHD | Coronary heart disease |
| CHOP | CCAAT-enhancer-binding protein homologous protein |
| DON | 6-diazo-5-oxo-norleucine |
| ER | Endoplasmic reticulum |
| GFAT | l-glutamine-d-fructose-6-phosphate amidotransferase |
| GIK | Glucose-insulin-potassium |
| HBP | Hexosamine biosynthetic pathway |
| HSP | Heat shock proteins |
| IL-6 | Interleurkin-6 |
| IPC | Ischemic preconditioning |
| IR | Ischemia-reperfusion |
| mPTP | Mitochondrial permeability transition pore |
| NAG | 1,2-dideoxy-2′-methyl-α-d-glucopyranoso-[2,1-d]-δ2′-thiazoline |
| NButGT | 1,2-dideoxy-2′-propyl-α-d-glucopyranoso-[2,1-d]-δ2′-thiazoline |
| NF-κB | Nuclear factor kappa-B |
| NO | Nitric oxide |
| O-GlcNAc | O-linked β-N-acetylglucosamine |
| OGT | Uridine-diphospho-N-acetylglucosamine:polypetptide-N-acetylglycosaminyltransferase |
| O-GlcNAcase | β-N-hexoamininidase |
| PUGNAc | O-(2-acetamido-2-deoxy-d-glucopyranosylidene)amino-N-phenylcarbamate |
| RISK | Reperfusion Injury Salvage Kinase pathway |
| RIC | Remote ischemic conditioining |
| SAFE | Survival activating factor enhancement |
| STEMI | ST-elevation myocardial infarction |
| TNF-α | Tumor necrosis factor-α |
| TTO4 | 2[(4-chlorophenyl)imino]tetrahydro-4-oxo-3-[2-tricyclo(3.3.1.13.7)dec-1-ylethel] |
| UDP-5SGlcNAc | Uridine diphospho-5-thio-N-acetylglucosamine |
| UDP-GlcNAc | Uridine-diphosphate-N-acetylglycosamine |
| VDAC | Voltage-dependent anion channel |
References
- Burns, R.J.; Gibbons, R.J.; Yi, Q.; Roberts, R.S.; Miller, T.D.; Schaer, G.L.; Anderson, J.L.; Yusuf, S. The relationships of left ventricular ejection fraction, end-systolic volume index and infarct size to six-month mortality after hospital discharge following myocardial infarction treated by thrombolysis. J. Am. Coll. Cardiol. 2002, 39, 30–36. [Google Scholar] [CrossRef] [Green Version]
- Lassen, J.F.; Botker, H.E.; Terkelsen, C.J. Timely and optimal treatment of patients with STEMI. Nat. Rev. Cardiol. 2013, 10, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Andersen, H.R.; Nielsen, T.T.; Rasmussen, K.; Thuesen, L.; Kelbaek, H.; Thayssen, P.; Abildgaard, U.; Pedersen, F.; Madsen, J.K.; Grande, P.; et al. A comparison of coronary angioplasty with fibrinolytic therapy in acute myocardial infarction. N. Engl. J. Med. 2003, 349, 733–742. [Google Scholar] [CrossRef] [PubMed]
- Keeley, E.C.; Boura, J.A.; Grines, C.L. Primary angioplasty versus intravenous thrombolytic therapy for acute myocardial infarction: A quantitative review of 23 randomised trials. Lancet 2003, 361, 13–20. [Google Scholar] [CrossRef]
- World Health Organisation. Available online: http://www.who.int/mediacentre/factsheets/fs317/en/ (accessed on 1 November 2018).
- Yellon, D.M.; Hausenloy, D.J. Myocardial reperfusion injury. N. Engl. J. Med. 2007, 357, 1121–1135. [Google Scholar] [CrossRef]
- Murry, C.E.; Jennings, R.B.; Reimer, K.A. Preconditioning with ischemia: A delay of lethal cell injury in ischemic myocardium. Circulation 1986, 74, 1124–1136. [Google Scholar] [CrossRef] [PubMed]
- Kharbanda, R.K.; Mortensen, U.M.; White, P.A.; Kristiansen, S.B.; Schmidt, M.R.; Hoschtitzky, J.A.; Vogel, M.; Sorensen, K.; Redington, A.N.; MacAllister, R. Transient limb ischemia induces remote ischemic preconditioning in vivo. Circulation 2002, 106, 2881–2883. [Google Scholar] [CrossRef] [PubMed]
- Przyklenk, K.; Bauer, B.; Ovize, M.; Kloner, R.A.; Whittaker, P. Regional ischemic ‘preconditioning’ protects remote virgin myocardium from subsequent sustained coronary occlusion. Circulation 1993, 87, 893–899. [Google Scholar] [CrossRef] [PubMed]
- Botker, H.E.; Kharbanda, R.; Schmidt, M.R.; Bottcher, M.; Kaltoft, A.K.; Terkelsen, C.J.; Munk, K.; Andersen, N.H.; Hansen, T.M.; Trautner, S.; et al. Remote ischaemic conditioning before hospital admission, as a complement to angioplasty, and effect on myocardial salvage in patients with acute myocardial infarction: A randomised trial. Lancet 2010, 375, 727–734. [Google Scholar] [CrossRef]
- Cheung, M.M.; Kharbanda, R.K.; Konstantinov, I.E.; Shimizu, M.; Frndova, H.; Li, J.; Holtby, H.M.; Cox, P.N.; Smallhorn, J.F.; Van Arsdell, G.S.; et al. Randomized controlled trial of the effects of remote ischemic preconditioning on children undergoing cardiac surgery: First clinical application in humans. J. Am. Coll. Cardiol. 2006, 47, 2277–2282. [Google Scholar] [CrossRef] [PubMed]
- Hausenloy, D.J.; Mwamure, P.K.; Venugopal, V.; Harris, J.; Barnard, M.; Grundy, E.; Ashley, E.; Vichare, S.; Di, S.C.; Kolvekar, S.; et al. Effect of remote ischaemic preconditioning on myocardial injury in patients undergoing coronary artery bypass graft surgery: A randomised controlled trial. Lancet 2007, 370, 575–579. [Google Scholar] [CrossRef]
- Hougaard, K.D.; Hjort, N.; Zeidler, D.; Sorensen, L.; Norgaard, A.; Hansen, T.M.; von Weitzel-Mudersbach, P.; Simonsen, C.Z.; Damgaard, D.; Gottrup, H.; et al. Remote ischemic perconditioning as an adjunct therapy to thrombolysis in patients with acute ischemic stroke: A randomized trial. Stroke 2014, 45, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Zachara, N.E.; O’Donnell, N.; Cheung, W.D.; Mercer, J.J.; Marth, J.D.; Hart, G.W. Dynamic O-GlcNAc modification of nucleocytoplasmic proteins in response to stress. A survival response of mammalian cells. J. Biol. Chem. 2004, 279, 30133–30142. [Google Scholar] [CrossRef] [PubMed]
- Zachara, N.E.; Hart, G.W. Cell signaling, the essential role of O-GlcNAc! Biochim. Biophys. Acta 2006, 1761, 599–617. [Google Scholar] [CrossRef] [PubMed]
- Laczy, B.; Marsh, S.A.; Brocks, C.A.; Wittmann, I.; Chatham, J.C. Inhibition of O-GlcNAcase in perfused rat hearts by NAG-thiazolines at the time of reperfusion is cardioprotective in an O-GlcNAc-dependent manner. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H1715–H1727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Pang, Y.; Chang, T.; Bounelis, P.; Chatham, J.C.; Marchase, R.B. Increased hexosamine biosynthesis and protein O-GlcNAc levels associated with myocardial protection against calcium paradox and ischemia. J. Mol. Cell. Cardiol. 2006, 40, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Marchase, R.B.; Chatham, J.C. Glutamine-induced protection of isolated rat heart from ischemia/reperfusion injury is mediated via the hexosamine biosynthesis pathway and increased protein O-GlcNAc levels. J. Mol. Cell. Cardiol. 2007, 42, 177–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Marchase, R.B.; Chatham, J.C. Increased O-GlcNAc levels during reperfusion lead to improved functional recovery and reduced calpain proteolysis. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H1391–H1399. [Google Scholar] [CrossRef] [Green Version]
- Jones, S.P.; Zachara, N.E.; Ngoh, G.A.; Hill, B.G.; Teshima, Y.; Bhatnagar, A.; Hart, G.W.; Marban, E. Cardioprotection by N-acetylglucosamine linkage to cellular proteins. Circulation 2008, 117, 1172–1182. [Google Scholar] [CrossRef]
- Paelestik, K.B.; Jespersen, N.R.; Jensen, R.V.; Johnsen, J.; Botker, H.E.; Kristiansen, S.B. Effects of hypoglycemia on myocardial susceptibility to ischemia-reperfusion injury and preconditioning in hearts from rats with and without type 2 diabetes. Cardiovasc. Diabetol. 2017, 16, 148. [Google Scholar] [CrossRef]
- Vibjerg, J.R.; Johnsen, J.; Buus, K.S.; Zachara, N.E.; Botker, H.E. Ischemic preconditioning increases myocardial O-GlcNAc glycosylation. Scand. Cardiovasc. J. 2013, 47, 168–174. [Google Scholar]
- Jensen, R.V.; Zachara, N.E.; Nielsen, P.H.; Kimose, H.H.; Kristiansen, S.B.; Botker, H.E. Impact of O-GlcNAc on cardioprotection by remote ischaemic preconditioning in non-diabetic and diabetic patients. Cardiovasc. Res. 2013, 97, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Champattanachai, V.; Marchase, R.B.; Chatham, J.C. Glucosamine protects neonatal cardiomyocytes from ischemia-reperfusion injury via increased protein-associated O-GlcNAc. Am. J. Physiol. Cell Physiol. 2007, 292, C178–C187. [Google Scholar] [CrossRef]
- Champattanachai, V.; Marchase, R.B.; Chatham, J.C. Glucosamine protects neonatal cardiomyocytes from ischemia-reperfusion injury via increased protein O-GlcNAc and increased mitochondrial Bcl-2. Am. J. Physiol. Cell Physiol. 2008, 294, C1509–C1520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ngoh, G.A.; Facundo, H.T.; Hamid, T.; Dillmann, W.; Zachara, N.E.; Jones, S.P. Unique hexosaminidase reduces metabolic survival signal and sensitizes cardiac myocytes to hypoxia/reoxygenation injury. Circ. Res. 2009, 104, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Zafir, A.; Readnower, R.; Long, B.W.; McCracken, J.; Aird, A.; Alvarez, A.; Cummins, T.D.; Li, Q.; Hill, B.G.; Bhatnagar, A.; et al. Protein O-GlcNAcylation is a novel cytoprotective signal in cardiac stem cells. Stem Cells 2013, 31, 765–775. [Google Scholar] [CrossRef]
- Ngoh, G.A.; Watson, L.J.; Facundo, H.T.; Dillmann, W.; Jones, S.P. Non-canonical glycosyltransferase modulates post-hypoxic cardiac myocyte death and mitochondrial permeability transition. J. Mol. Cell. Cardiol. 2008, 45, 313–325. [Google Scholar] [CrossRef] [Green Version]
- Ngoh, G.A.; Hamid, T.; Prabhu, S.D.; Jones, S.P. O-GlcNAc signaling attenuates ER stress-induced cardiomyocyte death. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H1711–H1719. [Google Scholar] [CrossRef] [Green Version]
- Milewski, S. Glucosamine-6-phosphate synthase—The multi-facets enzyme. Biochim. Biophys. Acta 2002, 1597, 173–192. [Google Scholar] [CrossRef]
- Nagy, T.; Champattanachai, V.; Marchase, R.B.; Chatham, J.C. Glucosamine inhibits angiotensin II-induced cytoplasmic Ca2+ elevation in neonatal cardiomyocytes via protein-associated O-linked N-acetylglucosamine. Am. J. Physiol. Cell Physiol. 2006, 290, C57–C65. [Google Scholar] [CrossRef]
- Gloster, T.M.; Zandberg, W.F.; Heinonen, J.E.; Shen, D.L.; Deng, L.; Vocadlo, D.J. Hijacking a biosynthetic pathway yields a glycosyltransferase inhibitor within cells. Nat. Chem. Biol. 2011, 7, 174–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, T.N.; Alborn, W.E.; Knierman, M.D.; Konrad, R.J. Alloxan is an inhibitor of O-GlcNAc-selective N-acetyl-β-d-glucosaminidase. Biochem. Biophys. Res. Commun. 2006, 350, 1038–1043. [Google Scholar] [CrossRef] [PubMed]
- Slawson, C.; Zachara, N.E.; Vosseller, K.; Cheung, W.D.; Lane, M.D.; Hart, G.W. Perturbations in O-linked β-N-acetylglucosamine protein modification cause severe defects in mitotic progression and cytokinesis. J. Biol. Chem. 2005, 280, 32944–32956. [Google Scholar] [CrossRef] [PubMed]
- Han, I.; Kudlow, J.E. Reduced O glycosylation of Sp1 is associated with increased proteasome susceptibility. Mol. Cell. Biol. 1997, 17, 2550–2558. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Tjian, R. O-glycosylation of eukaryotic transcription factors: Implications for mechanisms of transcriptional regulation. Cell 1988, 55, 125–133. [Google Scholar] [CrossRef]
- Vosseller, K.; Sakabe, K.; Wells, L.; Hart, G.W. Diverse regulation of protein function by O-GlcNAc: A nuclear and cytoplasmic carbohydrate post-translational modification. Curr. Opin. Chem. Biol. 2002, 6, 851–857. [Google Scholar] [CrossRef]
- Zhu, Y.; Shan, X.; Yuzwa, S.A.; Vocadlo, D.J. The emerging link between O-GlcNAc and Alzheimer disease. J. Biol. Chem. 2014, 289, 34472–34481. [Google Scholar] [CrossRef]
- Mazars, R.; Gonzalez-de-Peredo, A.; Cayrol, C.; Lavigne, A.C.; Vogel, J.L.; Ortega, N.; Lacroix, C.; Gautier, V.; Huet, G.; Ray, A.; et al. The THAP-zinc finger protein THAP1 associates with coactivator HCF-1 and O-GlcNAc transferase: A link between DYT6 and DYT3 dystonias. J. Biol. Chem. 2010, 285, 13364–13371. [Google Scholar] [CrossRef]
- Slawson, C.; Hart, G.W. O-GlcNAc signalling: Implications for cancer cell biology. Nat. Rev. Cancer 2011, 11, 678–684. [Google Scholar] [CrossRef]
- Lunde, I.G.; Aronsen, J.M.; Kvaloy, H.; Qvigstad, E.; Sjaastad, I.; Tonnessen, T.; Christensen, G.; Gronning-Wang, L.M.; Carlson, C.R. Cardiac O-GlcNAc signaling is increased in hypertrophy and heart failure. Physiol. Genom. 2012, 44, 162–172. [Google Scholar] [CrossRef] [Green Version]
- Marshall, S.; Bacote, V.; Traxinger, R.R. Discovery of a metabolic pathway mediating glucose-induced desensitization of the glucose transport system. Role of hexosamine biosynthesis in the induction of insulin resistance. J. Biol. Chem. 1991, 266, 4706–4712. [Google Scholar] [PubMed]
- McClain, D.A.; Paterson, A.J.; Roos, M.D.; Wei, X.; Kudlow, J.E. Glucose and glucosamine regulate growth factor gene expression in vascular smooth muscle cells. Proc. Natl. Acad. Sci. USA 1992, 89, 8150–8154. [Google Scholar] [CrossRef] [PubMed]
- Fulop, N.; Mason, M.M.; Dutta, K.; Wang, P.; Davidoff, A.J.; Marchase, R.B.; Chatham, J.C. Impact of Type 2 diabetes and aging on cardiomyocyte function and O-linked N-acetylglucosamine levels in the heart. Am. J. Physiol. Cell Physiol. 2007, 292, C1370–C1378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, S.Y.; Shin, J.H.; Hwang, J.S.; Kim, S.Y.; Shin, J.A.; Oh, E.S.; Oh, S.; Kim, J.B.; Lee, J.K.; Han, I.O. Glucosamine exerts a neuroprotective effect via suppression of inflammation in rat brain ischemia/reperfusion injury. Glia 2010, 58, 1881–1892. [Google Scholar] [CrossRef] [PubMed]
- Kawabata, K.I.; Netticadan, T.; Osada, M.; Tamura, K.; Dhalla, N.S. Mechanisms of ischemic preconditioning effects on Ca(2+) paradox-induced changes in heart. Am. J. Physiol. Heart Circ. Physiol. 2000, 278, H1008–H1015. [Google Scholar] [CrossRef] [PubMed]
- Temsah, R.M.; Kawabata, K.; Chapman, D.; Dhalla, N.S. Preconditioning prevents alterations in cardiac SR gene expression due to ischemia-reperfusion. Am. J. Physiol. Heart Circ. Physiol. 2002, 282, H1461–H1466. [Google Scholar] [CrossRef]
- Zimmerman, A.N.; Hulsmann, W.C. Paradoxical influence of calcium ions on the permeability of the cell membranes of the isolated rat heart. Nature 1966, 211, 646–647. [Google Scholar] [CrossRef] [PubMed]
- Hulsmann, W.C. Morphological changes of heart muscle caused by successive perfusion with calcium-free and calcium-containing solutions (calcium paradox). Cardiovasc. Res. 2000, 45, 119–120, 122. [Google Scholar] [CrossRef]
- Ngoh, G.A.; Watson, L.J.; Facundo, H.T.; Jones, S.P. Augmented O-GlcNAc signaling attenuates oxidative stress and calcium overload in cardiomyocytes. Amino Acids 2011, 40, 895–911. [Google Scholar] [CrossRef] [PubMed]
- Rengifo, J.; Gibson, C.J.; Winkler, E.; Collin, T.; Ehrlich, B.E. Regulation of the inositol 1,4,5-trisphosphate receptor type I by O-GlcNAc glycosylation. J. Neurosci. 2007, 27, 13813–13821. [Google Scholar] [CrossRef]
- Cho, H.J.; Mook-Jung, I. O-GlcNAcylation regulates endoplasmic reticulum exit sites through Sec31A modification in conventional secretory pathway. FASEB J. 2018, 32, 4641–4657. [Google Scholar] [CrossRef] [PubMed]
- Baines, C.P. The mitochondrial permeability transition pore and ischemia-reperfusion injury. Basic Res. Cardiol. 2009, 104, 181–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halestrap, A.P. A pore way to die: The role of mitochondria in reperfusion injury and cardioprotection. Biochem. Soc. Trans. 2010, 38, 841–860. [Google Scholar] [CrossRef]
- Hausenloy, D.J.; Yellon, D.M. The mitochondrial permeability transition pore: Its fundamental role in mediating cell death during ischaemia and reperfusion. J. Mol. Cell. Cardiol. 2003, 35, 339–341. [Google Scholar] [CrossRef]
- Goldberg, H.; Whiteside, C.; Fantus, I.G. O-linked β-N-acetylglucosamine supports p38 MAPK activation by high glucose in glomerular mesangial cells. Am. J. Physiol. Endocrinol. Metab. 2011, 301, E713–E726. [Google Scholar] [CrossRef] [PubMed]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Lima, V.V.; Spitler, K.; Choi, H.; Webb, R.C.; Tostes, R.C. O-GlcNAcylation and oxidation of proteins: Is signalling in the cardiovascular system becoming sweeter? Clin. Sci. 2012, 123, 473–486. [Google Scholar] [CrossRef]
- Singh, L.P.; Cheng, D.W.; Kowluru, R.; Levi, E.; Jiang, Y. Hexosamine induction of oxidative stress, hypertrophy and laminin expression in renal mesangial cells: Effect of the anti-oxidant α-lipoic acid. Cell Biochem. Funct. 2007, 25, 537–550. [Google Scholar] [CrossRef] [PubMed]
- Hirose, K.; Tsutsumi, Y.M.; Tsutsumi, R.; Shono, M.; Katayama, E.; Kinoshita, M.; Tanaka, K.; Oshita, S. Role of the O-linked β-N-acetylglucosamine in the cardioprotection induced by isoflurane. Anesthesiology 2011, 115, 955–962. [Google Scholar] [CrossRef]
- Miura, T.; Tanno, M. The mPTP and its regulatory proteins: Final common targets of signalling pathways for protection against necrosis. Cardiovasc. Res. 2012, 94, 181–189. [Google Scholar] [CrossRef]
- Woodfield, K.; Ruck, A.; Brdiczka, D.; Halestrap, A.P. Direct demonstration of a specific interaction between cyclophilin-D and the adenine nucleotide translocase confirms their role in the mitochondrial permeability transition. Biochem. J. 1998, 336 Pt 2, 287–290. [Google Scholar] [CrossRef] [Green Version]
- Qi, X.; Vallentin, A.; Churchill, E.; Mochly-Rosen, D. deltaPKC participates in the endoplasmic reticulum stress-induced response in cultured cardiac myocytes and ischemic heart. J. Mol. Cell. Cardiol. 2007, 43, 420–428. [Google Scholar] [CrossRef]
- Teshima, Y.; Akao, M.; Jones, S.P.; Marban, E. Uncoupling protein-2 overexpression inhibits mitochondrial death pathway in cardiomyocytes. Circ. Res. 2003, 93, 192–200. [Google Scholar] [CrossRef]
- Kim, I.; Xu, W.; Reed, J.C. Cell death and endoplasmic reticulum stress: Disease relevance and therapeutic opportunities. Nat. Rev. Drug Discov. 2008, 7, 1013–1030. [Google Scholar] [CrossRef]
- Oyadomari, S.; Mori, M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004, 11, 381–389. [Google Scholar] [CrossRef]
- Nishitoh, H. CHOP is a multifunctional transcription factor in the ER stress response. J. Biochem. 2012, 151, 217–219. [Google Scholar] [CrossRef]
- Tabas, I.; Ron, D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 2011, 13, 184–190. [Google Scholar] [CrossRef] [Green Version]
- Suh, H.N.; Lee, Y.J.; Kim, M.O.; Ryu, J.M.; Han, H.J. Glucosamine-induced Sp1 O-GlcNAcylation ameliorates hypoxia-induced SGLT dysfunction in primary cultured renal proximal tubule cells. J. Cell. Physiol. 2014, 229, 1557–1568. [Google Scholar] [CrossRef]
- Jang, I.; Kim, H.B.; Seo, H.; Kim, J.Y.; Choi, H.; Yoo, J.S.; Kim, J.W.; Cho, J.W. O-GlcNAcylation of eIF2α regulates the phospho-eIF2α-mediated ER stress response. Biochim. Biophys. Acta 2015, 1853, 1860–1869. [Google Scholar] [CrossRef] [Green Version]
- Vinten-Johansen, J. Involvement of neutrophils in the pathogenesis of lethal myocardial reperfusion injury. Cardiovasc. Res. 2004, 61, 481–497. [Google Scholar] [CrossRef] [Green Version]
- Cooper, M.; Lindholm, P.; Pieper, G.; Seibel, R.; Moore, G.; Nakanishi, A.; Dembny, K.; Komorowski, R.; Johnson, C.; Adams, M.; et al. Myocardial nuclear factor-κB activity and nitric oxide production in rejecting cardiac allografts. Transplantation 1998, 66, 838–844. [Google Scholar] [CrossRef]
- Sawa, Y.; Morishita, R.; Suzuki, K.; Kagisaki, K.; Kaneda, Y.; Maeda, K.; Kadoba, K.; Matsuda, H. A novel strategy for myocardial protection using in vivo transfection of cis element ‘decoy’ against NFκB binding site: Evidence for a role of NFκB in ischemia-reperfusion injury. Circulation 1997, 96, II-4. [Google Scholar]
- Xuan, Y.T.; Tang, X.L.; Banerjee, S.; Takano, H.; Li, R.C.; Han, H.; Qiu, Y.; Li, J.J.; Bolli, R. Nuclear factor-κB plays an essential role in the late phase of ischemic preconditioning in conscious rabbits. Circ. Res. 1999, 84, 1095–1109. [Google Scholar] [CrossRef]
- Morgan, E.N.; Boyle, E.M., Jr.; Yun, W.; Griscavage-Ennis, J.M.; Farr, A.L.; Canty, T.G., Jr.; Pohlman, T.H.; Verrier, E.D. An essential role for NF-κB in the cardioadaptive response to ischemia. Ann. Thorac. Surg. 1999, 68, 377–382. [Google Scholar] [CrossRef]
- Shen, Y.; Qiu, T.; Liu, X.H.; Zhang, L.; Wang, Z.S.; Zhou, J.Q. Renal ischemia-reperfusion injury attenuated by splenic ischemic preconditioning. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 2134–2142. [Google Scholar]
- Xiao, Q.; Ye, Q.; Wang, W.; Xiao, J.; Fu, B.; Xia, Z.; Zhang, X.; Liu, Z.; Zeng, X. Mild hypothermia pretreatment protects against liver ischemia reperfusion injury via the PI3K/AKT/FOXO3a pathway. Mol. Med. Rep. 2017, 16, 7520–7526. [Google Scholar] [CrossRef] [Green Version]
- Ji, Y.Y.; Wang, Z.D.; Wang, S.F.; Wang, B.T.; Yang, Z.A.; Zhou, X.R.; Lei, N.N.; Yue, W.N. Ischemic preconditioning ameliorates intestinal injury induced by ischemia-reperfusion in rats. World J. Gastroenterol. 2015, 21, 8081–8088. [Google Scholar] [CrossRef]
- Gouze, J.N.; Bianchi, A.; Becuwe, P.; Dauca, M.; Netter, P.; Magdalou, J.; Terlain, B.; Bordji, K. Glucosamine modulates IL-1-induced activation of rat chondrocytes at a receptor level, and by inhibiting the NF-κ B pathway. FEBS Lett. 2002, 510, 166–170. [Google Scholar] [CrossRef]
- Hua, J.; Sakamoto, K.; Nagaoka, I. Inhibitory actions of glucosamine, a therapeutic agent for osteoarthritis, on the functions of neutrophils. J. Leukoc. Biol. 2002, 71, 632–640. [Google Scholar]
- Ma, L.; Rudert, W.A.; Harnaha, J.; Wright, M.; Machen, J.; Lakomy, R.; Qian, S.; Lu, L.; Robbins, P.D.; Trucco, M.; et al. Immunosuppressive effects of glucosamine. J. Biol. Chem. 2002, 277, 39343–39349. [Google Scholar] [CrossRef]
- Zhang, G.X.; Yu, S.; Gran, B.; Rostami, A. Glucosamine abrogates the acute phase of experimental autoimmune encephalomyelitis by induction of Th2 response. J. Immunol. 2005, 175, 7202–7208. [Google Scholar] [CrossRef]
- Xing, D.; Feng, W.; Not, L.G.; Miller, A.P.; Zhang, Y.; Chen, Y.F.; Majid-Hassan, E.; Chatham, J.C.; Oparil, S. Increased protein O-GlcNAc modification inhibits inflammatory and neointimal responses to acute endoluminal arterial injury. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H335–H342. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Ma, X.; Li, D.; Hao, J. Thiamet G mediates neuroprotection in experimental stroke by modulating microglia/macrophage polarization and inhibiting NF-κB p65 signaling. J. Cereb. Blood Flow Metab. 2017, 37, 2938–2951. [Google Scholar] [CrossRef]
- Yang, S.; Zou, L.Y.; Bounelis, P.; Chaudry, I.; Chatham, J.C.; Marchase, R.B. Glucosamine administration during resuscitation improves organ function after trauma hemorrhage. Shock 2006, 25, 600–607. [Google Scholar] [CrossRef]
- Zou, L.; Yang, S.; Hu, S.; Chaudry, I.H.; Marchase, R.B.; Chatham, J.C. The protective effects of PUGNAc on cardiac function after trauma-hemorrhage are mediated via increased protein O-GlcNAc levels. Shock 2007, 27, 402–408. [Google Scholar] [CrossRef]
- Not, L.G.; Brocks, C.A.; Vamhidy, L.; Marchase, R.B.; Chatham, J.C. Increased O-linked β-N-acetylglucosamine levels on proteins improves survival, reduces inflammation and organ damage 24 h after trauma-hemorrhage in rats. Crit. Care Med. 2010, 38, 562–571. [Google Scholar] [CrossRef]
- Not, L.G.; Marchase, R.B.; Fulop, N.; Brocks, C.A.; Chatham, J.C. Glucosamine administration improves survival rate after severe hemorrhagic shock combined with trauma in rats. Shock 2007, 28, 345–352. [Google Scholar]
- Zou, L.; Yang, S.; Champattanachai, V.; Hu, S.; Chaudry, I.H.; Marchase, R.B.; Chatham, J.C. Glucosamine improves cardiac function following trauma-hemorrhage by increased protein O-GlcNAcylation and attenuation of NF-κB signaling. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H515–H523. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Su, K.; Yang, X.; Bowe, D.B.; Paterson, A.J.; Kudlow, J.E. O-GlcNAc modification is an endogenous inhibitor of the proteasome. Cell 2003, 115, 715–725. [Google Scholar] [CrossRef]
- Ju, Y.; Hua, J.; Sakamoto, K.; Ogawa, H.; Nagaoka, I. Modulation of TNF-α-induced endothelial cell activation by glucosamine, a naturally occurring amino monosaccharide. Int. J. Mol. Med. 2008, 22, 809–815. [Google Scholar]
- Xing, D.; Gong, K.; Feng, W.; Nozell, S.E.; Chen, Y.F.; Chatham, J.C.; Oparil, S. O-GlcNAc modification of NFκB p65 inhibits TNF-α-induced inflammatory mediator expression in rat aortic smooth muscle cells. PLoS ONE 2011, 6, e24021. [Google Scholar] [CrossRef]
- Hilgers, R.H.; Xing, D.; Gong, K.; Chen, Y.F.; Chatham, J.C.; Oparil, S. Acute O-GlcNAcylation prevents inflammation-induced vascular dysfunction. Am. J. Physiol. Heart Circ. Physiol. 2012, 303, H513–H522. [Google Scholar] [CrossRef] [Green Version]
- Whitley, D.; Goldberg, S.P.; Jordan, W.D. Heat shock proteins: A review of the molecular chaperones. J. Vasc. Surg. 1999, 29, 748–751. [Google Scholar] [CrossRef] [Green Version]
- Sohn, K.C.; Lee, K.Y.; Park, J.E.; Do, S.I. OGT functions as a catalytic chaperone under heat stress response: A unique defense role of OGT in hyperthermia. Biochem. Biophys. Res. Commun. 2004, 322, 1045–1051. [Google Scholar] [CrossRef]
- Kazemi, Z.; Chang, H.; Haserodt, S.; McKen, C.; Zachara, N.E. O-linked β-N-acetylglucosamine (O-GlcNAc) regulates stress-induced heat shock protein expression in a GSK-3β-dependent manner. J. Biol. Chem. 2010, 285, 39096–39107. [Google Scholar] [CrossRef]
- Tanimoto, T.; Parseghian, M.H.; Nakahara, T.; Kawai, H.; Narula, N.; Kim, D.; Nishimura, R.; Weisbart, R.H.; Chan, G.; Richieri, R.A.; et al. Cardioprotective Effects of HSP72 Administration on Ischemia-Reperfusion Injury. J. Am. Coll. Cardiol. 2017, 70, 1479–1492. [Google Scholar] [CrossRef]
- Song, Y.J.; Zhong, C.B.; Wang, X.B. Heat shock protein 70: A promising therapeutic target for myocardial ischemia-reperfusion injury. J. Cell. Physiol. 2018, 23, 1190–1207. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, C.; Zhang, C.; Li, J.; Guo, W.; Yan, D.; Yang, C.; Zhao, J.; Xia, T.; Wang, Y.; et al. Heat shock protein 70 inhibits cardiomyocyte necroptosis through repressing autophagy in myocardial ischemia/reperfusion injury. In Vitro Cell. Dev. Biol. Anim. 2016, 52, 690–698. [Google Scholar] [CrossRef]
- Konstantinov, I.E.; Arab, S.; Kharbanda, R.K.; Li, J.; Cheung, M.M.; Cherepanov, V.; Downey, G.P.; Liu, P.P.; Cukerman, E.; Coles, J.G.; et al. The remote ischemic preconditioning stimulus modifies inflammatory gene expression in humans. Physiol. Genom. 2004, 19, 143–150. [Google Scholar] [CrossRef] [Green Version]
- Wischmeyer, P.E.; Jayakar, D.; Williams, U.; Singleton, K.D.; Riehm, J.; Bacha, E.A.; Jeevanandam, V.; Christians, U.; Serkova, N. Single dose of glutamine enhances myocardial tissue metabolism, glutathione content, and improves myocardial function after ischemia-reperfusion injury. JPEN J. Parenter. Enter. Nutr. 2003, 27, 396–403. [Google Scholar] [CrossRef]
- Guinez, C.; Lemoine, J.; Michalski, J.C.; Lefebvre, T. 70-kDa-heat shock protein presents an adjustable lectinic activity towards O-linked N-acetylglucosamine. Biochem. Biophys. Res. Commun. 2004, 319, 21–26. [Google Scholar] [CrossRef]
- Hausenloy, D.J.; Tsang, A.; Mocanu, M.M.; Yellon, D.M. Ischemic preconditioning protects by activating prosurvival kinases at reperfusion. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H971–H976. [Google Scholar] [CrossRef]
- Hausenloy, D.J.; Mocanu, M.M.; Yellon, D.M. Cross-talk between the survival kinases during early reperfusion: Its contribution to ischemic preconditioning. Cardiovasc. Res. 2004, 63, 305–312. [Google Scholar] [CrossRef]
- Wang, S.; Huang, X.; Sun, D.; Xin, X.; Pan, Q.; Peng, S.; Liang, Z.; Luo, C.; Yang, Y.; Jiang, H.; et al. Extensive crosstalk between O-GlcNAcylation and phosphorylation regulates Akt signaling. PLoS ONE 2012, 7, e37427. [Google Scholar] [CrossRef]
- Vosseller, K.; Wells, L.; Lane, M.D.; Hart, G.W. Elevated nucleocytoplasmic glycosylation by O-GlcNAc results in insulin resistance associated with defects in Akt activation in 3T3-L1 adipocytes. Proc. Natl. Acad. Sci. USA 2002, 99, 5313–5318. [Google Scholar] [CrossRef] [Green Version]
- Kang, E.S.; Han, D.; Park, J.; Kwak, T.K.; Oh, M.A.; Lee, S.A.; Choi, S.; Park, Z.Y.; Kim, Y.; Lee, J.W. O-GlcNAc modulation at Akt1 Ser473 correlates with apoptosis of murine pancreatic β cells. Exp. Cell Res. 2008, 314, 2238–2248. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Gu, J.H.; Dai, C.L.; Gu, J.; Jin, X.; Sun, J.; Iqbal, K.; Liu, F.; Gong, C.X. O-GlcNAcylation regulates ischemia-induced neuronal apoptosis through AKT signaling. Sci. Rep. 2015, 5, 14500. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Chen, R.; Jia, P.; Fang, Y.; Liu, T.; Song, N.; Xu, X.; Ji, J.; Ding, X. Augmented O-GlcNAc signaling via glucosamine attenuates oxidative stress and apoptosis following contrast-induced acute kidney injury in rats. Free Radic. Biol. Med. 2017, 103, 121–132. [Google Scholar] [CrossRef]
- Ku, N.O.; Toivola, D.M.; Strnad, P.; Omary, M.B. Cytoskeletal keratin glycosylation protects epithelial tissue from injury. Nat. Cell Biol. 2010, 12, 876–885. [Google Scholar] [CrossRef] [Green Version]
- Lecour, S. Multiple protective pathways against reperfusion injury: A SAFE path without Aktion? J. Mol. Cell. Cardiol. 2009, 46, 607–609. [Google Scholar] [CrossRef]
- Smith, R.M.; Suleman, N.; McCarthy, J.; Sack, M.N. Classic ischemic but not pharmacologic preconditioning is abrogated following genetic ablation of the TNFα gene. Cardiovasc. Res. 2002, 55, 553–560. [Google Scholar] [CrossRef] [Green Version]
- Lambert, M.; Bastide, B.; Cieniewski-Bernard, C. Involvement of O-GlcNAcylation in the Skeletal Muscle Physiology and Physiopathology: Focus on Muscle Metabolism. Front. Endocrinol. 2018, 9, 578. [Google Scholar] [CrossRef]
- Darley-Usmar, V.M.; Ball, L.E.; Chatham, J.C. Protein O-linked β-N-acetylglucosamine: A novel effector of cardiomyocyte metabolism and function. J. Mol. Cell. Cardiol. 2012, 52, 538–549. [Google Scholar] [CrossRef]
- Ma, J.; Hart, G.W. Protein O-GlcNAcylation in diabetes and diabetic complications. Expert Rev. Proteom. 2013, 10, 365–380. [Google Scholar] [CrossRef]
- Lima, V.V.; Giachini, F.R.; Carneiro, F.S.; Carneiro, Z.N.; Fortes, Z.B.; Carvalho, M.H.; Webb, R.C.; Tostes, R.C. Increased vascular O-GlcNAcylation augments reactivity to constrictor stimuli—VASOACTIVE PEPTIDE SYMPOSIUM. J. Am. Soc. Hypertens. 2008, 2, 410–417. [Google Scholar] [CrossRef]
- Degrell, P.; Cseh, J.; Mohas, M.; Molnar, G.A.; Pajor, L.; Chatham, J.C.; Fulop, N.; Wittmann, I. Evidence of O-linked N-acetylglucosamine in diabetic nephropathy. Life Sci. 2009, 84, 389–393. [Google Scholar] [CrossRef]
- Akimoto, Y.; Kreppel, L.K.; Hirano, H.; Hart, G.W. Increased O-GlcNAc transferase in pancreas of rats with streptozotocin-induced diabetes. Diabetologia 2000, 43, 1239–1247. [Google Scholar] [CrossRef]
- Wang, Z.; Park, K.; Comer, F.; Hsieh-Wilson, L.C.; Saudek, C.D.; Hart, G.W. Site-specific GlcNAcylation of human erythrocyte proteins: Potential biomarker(s) for diabetes. Diabetes 2009, 58, 309–317. [Google Scholar] [CrossRef]
- Akimoto, Y.; Kawakami, H.; Yamamoto, K.; Munetomo, E.; Hida, T.; Hirano, H. Elevated expression of O-GlcNAc-modified proteins and O-GlcNAc transferase in corneas of diabetic Goto-Kakizaki rats. Investig. Ophthalmol. Vis. Sci. 2003, 44, 3802–3809. [Google Scholar] [CrossRef]
- Liu, Y.; Thornton, J.D.; Cohen, M.V.; Downey, J.M.; Schaffer, S.W. Streptozotocin-induced non-insulin-dependent diabetes protects the heart from infarction. Circulation 1993, 88, 1273–1278. [Google Scholar] [CrossRef]
- Povlsen, J.A.; Lofgren, B.; Dalgas, C.; Birkler, R.I.; Johannsen, M.; Stottrup, N.B.; Botker, H.E. Protection against myocardial ischemia-reperfusion injury at onset of type 2 diabetes in Zucker diabetic fatty rats is associated with altered glucose oxidation. PLoS ONE 2013, 8, e64093. [Google Scholar] [CrossRef]
- Galagudza, M.M.; Nekrasova, M.K.; Syrenskii, A.V.; Nifontov, E.M. Resistance of the myocardium to ischemia and the efficacy of ischemic preconditioning in experimental diabetes mellitus. Neurosci. Behav. Physiol. 2007, 37, 489–493. [Google Scholar] [CrossRef]
- Kristiansen, S.B.; Lofgren, B.; Stottrup, N.B.; Khatir, D.; Nielsen-Kudsk, J.E.; Nielsen, T.T.; Botker, H.E.; Flyvbjerg, A. Ischaemic preconditioning does not protect the heart in obese and lean animal models of type 2 diabetes. Diabetologia 2004, 47, 1716–1721. [Google Scholar] [CrossRef]
- Tsang, A.; Hausenloy, D.J.; Mocanu, M.M.; Carr, R.D.; Yellon, D.M. Preconditioning the diabetic heart: The importance of Akt phosphorylation. Diabetes 2005, 54, 2360–2364. [Google Scholar] [CrossRef]
- Hjortbak, M.V.; Hjort, J.; Povlsen, J.A.; Jensen, R.V.; Stottrup, N.B.; Laursen, M.R.; Jespersen, N.R.; Lofgren, B.; Botker, H.E. Influence of diabetes mellitus duration on the efficacy of ischemic preconditioning in a Zucker diabetic fatty rat model. PLoS ONE 2018, 13, e0192981. [Google Scholar] [CrossRef]
- Kristensen, J.; Maeng, M.; Mortensen, U.M.; Berg, J.; Rehling, M.; Nielsen, T.T. Lack of cardioprotection from metabolic support with glutamine or glutamate in a porcine coronary occlusion model. Scand. Cardiovasc. J. 2005, 39, 115–120. [Google Scholar] [CrossRef]
- Aghazadeh-Habashi, A.; Jamali, F. The glucosamine controversy; a pharmacokinetic issue. J. Pharm. Pharm. Sci. 2011, 14, 264–273. [Google Scholar] [CrossRef]
- Solskov, L.; Lofgren, B.; Kristiansen, S.B.; Jessen, N.; Pold, R.; Nielsen, T.T.; Botker, H.E.; Schmitz, O.; Lund, S. Metformin induces cardioprotection against ischaemia/reperfusion injury in the rat heart 24 h after administration. Basic Clin. Pharmacol. Toxicol. 2008, 103, 82–87. [Google Scholar] [CrossRef]
- Kim, Y.S.; Kim, M.; Choi, M.Y.; Lee, D.H.; Roh, G.S.; Kim, H.J.; Kang, S.S.; Cho, G.J.; Kim, S.J.; Yoo, J.M.; et al. Metformin protects against retinal cell death in diabetic mice. Biochem. Biophys. Res. Commun. 2017, 492, 397–403. [Google Scholar] [CrossRef]
- Ellenberger, C.; Sologashvili, T.; Kreienbuhl, L.; Cikirikcioglu, M.; Diaper, J.; Licker, M. Myocardial Protection by Glucose-Insulin-Potassium in Moderate- to High-Risk Patients Undergoing Elective On-Pump Cardiac Surgery: A Randomized Controlled Trial. Anesth. Analg. 2018, 126, 1133–1141. [Google Scholar] [CrossRef]
- Ahmad, S.; Ahmad, R.A.; Qureshi, B.A.; Baig, M.A.R. Myocardial protection with Glucose-Insulin-Potassium infusion during adult cardiac surgery. Pak. J. Med. Sci. 2017, 33, 325–329. [Google Scholar] [CrossRef]
- Selker, H.P.; Udelson, J.E.; Massaro, J.M.; Ruthazer, R.; D’Agostino, R.B.; Griffith, J.L.; Sheehan, P.R.; Desvigne-Nickens, P.; Rosenberg, Y.; Tian, X.; et al. One-year outcomes of out-of-hospital administration of intravenous glucose, insulin, and potassium (GIK) in patients with suspected acute coronary syndromes (from the IMMEDIATE [Immediate Myocardial Metabolic Enhancement During Initial Assessment and Treatment in Emergency Care] Trial). Am. J. Cardiol. 2014, 113, 1599–1605. [Google Scholar]
- Yang, Y.M.; Zhu, J.; Tan, H.Q.; Liang, Y.; Zhang, Y.; Li, Y.; Li, J.D.; Liu, L.S. [Effect of high dose glucose-insulin-potassium infusion in patients with acute ST-segment elevation myocardial infarction: Analysis of 7510 patients in China as part of CREATE-ECLA study]. Zhonghua Yi Xue Za Zhi 2008, 88, 1806–1810. [Google Scholar]
- Diaz, R.; Goyal, A.; Mehta, S.R.; Afzal, R.; Xavier, D.; Pais, P.; Chrolavicius, S.; Zhu, J.; Kazmi, K.; Liu, L.; et al. Glucose-insulin-potassium therapy in patients with ST-segment elevation myocardial infarction. JAMA 2007, 298, 2399–2405. [Google Scholar] [CrossRef]
- Rasoul, S.; Ottervanger, J.P.; Timmer, J.R.; Svilaas, T.; Henriques, J.P.; Dambrink, J.H.; van der Horst, I.C.; Zijlstra, F. One year outcomes after glucose-insulin-potassium in ST elevation myocardial infarction. The Glucose-insulin-potassium study II. Int. J. Cardiol. 2007, 122, 52–55. [Google Scholar] [CrossRef]
- Howell, N.J.; Ashrafian, H.; Drury, N.E.; Ranasinghe, A.M.; Contractor, H.; Isackson, H.; Calvert, M.; Williams, L.K.; Freemantle, N.; Quinn, D.W.; et al. Glucose-insulin-potassium reduces the incidence of low cardiac output episodes after aortic valve replacement for aortic stenosis in patients with left ventricular hypertrophy: Results from the Hypertrophy, Insulin, Glucose, and Electrolytes (HINGE) trial. Circulation 2011, 123, 170–177. [Google Scholar] [CrossRef]
- Chun, W.J.; Nah, D.Y.; Bae, J.H.; Chung, J.W.; Lee, H.; Moon, I.S. Glucose-insulin-potassium solution protects ventricular myocytes of neonatal rat in an in vitro coverslip ischemia/reperfusion model. Korean Circ. J. 2015, 45, 234–241. [Google Scholar] [CrossRef]
- Sack, M.N.; Yellon, D.M. Insulin therapy as an adjunct to reperfusion after acute coronary ischemia: A proposed direct myocardial cell survival effect independent of metabolic modulation. J. Am. Coll. Cardiol. 2003, 41, 1404–1407. [Google Scholar] [CrossRef]
- Jonassen, A.K.; Sack, M.N.; Mjos, O.D.; Yellon, D.M. Myocardial protection by insulin at reperfusion requires early administration and is mediated via Akt and p70s6 kinase cell-survival signaling. Circ. Res. 2001, 89, 1191–1198. [Google Scholar] [CrossRef]
- Julier, K.; da Silva, R.; Garcia, C.; Bestmann, L.; Frascarolo, P.; Zollinger, A.; Chassot, P.G.; Schmid, E.R.; Turina, M.I.; von Segesser, L.K.; et al. Preconditioning by sevoflurane decreases biochemical markers for myocardial and renal dysfunction in coronary artery bypass graft surgery: A double-blinded, placebo-controlled, multicenter study. Anesthesiology 2003, 98, 1315–1327. [Google Scholar] [CrossRef]
- Landoni, G.; Fochi, O.; Torri, G. Cardiac protection by volatile anaesthetics: A review. Curr. Vasc. Pharmacol. 2008, 6, 108–111. [Google Scholar] [CrossRef]
- Belhomme, D.; Peynet, J.; Louzy, M.; Launay, J.M.; Kitakaze, M.; Menasche, P. Evidence for preconditioning by isoflurane in coronary artery bypass graft surgery. Circulation 1999, 100, II340–II344. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jensen, R.V.; Andreadou, I.; Hausenloy, D.J.; Bøtker, H.E. The Role of O-GlcNAcylation for Protection against Ischemia-Reperfusion Injury. Int. J. Mol. Sci. 2019, 20, 404. https://doi.org/10.3390/ijms20020404
Jensen RV, Andreadou I, Hausenloy DJ, Bøtker HE. The Role of O-GlcNAcylation for Protection against Ischemia-Reperfusion Injury. International Journal of Molecular Sciences. 2019; 20(2):404. https://doi.org/10.3390/ijms20020404
Chicago/Turabian StyleJensen, Rebekka Vibjerg, Ioanna Andreadou, Derek J. Hausenloy, and Hans Erik Bøtker. 2019. "The Role of O-GlcNAcylation for Protection against Ischemia-Reperfusion Injury" International Journal of Molecular Sciences 20, no. 2: 404. https://doi.org/10.3390/ijms20020404
APA StyleJensen, R. V., Andreadou, I., Hausenloy, D. J., & Bøtker, H. E. (2019). The Role of O-GlcNAcylation for Protection against Ischemia-Reperfusion Injury. International Journal of Molecular Sciences, 20(2), 404. https://doi.org/10.3390/ijms20020404