The Yin-Yang Regulation of Reactive Oxygen Species and MicroRNAs in Cancer



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Significance of ROS in Cancer Development
2.1. ROS in Cancer Initiation
2.2. ROS in Cancer Cell Proliferation
2.3. ROS in Cancer Metastasis
2.4. ROS in Cancer Stem Cells
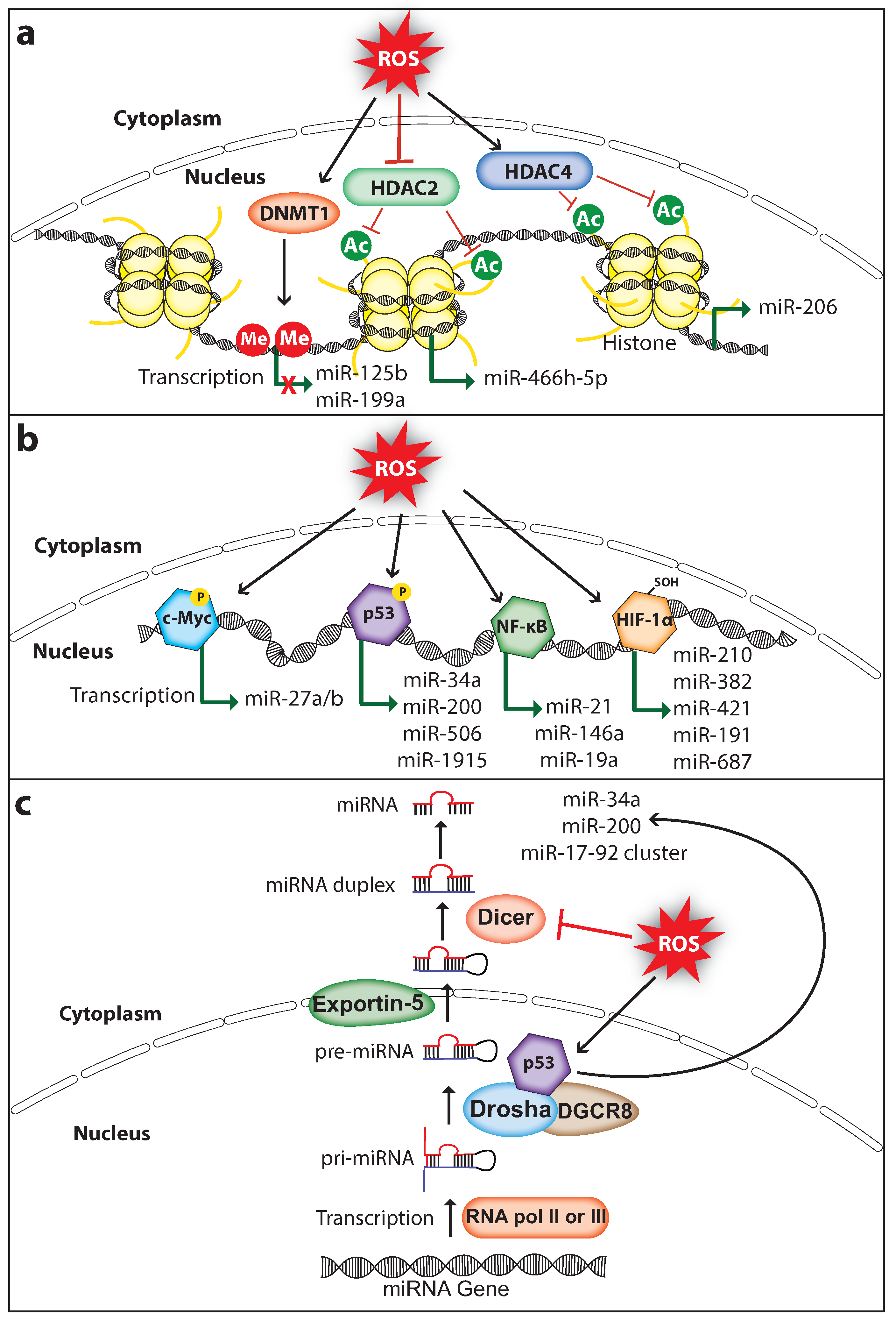
3. ROS Regulate MiRNA Expression
3.1. Regulation of MiRNA Expression via Epigenetic Modifications
3.2. Regulation of MiRNA Expression via Transcription Factors
3.2.1. C-Myc
3.2.2. P53
3.2.3. NFκB
3.2.4. HIF-1α
3.3. Regulation of MiRNA Processing
4. MiRNAs Regulate ROS Homeostasis
4.1. Regulation of ROS Producer
4.2. Regulation of Mitochondrial Functions
4.3. Regulation of Antioxidants
4.4. Regulation of NRF2/KEAP1 System
5. The Interplay of ROS and MiRNAs in Cancer
6. Challenges in Using Antioxidants for Anti-Cancer Therapy
7. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Snezhkina, A.V.; Kudryavtseva, A.V.; Kardymon, O.L.; Savvateeva, M.V.; Melnikova, N.V.; Krasnov, G.S.; Dmitriev, A.A. ROS Generation and antioxidant defense systems in normal and malignant cells. Oxidative Med. Cell. Longev. 2019, 2019, 6175804. [Google Scholar] [CrossRef] [PubMed]
- Di Meo, S.; Reed, T.T.; Venditti, P.; Victor, V.M. Role of ROS and RNS sources in physiological and pathological conditions. Oxidative Med. Cell. Longev. 2016, 2016, 1245049. [Google Scholar] [CrossRef] [PubMed]
- Brieger, K.; Schiavone, S.; Miller, F.J., Jr.; Krause, K.H. Reactive oxygen species: From health to disease. Swiss Med Wkly. 2012, 142, 13659. [Google Scholar] [CrossRef] [PubMed]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Jena, N.R. DNA damage by reactive species: Mechanisms, mutation and repair. J. Biosci. 2012, 37, 503–517. [Google Scholar] [CrossRef] [PubMed]
- Poulsen, H.E.; Specht, E.; Broedbaek, K.; Henriksen, T.; Ellervik, C.; Mandrup-Poulsen, T.; Tonnesen, M.; Nielsen, P.E.; Andersen, H.U.; Weimann, A. RNA modifications by oxidation: A novel disease mechanism? Free. Radic. Biol. Med. 2012, 52, 1353–1361. [Google Scholar] [CrossRef] [PubMed]
- Fimognari, C. Role of oxidative RNA damage in chronic-degenerative diseases. Oxidative Med. Cell. Longev. 2015, 2015, 358713. [Google Scholar] [CrossRef]
- Ma, Y.; Zhang, L.; Rong, S.; Qu, H.; Zhang, Y.; Chang, D.; Pan, H.; Wang, W. Relation between gastric cancer and protein oxidation, DNA damage, and lipid peroxidation. Oxidative Med. Cell. Longev. 2013, 2013, 543760. [Google Scholar] [CrossRef]
- Barrera, G. Oxidative stress and lipid peroxidation products in cancer progression and therapy. ISRN Oncol. 2012, 2012, 137289. [Google Scholar] [CrossRef]
- Bartel, D.P. Metazoan MicroRNAs. Cell 2018, 173, 20–51. [Google Scholar] [CrossRef]
- Peng, Y.; Croce, C.M. The role of MicroRNAs in human cancer. Signal Transduct. Target. Ther. 2016, 1, 15004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thulasingam, S.; Massilamany, C.; Gangaplara, A.; Dai, H.; Yarbaeva, S.; Subramaniam, S.; Riethoven, J.J.; Eudy, J.; Lou, M.; Reddy, J. MiR-27b, an oxidative stress-responsive microRNA modulates nuclear factor-kB pathway in RAW 264.7 cells. Mol. Cell. Biochem. 2011, 352, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; He, X.; Zhang, L.; Liu, P. MicroRNA26a protects vascular smooth muscle cells against H2O2 induced injury through activation of the PTEN/AKT/mTOR pathway. Int. J. Mol. Med. 2018, 42, 1367–1378. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Zhou, M.; Zhou, W. MicroRNA-30e regulates TGF-beta-mediated NADPH oxidase 4-dependent oxidative stress by Snai1 in atherosclerosis. Int. J. Mol. Med. 2019, 43, 1806–1816. [Google Scholar] [CrossRef]
- Van Houten, B.; Santa-Gonzalez, G.A.; Camargo, M. DNA repair after oxidative stress: Current challenges. Curr. Opin. Toxicol. 2018, 7, 9–16. [Google Scholar] [CrossRef]
- Anastasiadou, E.; Jacob, L.S.; Slack, F.J. Non-coding RNA networks in cancer. Nat. Rev. Cancer 2018, 18, 5–18. [Google Scholar] [CrossRef]
- Davies, M.J. Protein oxidation and peroxidation. Biochem. J. 2016, 473, 805–825. [Google Scholar] [CrossRef] [Green Version]
- Gangwar, A.; Paul, S.; Ahmad, Y.; Bhargava, K. Competing trends of ROS and RNS-mediated protein modifications during hypoxia as an alternate mechanism of NO benefits. Biochimie 2018, 148, 127–138. [Google Scholar] [CrossRef]
- Liou, G.Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef] [Green Version]
- Zhong, H.; Yin, H. Role of lipid peroxidation derived 4-hydroxynonenal (4-HNE) in cancer: Focusing on mitochondria. Redox Biol. 2015, 4, 193–199. [Google Scholar] [CrossRef]
- Liao, Z.; Chua, D.; Tan, N.S. Reactive oxygen species: A volatile driver of field cancerization and metastasis. Mol. Cancer 2019, 18, 65. [Google Scholar] [CrossRef] [PubMed]
- Lingappan, K. NF-kappaB in oxidative stress. Curr. Opin. Toxicol. 2018, 7, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Verbon, E.H.; Post, J.A.; Boonstra, J. The influence of reactive oxygen species on cell cycle progression in mammalian cells. Gene 2012, 511, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Ramos, R.; Lopez-Carrillo, L.; Rios-Perez, A.D.; De Vizcaya-Ruiz, A.; Cebrian, M.E. Sodium arsenite induces ROS generation, DNA oxidative damage, HO-1 and c-Myc proteins, NF-kappaB activation and cell proliferation in human breast cancer MCF-7 cells. Mutat. Res. 2009, 674, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Menon, S.G.; Coleman, M.C.; Walsh, S.A.; Spitz, D.R.; Goswami, P.C. Differential susceptibility of nonmalignant human breast epithelial cells and breast cancer cells to thiol antioxidant-induced G(1)-delay. Antioxid. Redox Signal. 2005, 7, 711–718. [Google Scholar] [CrossRef] [PubMed]
- Vanharanta, S.; Massague, J. Origins of metastatic traits. Cancer cell 2013, 24, 410–421. [Google Scholar] [CrossRef]
- Piskounova, E.; Agathocleous, M.; Murphy, M.M.; Hu, Z.; Huddlestun, S.E.; Zhao, Z.; Leitch, A.M.; Johnson, T.M.; DeBerardinis, R.J.; Morrison, S.J. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 2015, 527, 186–191. [Google Scholar] [CrossRef] [Green Version]
- Hitchler, M.J.; Oberley, L.W.; Domann, F.E. Epigenetic silencing of SOD2 by histone modifications in human breast cancer cells. Free Radic. Biol. Med. 2008, 45, 1573–1580. [Google Scholar] [CrossRef] [Green Version]
- Lewis, A.; Du, J.; Liu, J.; Ritchie, J.M.; Oberley, L.W.; Cullen, J.J. Metastatic progression of pancreatic cancer: Changes in antioxidant enzymes and cell growth. Clin. Exp. Metastasis 2005, 22, 523–532. [Google Scholar] [CrossRef]
- Roy, R.; Morad, G.; Jedinak, A.; Moses, M.A. Metalloproteinases and their roles in human cancer. Anat. Rec. (Hoboken) 2019. [Google Scholar] [CrossRef]
- Kang, K.A.; Ryu, Y.S.; Piao, M.J.; Shilnikova, K.; Kang, H.K.; Yi, J.M.; Boulanger, M.; Paolillo, R.; Bossis, G.; Yoon, S.Y.; et al. DUOX2-mediated production of reactive oxygen species induces epithelial mesenchymal transition in 5-fluorouracil resistant human colon cancer cells. Redox. Biol. 2018, 17, 224–235. [Google Scholar] [CrossRef] [PubMed]
- Radisky, D.C.; Levy, D.D.; Littlepage, L.E.; Liu, H.; Nelson, C.M.; Fata, J.E.; Leake, D.; Godden, E.L.; Albertson, D.G.; Nieto, M.A.; et al. Rac1b and reactive oxygen species mediate MMP-3-induced EMT and genomic instability. Nature 2005, 436, 123–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Wetering, S.; van Buul, J.D.; Quik, S.; Mul, F.P.; Anthony, E.C.; ten Klooster, J.P.; Collard, J.G.; Hordijk, P.L. Reactive oxygen species mediate Rac-induced loss of cell-cell adhesion in primary human endothelial cells. J. Cell Sci. 2002, 115, 1837–1846. [Google Scholar] [PubMed]
- Kuai, W.X.; Wang, Q.; Yang, X.Z.; Zhao, Y.; Yu, R.; Tang, X.J. Interleukin-8 associates with adhesion, migration, invasion and chemosensitivity of human gastric cancer cells. World J. Gastroenterol. 2012, 18, 979–985. [Google Scholar] [CrossRef]
- Ghislin, S.; Obino, D.; Middendorp, S.; Boggetto, N.; Alcaide-Loridan, C.; Deshayes, F. LFA-1 and ICAM-1 expression induced during melanoma-endothelial cell co-culture favors the transendothelial migration of melanoma cell lines in vitro. BMC Cancer 2012, 12, 455. [Google Scholar] [CrossRef] [PubMed]
- Sawada, J.; Li, F.; Komatsu, M. R-Ras Inhibits VEGF-Induced p38MAPK Activation and HSP27 Phosphorylation in Endothelial Cells. J. Vasc. Res. 2015, 52, 347–359. [Google Scholar] [CrossRef]
- Shi, X.; Zhang, Y.; Zheng, J.; Pan, J. Reactive oxygen species in cancer stem cells. Antioxid. Redox Signal. 2012, 16, 1215–1228. [Google Scholar] [CrossRef]
- Diehn, M.; Cho, R.W.; Lobo, N.A.; Kalisky, T.; Dorie, M.J.; Kulp, A.N.; Qian, D.; Lam, J.S.; Ailles, L.E.; Wong, M.; et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 2009, 458, 780–783. [Google Scholar] [CrossRef]
- Pasto, A.; Bellio, C.; Pilotto, G.; Ciminale, V.; Silic-Benussi, M.; Guzzo, G.; Rasola, A.; Frasson, C.; Nardo, G.; Zulato, E.; et al. Cancer stem cells from epithelial ovarian cancer patients privilege oxidative phosphorylation, and resist glucose deprivation. Oncotarget 2014, 5, 4305–4319. [Google Scholar] [CrossRef] [Green Version]
- Jang, Y.Y.; Sharkis, S.J. A low level of reactive oxygen species selects for primitive hematopoietic stem cells that may reside in the low-oxygenic niche. Blood 2007, 110, 3056–3063. [Google Scholar] [CrossRef] [Green Version]
- He, J.; Xu, Q.; Jing, Y.; Agani, F.; Qian, X.; Carpenter, R.; Li, Q.; Wang, X.R.; Peiper, S.S.; Lu, Z.; et al. Reactive oxygen species regulate ERBB2 and ERBB3 expression via miR-199a/125b and DNA methylation. EMBO Rep. 2012, 13, 1116–1122. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Jing, Y.; Li, W.; Qian, X.; Xu, Q.; Li, F.S.; Liu, L.Z.; Jiang, B.H.; Jiang, Y. Roles and mechanism of miR-199a and miR-125b in tumor angiogenesis. PLoS ONE 2013, 8, e56647. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Seto, E. HDACs and HDAC inhibitors in cancer development and therapy. Cold Spring Harb. Perspect. Med. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Ago, T.; Liu, T.; Zhai, P.; Chen, W.; Li, H.; Molkentin, J.D.; Vatner, S.F.; Sadoshima, J. A redox-dependent pathway for regulating class II HDACs and cardiac hypertrophy. Cell 2008, 133, 978–993. [Google Scholar] [CrossRef] [PubMed]
- Ciesla, M.; Marona, P.; Kozakowska, M.; Jez, M.; Seczynska, M.; Loboda, A.; Bukowska-Strakova, K.; Szade, A.; Walawender, M.; Kusior, M.; et al. Heme oxygenase-1 controls an HDAC4-miR-206 pathway of oxidative stress in rhabdomyosarcoma. Cancer Res. 2016, 76, 5707–5718. [Google Scholar] [CrossRef]
- Singh, A.; Happel, C.; Manna, S.K.; Acquaah-Mensah, G.; Carrerero, J.; Kumar, S.; Nasipuri, P.; Krausz, K.W.; Wakabayashi, N.; Dewi, R.; et al. Transcription factor NRF2 regulates miR-1 and miR-206 to drive tumorigenesis. J. Clin. Investig. 2013, 123, 2921–2934. [Google Scholar] [CrossRef] [Green Version]
- Druz, A.; Betenbaugh, M.; Shiloach, J. Glucose depletion activates mmu-miR-466h-5p expression through oxidative stress and inhibition of histone deacetylation. Nucleic Acids Res. 2012, 40, 7291–7302. [Google Scholar] [CrossRef] [Green Version]
- Druz, A.; Chu, C.; Majors, B.; Santuary, R.; Betenbaugh, M.; Shiloach, J. A novel microRNA mmu-miR-466h affects apoptosis regulation in mammalian cells. Biotechnol. Bioeng. 2011, 108, 1651–1661. [Google Scholar] [CrossRef] [Green Version]
- Dando, I.; Cordani, M.; Dalla Pozza, E.; Biondani, G.; Donadelli, M.; Palmieri, M. Antioxidant Mechanisms and ROS-Related MicroRNAs in Cancer Stem Cells. Oxidative Med. Cell. Longev. 2015, 2015, 425708. [Google Scholar] [CrossRef]
- Benassi, B.; Fanciulli, M.; Fiorentino, F.; Porrello, A.; Chiorino, G.; Loda, M.; Zupi, G.; Biroccio, A. c-Myc phosphorylation is required for cellular response to oxidative stress. Mol. cell 2006, 21, 509–519. [Google Scholar] [CrossRef]
- Huang, S.; He, X.; Ding, J.; Liang, L.; Zhao, Y.; Zhang, Z.; Yao, X.; Pan, Z.; Zhang, P.; Li, J.; et al. Upregulation of miR-23a approximately 27a approximately 24 decreases transforming growth factor-beta-induced tumor-suppressive activities in human hepatocellular carcinoma cells. Int. J. Cancer 2008, 123, 972–978. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Li, T.W.; Zhou, Y.; Peng, H.; Liu, T.; Zandi, E.; Martinez-Chantar, M.L.; Mato, J.M.; Lu, S.C. Activation of a novel c-Myc-miR27-prohibitin 1 circuitry in cholestatic liver injury inhibits glutathione synthesis in mice. Antioxid. Redox Signal. 2015, 22, 259–274. [Google Scholar] [CrossRef] [PubMed]
- Yeo, C.Q.X.; Alexander, I.; Lin, Z.; Lim, S.; Aning, O.A.; Kumar, R.; Sangthongpitag, K.; Pendharkar, V.; Ho, V.H.B.; Cheok, C.F. p53 Maintains genomic stability by preventing interference between transcription and replication. Cell Rep. 2016, 15, 132–146. [Google Scholar] [CrossRef] [PubMed]
- Navarro, F.; Lieberman, J. miR-34 and p53: New insights into a complex functional relationship. PLoS ONE 2015, 10, e0132767. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, K.; Shi, Y.; Shao, C. The tango of ROS and p53 in tissue stem cells. Cell Death Differ. 2018, 25, 639–641. [Google Scholar] [CrossRef] [Green Version]
- Peuget, S.; Bonacci, T.; Soubeyran, P.; Iovanna, J.; Dusetti, N.J. Oxidative stress-induced p53 activity is enhanced by a redox-sensitive TP53INP1 SUMOylation. Cell Death Differ. 2014, 21, 1107–1118. [Google Scholar] [CrossRef] [Green Version]
- Maillet, A.; Pervaiz, S. Redox regulation of p53, redox effectors regulated by p53: A subtle balance. Antioxid. Redox Signal. 2012, 16, 1285–1294. [Google Scholar] [CrossRef]
- Haffo, L.; Lu, J.; Bykov, V.J.N.; Martin, S.S.; Ren, X.; Coppo, L.; Wiman, K.G.; Holmgren, A. Inhibition of the glutaredoxin and thioredoxin systems and ribonucleotide reductase by mutant p53-targeting compound APR-246. Sci. Rep. 2018, 8, 12671. [Google Scholar] [CrossRef]
- Xiao, Y.; Yan, W.; Lu, L.; Wang, Y.; Lu, W.; Cao, Y.; Cai, W. p38/p53/miR-200a-3p feedback loop promotes oxidative stress-mediated liver cell death. Cell Cycle 2015, 14, 1548–1558. [Google Scholar] [CrossRef] [Green Version]
- Magenta, A.; Cencioni, C.; Fasanaro, P.; Zaccagnini, G.; Greco, S.; Sarra-Ferraris, G.; Antonini, A.; Martelli, F.; Capogrossi, M.C. miR-200c is upregulated by oxidative stress and induces endothelial cell apoptosis and senescence via ZEB1 inhibition. Cell Death Differ. 2011, 18, 1628–1639. [Google Scholar] [CrossRef] [Green Version]
- Gibbons, D.L.; Lin, W.; Creighton, C.J.; Rizvi, Z.H.; Gregory, P.A.; Goodall, G.J.; Thilaganathan, N.; Du, L.; Zhang, Y.; Pertsemlidis, A.; et al. Contextual extracellular cues promote tumor cell EMT and metastasis by regulating miR-200 family expression. Genes Dev. 2009, 23, 2140–2151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, X.; Wang, Z.; Fillmore, R.; Xi, Y. MiR-200, a new star miRNA in human cancer. Cancer Lett. 2014, 344, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Yin, M.; Ren, X.; Zhang, X.; Luo, Y.; Wang, G.; Huang, K.; Feng, S.; Bao, X.; Huang, K.; He, X.; et al. Selective killing of lung cancer cells by miRNA-506 molecule through inhibiting NF-kappaB p65 to evoke reactive oxygen species generation and p53 activation. Oncogene 2015, 34, 691–703. [Google Scholar] [CrossRef]
- Wan, Y.; Cui, R.; Gu, J.; Zhang, X.; Xiang, X.; Liu, C.; Qu, K.; Lin, T. Identification of Four Oxidative Stress-Responsive MicroRNAs, miR-34a-5p, miR-1915-3p, miR-638, and miR-150-3p, in Hepatocellular Carcinoma. Oxidative Med. Cell. Longev. 2017, 2017, 5189138. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-kappaB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Markopoulos, G.S.; Roupakia, E.; Tokamani, M.; Alabasi, G.; Sandaltzopoulos, R.; Marcu, K.B.; Kolettas, E. Roles of NF-kappaB Signaling in the regulation of miRNAs impacting on inflammation in cancer. Biomedicines 2018, 6, 40. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Xin, S.; He, Z.; Che, X.; Wang, J.; Xiao, X.; Chen, J.; Song, X. MicroRNA-21 (miR-21) post-transcriptionally downregulates tumor suppressor PDCD4 and promotes cell transformation, proliferation, and metastasis in renal cell carcinoma. Cell. Physiol. Biochem. 2014, 33, 1631–1642. [Google Scholar] [CrossRef]
- Li, Z.; Deng, X.; Kang, Z.; Wang, Y.; Xia, T.; Ding, N.; Yin, Y. Elevation of miR-21, through targeting MKK3, may be involved in ischemia pretreatment protection from ischemia-reperfusion induced kidney injury. J. Nephrol. 2016, 29, 27–36. [Google Scholar] [CrossRef]
- Yang, C.H.; Yue, J.; Pfeffer, S.R.; Fan, M.; Paulus, E.; Hosni-Ahmed, A.; Sims, M.; Qayyum, S.; Davidoff, A.M.; Handorf, C.R.; et al. MicroRNA-21 promotes glioblastoma tumorigenesis by down-regulating insulin-like growth factor-binding protein-3 (IGFBP3). J. Biol. Chem. 2014, 289, 25079–25087. [Google Scholar] [CrossRef]
- Peralta-Zaragoza, O.; Deas, J.; Meneses-Acosta, A.; De la, O.G.F.; Fernandez-Tilapa, G.; Gomez-Ceron, C.; Benitez-Boijseauneau, O.; Burguete-Garcia, A.; Torres-Poveda, K.; Bermudez-Morales, V.H.; et al. Relevance of miR-21 in regulation of tumor suppressor gene PTEN in human cervical cancer cells. BMC Cancer 2016, 16, 215. [Google Scholar] [CrossRef]
- Feng, Y.H.; Tsao, C.J. Emerging role of microRNA-21 in cancer. Biomed. Rep. 2016, 5, 395–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jajoo, S.; Mukherjea, D.; Kaur, T.; Sheehan, K.E.; Sheth, S.; Borse, V.; Rybak, L.P.; Ramkumar, V. Essential role of NADPH oxidase-dependent reactive oxygen species generation in regulating microRNA-21 expression and function in prostate cancer. Antioxid. Redox Signal. 2013, 19, 1863–1876. [Google Scholar] [CrossRef] [PubMed]
- Ling, M.; Li, Y.; Xu, Y.; Pang, Y.; Shen, L.; Jiang, R.; Zhao, Y.; Yang, X.; Zhang, J.; Zhou, J.; et al. Regulation of miRNA-21 by reactive oxygen species-activated ERK/NF-kappaB in arsenite-induced cell transformation. Free Radic. Biol. Med. 2012, 52, 1508–1518. [Google Scholar] [CrossRef] [PubMed]
- Mohr, S.; Doebele, C.; Comoglio, F.; Berg, T.; Beck, J.; Bohnenberger, H.; Alexe, G.; Corso, J.; Strobel, P.; Wachter, A.; et al. Hoxa9 and Meis1 cooperatively induce addiction to syk signaling by suppressing miR-146a in acute myeloid leukemia. Cancer Cell 2017, 31, 549–562. [Google Scholar] [CrossRef]
- Hu, H.Y.; Li, K.P.; Wang, X.J.; Liu, Y.; Lu, Z.G.; Dong, R.H.; Guo, H.B.; Zhang, M.X. Set9, NF-kappaB, and microRNA-21 mediate berberine-induced apoptosis of human multiple myeloma cells. Acta Pharmacol. Sin. 2013, 34, 157–166. [Google Scholar] [CrossRef]
- Hong, J.; Wang, Y.; Hu, B.C.; Xu, L.; Liu, J.Q.; Chen, M.H.; Wang, J.Z.; Han, F.; Zheng, Y.; Chen, X.; et al. Transcriptional downregulation of microRNA-19a by ROS production and NF-kappaB deactivation governs resistance to oxidative stress-initiated apoptosis. Oncotarget 2017, 8, 70967–70981. [Google Scholar] [CrossRef]
- Gerri, C.; Marin-Juez, R.; Marass, M.; Marks, A.; Maischein, H.M.; Stainier, D.Y.R. Hif-1alpha regulates macrophage-endothelial interactions during blood vessel development in zebrafish. Nat. Commun. 2017, 8, 15492. [Google Scholar] [CrossRef]
- Koh, M.Y.; Lemos, R. Jr.; Liu, X.; Powis, G. The hypoxia-associated factor switches cells from HIF-1alpha- to HIF-2alpha-dependent signaling promoting stem cell characteristics, aggressive tumor growth and invasion. Cancer Res. 2011, 71, 4015–4027. [Google Scholar] [CrossRef]
- Serocki, M.; Bartoszewska, S.; Janaszak-Jasiecka, A.; Ochocka, R.J.; Collawn, J.F.; Bartoszewski, R. miRNAs regulate the HIF switch during hypoxia: A novel therapeutic target. Angiogenesis 2018, 21, 183–202. [Google Scholar] [CrossRef]
- Wang, H.; Flach, H.; Onizawa, M.; Wei, L.; McManus, M.T.; Weiss, A. Negative regulation of Hif1a expression and TH17 differentiation by the hypoxia-regulated microRNA miR-210. Nat. Immunol. 2014, 15, 393–401. [Google Scholar] [CrossRef]
- Seok, J.K.; Lee, S.H.; Kim, M.J.; Lee, Y.M. MicroRNA-382 induced by HIF-1alpha is an angiogenic miR targeting the tumor suppressor phosphatase and tensin homolog. Nucleic Acids Res. 2014, 42, 8062–8072. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.; Liu, X.; Lin, F.; Li, P.; Liu, K.; Geng, R.; Dai, C.; Lin, Y.; Tang, W.; Wu, Z.; et al. MicroRNA-421 regulated by HIF-1alpha promotes metastasis, inhibits apoptosis, and induces cisplatin resistance by targeting E-cadherin and caspase-3 in gastric cancer. Oncotarget 2016, 7, 24466–24482. [Google Scholar] [CrossRef] [PubMed]
- Nagpal, N.; Ahmad, H.M.; Chameettachal, S.; Sundar, D.; Ghosh, S.; Kulshreshtha, R. HIF-inducible miR-191 promotes migration in breast cancer through complex regulation of TGFbeta-signaling in hypoxic microenvironment. Sci. Rep. 2015, 5, 9650. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, K.; Wei, Q.; Pabla, N.; Dong, G.; Mi, Q.S.; Liang, M.; Mei, C.; Dong, Z. MicroRNA-687 Induced by hypoxia-inducible factor-1 targets phosphatase and tensin homolog in renal ischemia-reperfusion injury. JASN 2015, 26, 1588–1596. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Sonveaux, P.; Rabbani, Z.N.; Liu, S.; Yan, B.; Huang, Q.; Vujaskovic, Z.; Dewhirst, M.W.; Li, C.Y. Regulation of HIF-1alpha stability through S-nitrosylation. Mol. Cell 2007, 26, 63–74. [Google Scholar] [CrossRef]
- Lim, J.H.; Lee, Y.M.; Chun, Y.S.; Chen, J.; Kim, J.E.; Park, J.W. Sirtuin 1 modulates cellular responses to hypoxia by deacetylating hypoxia-inducible factor 1alpha. Mol. Cell 2010, 38, 864–878. [Google Scholar] [CrossRef]
- Macfarlane, L.A.; Murphy, P.R. MicroRNA: Biogenesis, function and role in cancer. Curr. Genom. 2010, 11, 537–561. [Google Scholar] [CrossRef]
- Abdi, J.; Rastgoo, N.; Li, L.; Chen, W.; Chang, H. Role of tumor suppressor p53 and micro-RNA interplay in multiple myeloma pathogenesis. J. Hematol. Oncol. 2017, 10, 169. [Google Scholar] [CrossRef]
- Ungvari, Z.; Tucsek, Z.; Sosnowska, D.; Toth, P.; Gautam, T.; Podlutsky, A.; Csiszar, A.; Losonczy, G.; Valcarcel-Ares, M.N.; Sonntag, W.E.; et al. Aging-induced dysregulation of dicer1-dependent microRNA expression impairs angiogenic capacity of rat cerebromicrovascular endothelial cells. J. Gerontol. Ser. A Biol. Sci. Med Sci. 2013, 68, 877–891. [Google Scholar] [CrossRef]
- Shilo, S.; Roy, S.; Khanna, S.; Sen, C.K. Evidence for the involvement of miRNA in redox regulated angiogenic response of human microvascular endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 471–477. [Google Scholar] [CrossRef]
- Iliopoulos, D.; Hirsch, H.A.; Struhl, K. An epigenetic switch involving NF-kappaB, Lin28, Let-7 MicroRNA, and IL6 links inflammation to cell transformation. Cell 2009, 139, 693–706. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.X.; Gao, J.; Ding, S.L.; Wang, K.; Jiao, J.Q.; Wang, Y.; Sun, T.; Zhou, L.Y.; Long, B.; Zhang, X.J.; et al. Oxidative modification of miR-184 enables it to target Bcl-xL and Bcl-w. Mol. Cell 2015, 59, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Panday, A.; Sahoo, M.K.; Osorio, D.; Batra, S. NADPH oxidases: An overview from structure to innate immunity-associated pathologies. Cell. Mol. Immunol. 2015, 12, 5–23. [Google Scholar] [CrossRef] [PubMed]
- Li, S.Z.; Hu, Y.Y.; Zhao, J.; Zhao, Y.B.; Sun, J.D.; Yang, Y.F.; Ji, C.C.; Liu, Z.B.; Cao, W.D.; Qu, Y.; et al. MicroRNA-34a induces apoptosis in the human glioma cell line, A172, through enhanced ROS production and NOX2 expression. Biochem. Biophys. Res. Commun. 2014, 444, 6–12. [Google Scholar] [CrossRef]
- Liu, W.; Zabirnyk, O.; Wang, H.; Shiao, Y.H.; Nickerson, M.L.; Khalil, S.; Anderson, L.M.; Perantoni, A.O.; Phang, J.M. miR-23b targets proline oxidase, a novel tumor suppressor protein in renal cancer. Oncogene 2010, 29, 4914–4924. [Google Scholar] [CrossRef]
- Gordillo, G.M.; Biswas, A.; Khanna, S.; Pan, X.; Sinha, M.; Roy, S.; Sen, C.K. Dicer knockdown inhibits endothelial cell tumor growth via microRNA 21a-3p targeting of Nox-4. J. Biol. Chem. 2014, 289, 9027–9038. [Google Scholar] [CrossRef]
- Kim, J.H.; Park, S.G.; Song, S.Y.; Kim, J.K.; Sung, J.H. Reactive oxygen species-responsive miR-210 regulates proliferation and migration of adipose-derived stem cells via PTPN2. Cell Death Dis. 2013, 4, e588. [Google Scholar] [CrossRef]
- Chen, Z.; Li, Y.; Zhang, H.; Huang, P.; Luthra, R. Hypoxia-regulated microRNA-210 modulates mitochondrial function and decreases ISCU and COX10 expression. Oncogene 2010, 29, 4362–4368. [Google Scholar] [CrossRef] [Green Version]
- Venkataraman, S.; Alimova, I.; Fan, R.; Harris, P.; Foreman, N.; Vibhakar, R. MicroRNA 128a increases intracellular ROS level by targeting Bmi-1 and inhibits medulloblastoma cancer cell growth by promoting senescence. PLoS ONE 2010, 5, e10748. [Google Scholar] [CrossRef]
- Sripada, L.; Singh, K.; Lipatova, A.V.; Singh, A.; Prajapati, P.; Tomar, D.; Bhatelia, K.; Roy, M.; Singh, R.; Godbole, M.M.; et al. hsa-miR-4485 regulates mitochondrial functions and inhibits the tumorigenicity of breast cancer cells. J. Mol. Med. 2017, 95, 641–651. [Google Scholar] [CrossRef]
- Glorieux, C.; Calderon, P.B. Catalase, a remarkable enzyme: Targeting the oldest antioxidant enzyme to find a new cancer treatment approach. Biol. Chem. 2017, 398, 1095–1108. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Ng, W.L.; Wang, P.; Tian, L.; Werner, E.; Wang, H.; Doetsch, P.; Wang, Y. MicroRNA-21 modulates the levels of reactive oxygen species by targeting SOD3 and TNFalpha. Cancer Res. 2012, 72, 4707–4713. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Wu, J.; Pan, C.; Wang, H.; Ying, X.; Zhou, Y.; Yu, H.; Zuo, Y.; Pan, Z.; Liu, R.Y.; et al. Genetic and epigenetic down-regulation of microRNA-212 promotes colorectal tumor metastasis via dysregulation of MnSOD. Gastroenterology 2013, 145, 426–436. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Wells, A.; Padilla, M.T.; Kato, K.; Kim, K.C.; Lin, Y. A signaling pathway consisting of miR-551b, catalase and MUC1 contributes to acquired apoptosis resistance and chemoresistance. Carcinogenesis 2014, 35, 2457–2466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Chen, W.; Bai, L.; Chen, W.; Padilla, M.T.; Lin, A.S.; Shi, S.; Wang, X.; Lin, Y. Receptor-interacting protein 1 increases chemoresistance by maintaining inhibitor of apoptosis protein levels and reducing reactive oxygen species through a microRNA-146a-mediated catalase pathway. J. Biol. Chem. 2014, 289, 5654–5663. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Zhu, C.F.; Ma, M.Z.; Chen, G.; Song, M.; Zeng, Z.L.; Lu, W.H.; Yang, J.; Wen, S.; Chiao, P.J.; et al. Micro-RNA-155 is induced by K-Ras oncogenic signal and promotes ROS stress in pancreatic cancer. Oncotarget 2015, 6, 21148–21158. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef]
- Cortez, M.A.; Valdecanas, D.; Zhang, X.; Zhan, Y.; Bhardwaj, V.; Calin, G.A.; Komaki, R.; Giri, D.K.; Quini, C.C.; Wolfe, T.; et al. Therapeutic delivery of miR-200c enhances radiosensitivity in lung cancer. J. Am. Soc. Gene Ther. 2014, 22, 1494–1503. [Google Scholar] [CrossRef]
- Yang, M.; Yao, Y.; Eades, G.; Zhang, Y.; Zhou, Q. MiR-28 regulates Nrf2 expression through a Keap1-independent mechanism. Breast Cancer Res. Treat. 2011, 129, 983–991. [Google Scholar] [CrossRef]
- Singh, B.; Ronghe, A.M.; Chatterjee, A.; Bhat, N.K.; Bhat, H.K. MicroRNA-93 regulates NRF2 expression and is associated with breast carcinogenesis. Carcinogenesis 2013, 34, 1165–1172. [Google Scholar] [CrossRef] [Green Version]
- Papp, D.; Lenti, K.; Modos, D.; Fazekas, D.; Dul, Z.; Turei, D.; Foldvari-Nagy, L.; Nussinov, R.; Csermely, P.; Korcsmaros, T. The NRF2-related interactome and regulome contain multifunctional proteins and fine-tuned autoregulatory loops. FEBS Lett. 2012, 586, 1795–1802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabaria, S.; Choi, D.C.; Chaudhuri, A.D.; Jain, M.R.; Li, H.; Junn, E. MicroRNA-7 activates Nrf2 pathway by targeting Keap1 expression. Free Radic. Biol. Med. 2015, 89, 548–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eades, G.; Yang, M.; Yao, Y.; Zhang, Y.; Zhou, Q. miR-200a regulates Nrf2 activation by targeting Keap1 mRNA in breast cancer cells. J. Biol. Chem. 2011, 286, 40725–40733. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Lee, K.S.; Lee, D.K.; Kim, J.; Kwak, S.N.; Ha, K.S.; Choe, J.; Won, M.H.; Cho, B.R.; Jeoung, D.; et al. Hypoxia-responsive microRNA-101 promotes angiogenesis via heme oxygenase-1/vascular endothelial growth factor axis by targeting cullin 3. Antioxid. Redox Signal. 2014, 21, 2469–2482. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Zhu, H.; Wang, C.; Zhu, X.; Liu, G.; Chen, C.; Cui, Z. microRNA-455 targets cullin 3 to activate Nrf2 signaling and protect human osteoblasts from hydrogen peroxide. Oncotarget 2017, 8, 59225–59234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Huang, K.; You, Y.; Fu, X.; Hu, L.; Song, L.; Meng, Y. Hypoxia-induced miR-210 in epithelial ovarian cancer enhances cancer cell viability via promoting proliferation and inhibiting apoptosis. Int. J. Oncol. 2014, 44, 2111–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tay, Y.; Rinn, J.; Pandolfi, P.P. The multilayered complexity of ceRNA crosstalk and competition. Nature 2014, 505, 344–352. [Google Scholar] [CrossRef] [Green Version]
- Vinceti, M.; Filippini, T.; Del Giovane, C.; Dennert, G.; Zwahlen, M.; Brinkman, M.; Zeegers, M.P.; Horneber, M.; D’Amico, R.; Crespi, C.M. Selenium for preventing cancer. Cochrane Database Syst. Rev. 2018, 1, CD005195. [Google Scholar] [CrossRef]
- Harvie, M. Nutritional supplements and cancer: Potential benefits and proven harms. In American Society of Clinical Oncology Educational Book; American Society of Clinical Oncology: Alexandria, VA, USA, 2014. [Google Scholar] [CrossRef]
- Druesne-Pecollo, N.; Latino-Martel, P.; Norat, T.; Barrandon, E.; Bertrais, S.; Galan, P.; Hercberg, S. Beta-carotene supplementation and cancer risk: A systematic review and metaanalysis of randomized controlled trials. Int. J. Cancer 2010, 127, 172–184. [Google Scholar] [CrossRef]
- Jacobs, C.; Hutton, B.; Ng, T.; Shorr, R.; Clemons, M. Is there a role for oral or intravenous ascorbate (vitamin C) in treating patients with cancer? A systematic review. Oncologist 2015, 20, 210–223. [Google Scholar] [CrossRef] [PubMed]
- Sablina, A.A.; Budanov, A.V.; Ilyinskaya, G.V.; Agapova, L.S.; Kravchenko, J.E.; Chumakov, P.M. The antioxidant function of the p53 tumor suppressor. Nature Med. 2005, 11, 1306–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schubert, R.; Erker, L.; Barlow, C.; Yakushiji, H.; Larson, D.; Russo, A.; Mitchell, J.B.; Wynshaw-Boris, A. Cancer chemoprevention by the antioxidant tempol in Atm-deficient mice. Hum. Mol. Genet. 2004, 13, 1793–1802. [Google Scholar] [CrossRef] [PubMed]
- Sayin, V.I.; Ibrahim, M.X.; Larsson, E.; Nilsson, J.A.; Lindahl, P.; Bergo, M.O. Antioxidants accelerate lung cancer progression in mice. Sci. Transl. Med. 2014, 6, 221ra215. [Google Scholar] [CrossRef] [PubMed]
- Harris, I.S.; Treloar, A.E.; Inoue, S.; Sasaki, M.; Gorrini, C.; Lee, K.C.; Yung, K.Y.; Brenner, D.; Knobbe-Thomsen, C.B.; Cox, M.A.; et al. Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell 2015, 27, 211–222. [Google Scholar] [CrossRef]
- Wang, H.; Liu, X.; Long, M.; Huang, Y.; Zhang, L.; Zhang, R.; Zheng, Y.; Liao, X.; Wang, Y.; Liao, Q.; et al. NRF2 activation by antioxidant antidiabetic agents accelerates tumor metastasis. Sci. Transl. Med. 2016, 8, 334ra351. [Google Scholar] [CrossRef]
- Qu, Y.; Wang, J.; Ray, P.S.; Guo, H.; Huang, J.; Shin-Sim, M.; Bukoye, B.A.; Liu, B.; Lee, A.V.; Lin, X.; et al. Thioredoxin-like 2 regulates human cancer cell growth and metastasis via redox homeostasis and NF-kappaB signaling. J. Clin. Investig. 2011, 121, 212–225. [Google Scholar] [CrossRef]
- Kamarajugadda, S.; Cai, Q.; Chen, H.; Nayak, S.; Zhu, J.; He, M.; Jin, Y.; Zhang, Y.; Ai, L.; Martin, S.S.; et al. Manganese superoxide dismutase promotes anoikis resistance and tumor metastasis. Cell Death Dis. 2013, 4, e504. [Google Scholar] [CrossRef]
- Glasauer, A.; Sena, L.A.; Diebold, L.P.; Mazar, A.P.; Chandel, N.S. Targeting SOD1 reduces experimental non-small-cell lung cancer. J. Clin. Investig. 2014, 124, 117–128. [Google Scholar] [CrossRef]
- Nguyen, A.; Loo, J.M.; Mital, R.; Weinberg, E.M.; Man, F.Y.; Zeng, Z.; Paty, P.B.; Saltz, L.; Janjigian, Y.Y.; de Stanchina, E.; et al. PKLR promotes colorectal cancer liver colonization through induction of glutathione synthesis. J. Clin. Investig. 2016, 126, 681–694. [Google Scholar] [CrossRef]
- DeNicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011, 475, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, H.; Motohashi, H. NRF2 addiction in cancer cells. Cancer Sci. 2018, 109, 900–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Gal, K.; Ibrahim, M.X.; Wiel, C.; Sayin, V.I.; Akula, M.K.; Karlsson, C.; Dalin, M.G.; Akyurek, L.M.; Lindahl, P.; Nilsson, J.; et al. Antioxidants can increase melanoma metastasis in mice. Sci. Transl. Med. 2015, 7, 308re308. [Google Scholar] [CrossRef] [PubMed]
- Patra, K.C.; Hay, N. The pentose phosphate pathway and cancer. Trends Biochem. Sci. 2014, 39, 347–354. [Google Scholar] [CrossRef] [Green Version]
- Fan, J.; Ye, J.; Kamphorst, J.J.; Shlomi, T.; Thompson, C.B.; Rabinowitz, J.D. Quantitative flux analysis reveals folate-dependent NADPH production. Nature 2014, 510, 298–302. [Google Scholar] [CrossRef] [Green Version]
- Ren, J.G.; Seth, P.; Clish, C.B.; Lorkiewicz, P.K.; Higashi, R.M.; Lane, A.N.; Fan, T.W.; Sukhatme, V.P. Knockdown of malic enzyme 2 suppresses lung tumor growth, induces differentiation and impacts PI3K/AKT signaling. Sci. Rep. 2014, 4, 5414. [Google Scholar] [CrossRef]
- Mollaei, H.; Safaralizadeh, R.; Rostami, Z. MicroRNA replacement therapy in cancer. J. Cell. Physiol. 2019, 234, 12369–12384. [Google Scholar] [CrossRef]
- Bader, A.G.; Brown, D.; Winkler, M. The promise of microRNA replacement therapy. Cancer Res. 2010, 70, 7027–7030. [Google Scholar] [CrossRef]
- Ling, H.; Fabbri, M.; Calin, G.A. MicroRNAs and other non-coding RNAs as targets for anticancer drug development. Nature reviews. Drug Discov. 2013, 12, 847–865. [Google Scholar] [CrossRef] [Green Version]
- Hanna, J.; Hossain, G.S.; Kocerha, J. The Potential for microRNA Therapeutics and Clinical Research. Front. Genet. 2019, 10, 478. [Google Scholar] [CrossRef] [Green Version]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
R. Babu, K.; Tay, Y. The Yin-Yang Regulation of Reactive Oxygen Species and MicroRNAs in Cancer. Int. J. Mol. Sci. 2019, 20, 5335. https://doi.org/10.3390/ijms20215335
R. Babu K, Tay Y. The Yin-Yang Regulation of Reactive Oxygen Species and MicroRNAs in Cancer. International Journal of Molecular Sciences. 2019; 20(21):5335. https://doi.org/10.3390/ijms20215335
Chicago/Turabian StyleR. Babu, Kamesh, and Yvonne Tay. 2019. "The Yin-Yang Regulation of Reactive Oxygen Species and MicroRNAs in Cancer" International Journal of Molecular Sciences 20, no. 21: 5335. https://doi.org/10.3390/ijms20215335
APA StyleR. Babu, K., & Tay, Y. (2019). The Yin-Yang Regulation of Reactive Oxygen Species and MicroRNAs in Cancer. International Journal of Molecular Sciences, 20(21), 5335. https://doi.org/10.3390/ijms20215335