Synthesis and Evaluation of Anticancer Activity of New 4-Acyloxy Derivatives of Robustic Acid

 ,
,
Abstract
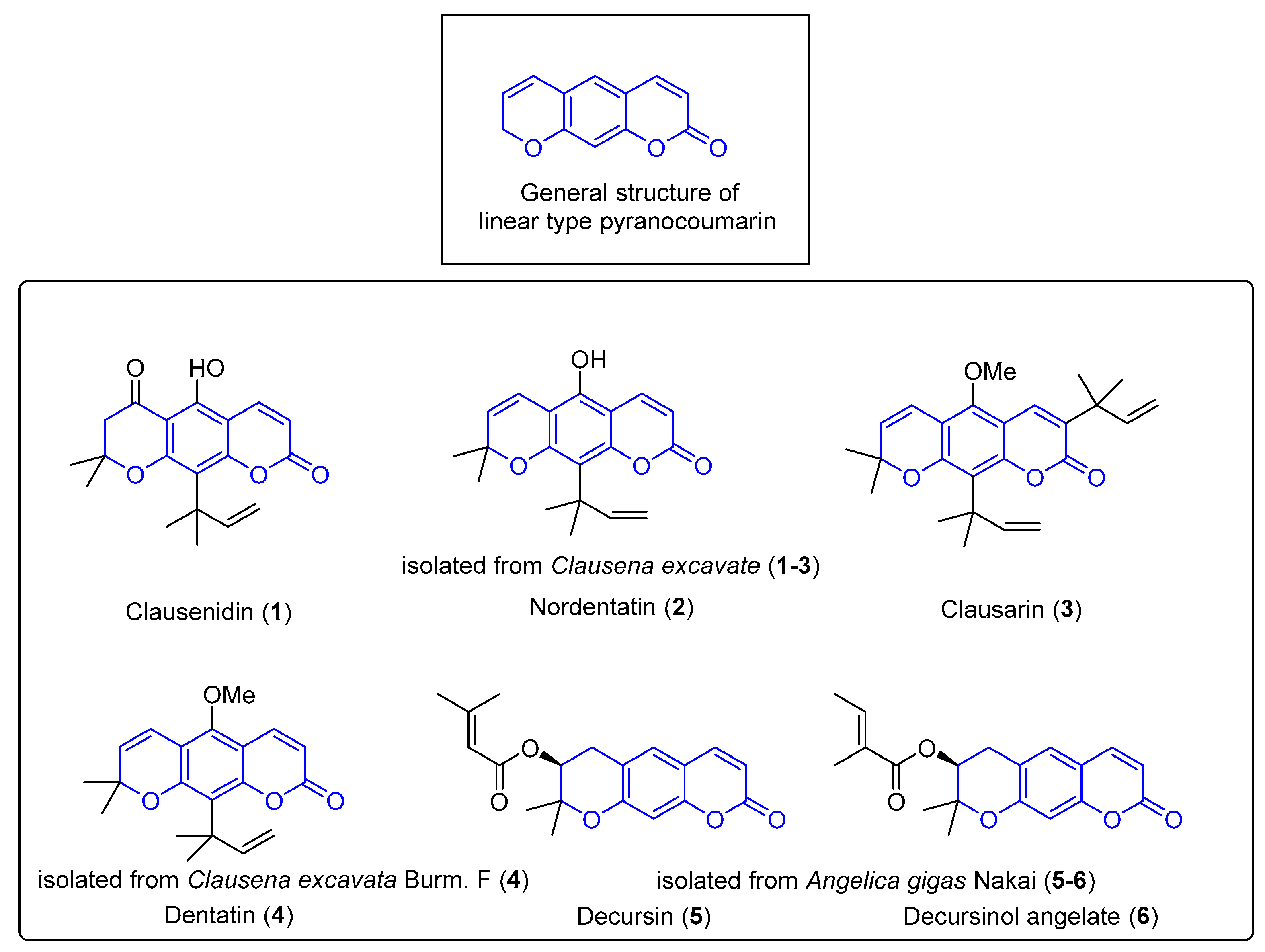
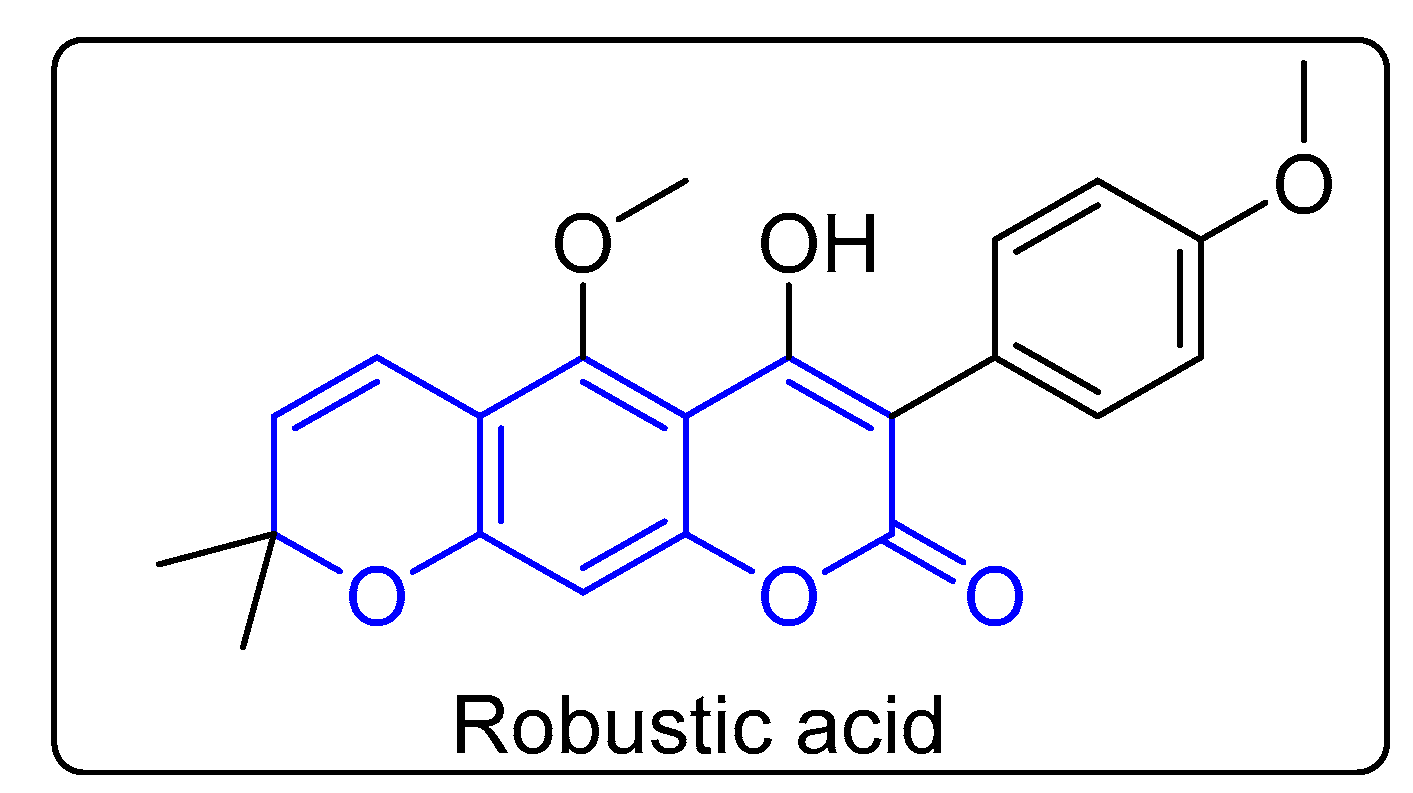
:1. Introduction
2. Results and Discussion
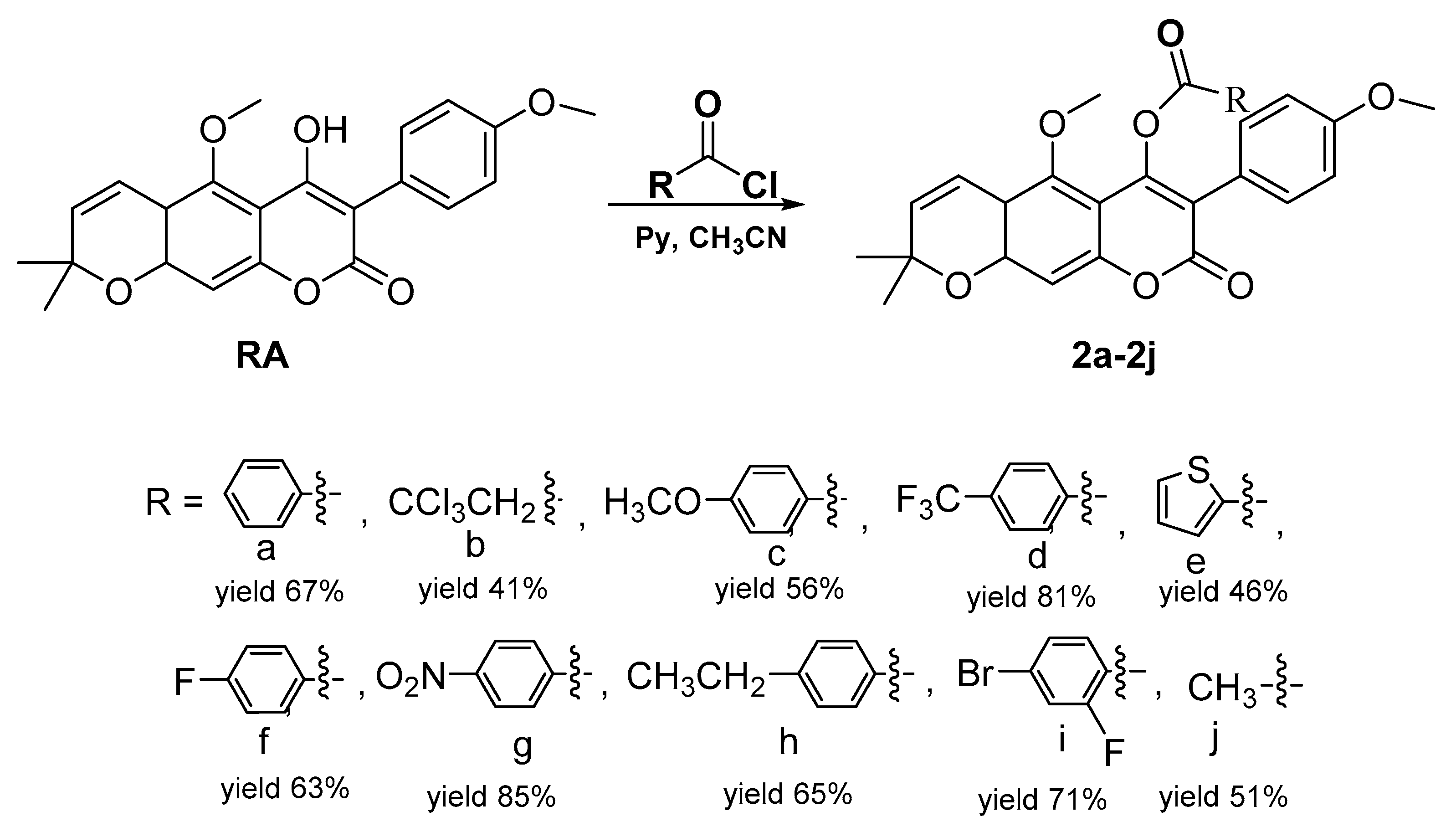
2.1. Chemistry
2.2. Biological Study
2.2.1. Anti-Proliferative Activity Screening
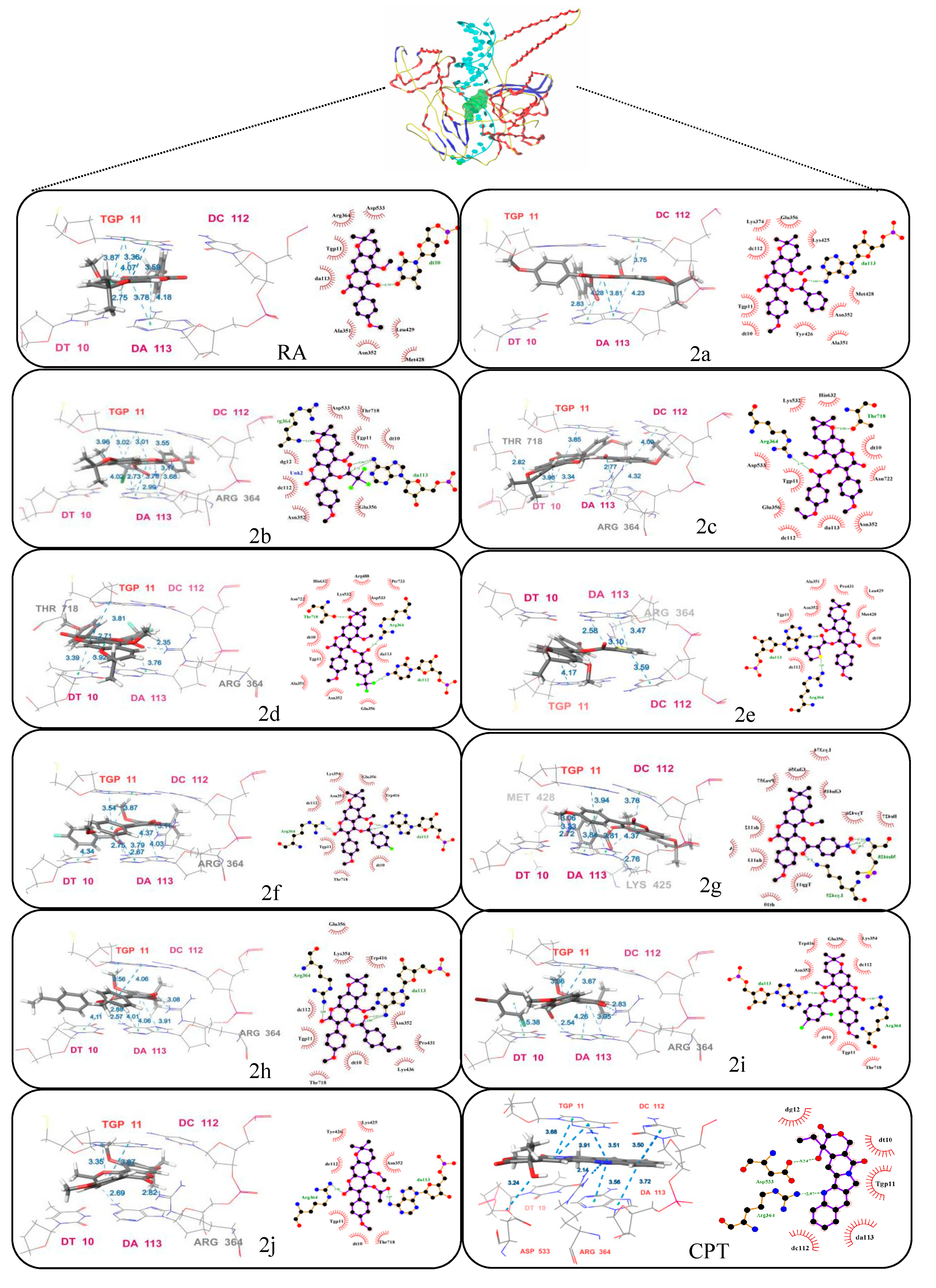
2.2.2. Evaluation of DNA Topoisomerase I (Topo I) Inhibitory Activity and Molecular Docking Study
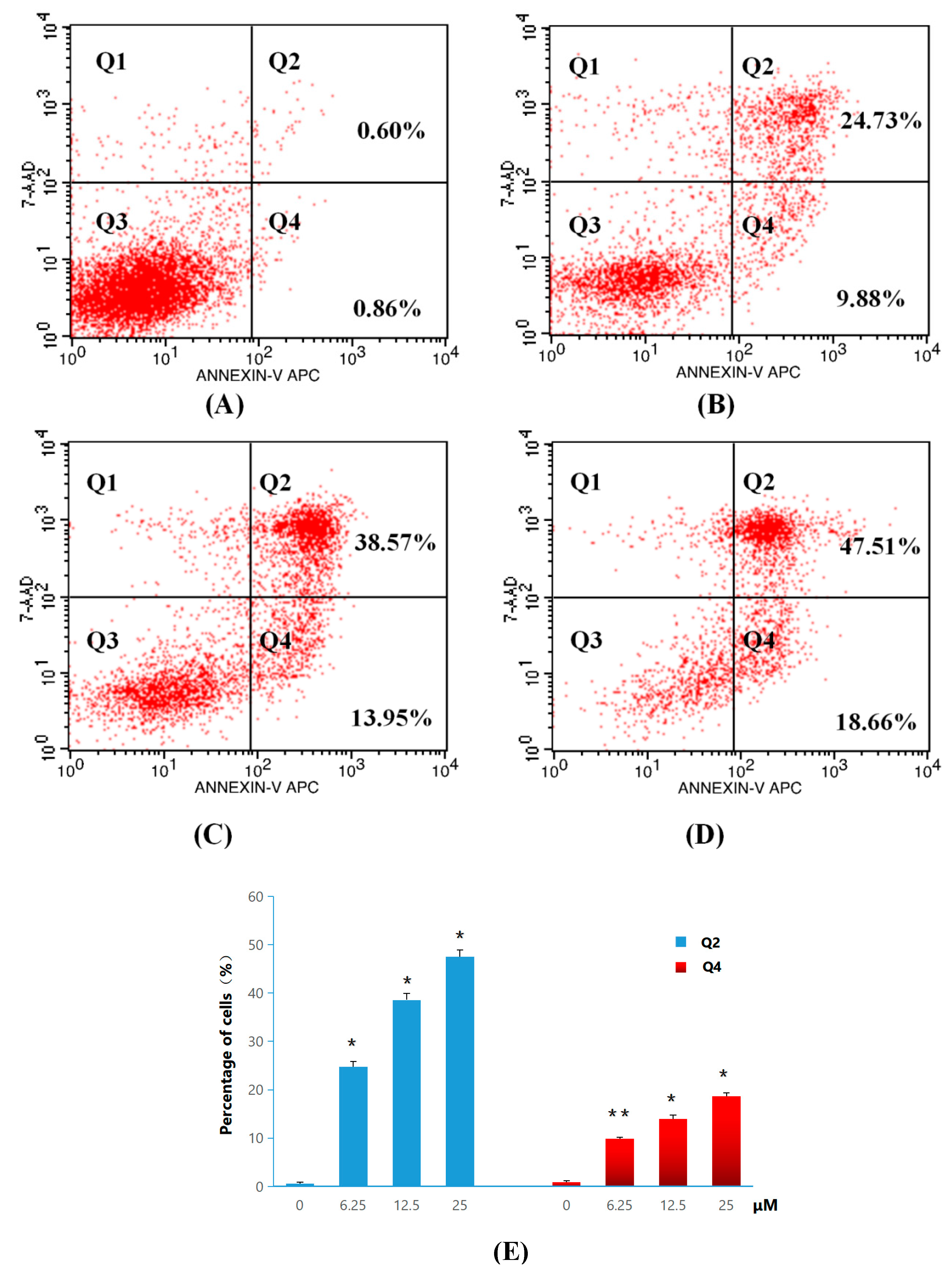
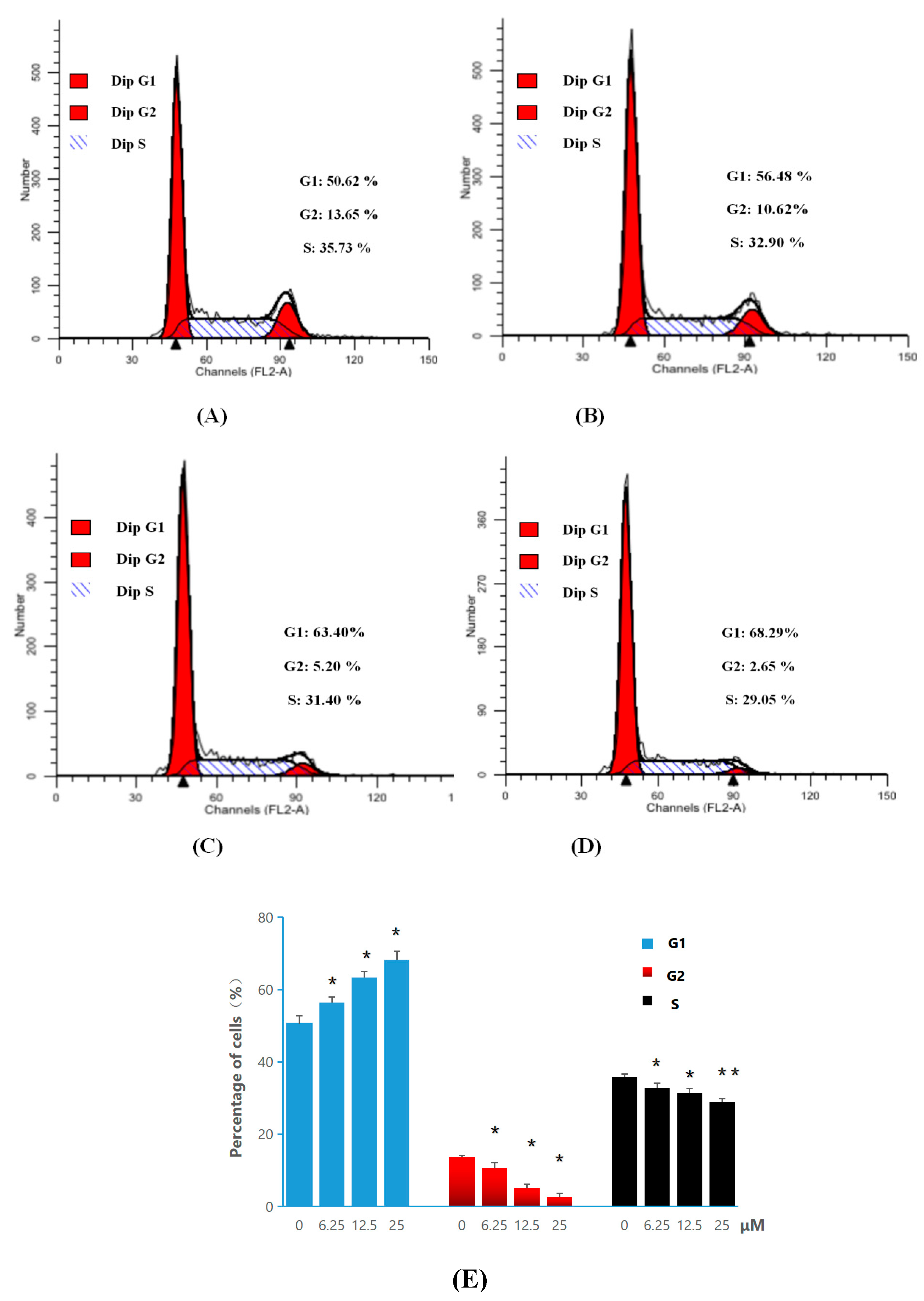
2.2.3. Apoptosis and Cell-Cycle Analysis
3. Materials and Methods
3.1. Synthesis Methods
General Procedure for the Synthesis of 4-Acyloxy Derivatives of Robustic Acid 2a–2j
3.2. Biological Activity
3.2.1. Antiproliferative Activity
3.2.2. Topo I Inhibitory Activity
3.2.3. General Procedure for AO/EB Staining
3.2.4. General Procedure for Apoptosis Ratio Determination
3.2.5. General Procedure for Cell Cycle Analysis
3.3. Molecular Docking
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Klenkar, J.; Molnar, M. Natural and synthetic coumarins as potential anticancer agents. J. Chem. Pharm. Res. 2015, 7, 1223–1238. [Google Scholar]
- Xia, L.X.; Wang, Y.B.; Huang, W.L.; Qian, H. Research advance of anticancer drugs with coumarin structures. Chin. J. New Drugs 2013, 23, 2392–2404. [Google Scholar]
- Symeonidis, T.; Fylaktakidou, K.C.; Hadjipavlou-Litina, D.J.; Litinas, K.E. Synthesis and anti-inflammatory evaluation of novel angularly or linearly fused coumarins. Eur. J. Med. Chem. 2009, 44, 5012–5017. [Google Scholar] [CrossRef] [PubMed]
- Kongkathip, B.; Kongkathip, N.; Sunthitikawinsakul, A.; Napaswat, C.; Yoosook, C. Anti-HIV-1 constituents from Clausena excavata: Part II. Carbazoles and a pyranocoumarin. Phytother. Res. 2005, 19, 728–731. [Google Scholar] [CrossRef] [PubMed]
- Su, C.R.; Yeh, S.F.; Liu, C.M.; Damu, A.G.; Kuo, T.; Chiang, P.; Bastow, K.F.; Lee, K.; Wu, T. Anti-HBV and cytotoxic activities of pyranocoumarin derivatives. Bioorg. Med. Chem. 2009, 17, 6137–6143. [Google Scholar] [CrossRef]
- Wu, J.; Fong, W.; Zhang, J.; Leung, C.; Kwong, H.; Yang, M.; Li, D.; Cheung, H. Reversal of multidrug resistance in cancer cells by pyranocoumarins isolated from Radix Peucedani. Eur. J. Pharmacal. 2003, 473, 9–17. [Google Scholar] [CrossRef]
- Lima, V.; Silva, C.B.; Mafezoli, J.; Bezerra, M.M.; Moraes, M.O.; Mourão, G.S.; Silva, J.N.; Oliveira, M.C.F. Antinociceptive activity of the pyranocoumarin seselin in mice. Fitoterapla 2006, 77, 574–578. [Google Scholar] [CrossRef]
- Lee, H.J.; Lee, H.J.; Lee, E.O.; Lee, J.H.; Lee, K.S.; Kim, K.H.; Kim, S.H.; Lü, J. In vivo Anti-Cancer Activity of Korean Angelica Gigas and its Major Pyranocoumarin Decursin. Am. J. Chin. Med. 2009, 37, 127–142. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Li, L.; Jiang, C.; Xing, C.; Kim, S.; Lu, J. Anti-cancer and Other Bioactivities of Korean Angelica gigas Nakai (AGN) and Its Major Pyranocoumarin Compounds. Anti Cancer Agents Med. Chem. (Former. Curr. Med. Chem. Anti Cancer Agents) 2012, 12, 1239–1254. [Google Scholar] [CrossRef]
- Huang, L.; Feng, Z.L.; Wang, Y.T.; Lin, L.G. Anticancer carbazole alkaloids and coumarins from Clausena plants: A review. Chin. J. Nat. Med. 2017, 15, 881–888. [Google Scholar] [CrossRef]
- Peng, W.W.; Song, W.W.; Tan, N.H. Research Progress On Coumarins from Clausena and Their Pharmacological. Nat. Prod. Res. Dev. 2017, 8, 1428–1438. [Google Scholar]
- Arbab, I.A.; Looi, C.Y.; Abdul, A.B.; Cheah, F.K.; Wong, W.F.; Sukari, M.A.; Abdullah, R.; Mohan, S.; Syam, S.; Arya, A. Dentatin Induces Apoptosis in Prostate Cancer Cells via Bcl-2, Bcl-xL, Survivin Downregulation, Caspase-9, -3/7 Activation, and NF-κB Inhibition. Evid. Based Compl. Altern. Med. 2012, 2012, 856029. [Google Scholar] [CrossRef] [PubMed]
- Arbab, I.A.; Abdul, A.B.; Sukari, M.A.; Abdullah, R.; Syam, S.; Kamalidehghan, B.; Ibrahim, M.Y.; Taha, M.M.; Abdelwahab, S.I.; Ali, H.M. Dentatin isolated from Clausena excavata induces apoptosis in MCF-7 cells through the intrinsic pathway with involvement of NF-κ B signalling and G0/G1 cell cycle arrest: A bioassay-guided approach. J. Ethnopharmacol. 2013, 145, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Andas, A.R.J.; Ahmad Bustamam, A.; Heshu Sulaiman, R.; Mohd Aspollah, S.; Siddig Ibrahim, A.; Nozlena Abdul, S.; Theebaa, A.; Ismail Adam, A. Dentatin from Clausena excavata Induces Apoptosis in HepG2 Cells via Mitochondrial Mediated Signaling. Asian Pac. J. Cancer Prev. 2015, 16, 4311. [Google Scholar] [CrossRef] [PubMed]
- Ahn, K.S.; Sim, W.S.; Kim, I.H. Decursin: A cytotoxic agent and protein kinase C activator from the root of Angelica gigas. Planta Med. 1996, 62, 7–9. [Google Scholar] [CrossRef]
- Ahn, K.S.; Sim, W.S.; Lee, I.K.; Seu, Y.B.; Kim, I.H. Decursinol angelate: A cytotoxic and protein kinase C activating agent from the root of Angelica gigas. Planta Med. 1997, 63, 360–361. [Google Scholar] [CrossRef]
- Wei, J.H.; Huo, L.N.; Huang, M.; Chen, R.; Feng, X. Robustic Acid and its Extraction Method and Application. CN2014105222249, 30 September 2014. [Google Scholar]
- Editorial Committee of the National Chinese Medicine Administrative Bureau. Chinese Materia Medica; Shanghai Scientific and Technical Education Publishing House: Shanghai, China, 2005; Volume 4. [Google Scholar]
- Pommier, Y. Diversity of DNA topoisomerases I and inhibitors. Biochimie 1998, 80, 255–270. [Google Scholar] [CrossRef]
- Ismail, M.M.F.; Rateb, H.S.; Hussein, M.M.M. Synthesis and docking studies of novel benzopyran-2-ones with anticancer activity. Eur. J. Med. Chem. 2010, 45, 3950–3959. [Google Scholar] [CrossRef]
- Zhao, L.X.; Moon, Y.S.; Basnet, A.; Kim, E.K.; Jahng, Y.; Park, J.G.; Jeong, T.C.; Cho, W.J.; Choi, S.U.; Chong, O.L. Synthesis, topoisomerase I inhibition and structure–activity relationship study of 2,4,6-trisubstituted pyridine derivatives. Bioorg. Med. Chem. Lett. 2004, 14, 1333–1337. [Google Scholar] [CrossRef]
- Hevener, K.E.; Zhao, W.; Ball, D.M.; Babaoglu, K.; Qi, J.; White, S.W.; Lee, R.E. Validation of Molecular Docking Programs for Virtual Screening against Dihydropteroate Synthase. J. Chem. Inf. Model. 2009, 2, 444–460. [Google Scholar] [CrossRef]
- Lill, M. Virtual screening in drug design. In Silico Models for Drug Discovery; Humana Press: Totowa, NJ, USA, 2013; Volume 993, pp. 1–12. [Google Scholar]
- Olivares, E.M.; Lwande, W.; Monache, F.D.; Bettolo, G.B.M. A pyrano-isoflavone from seeds of Milletia thonningii. Phytochemistry 1982, 21, 1763–1765. [Google Scholar] [CrossRef]
- Falshaw, C.P.; Harmer, R.A.; Ollis, W.D.; Wheeler, R.E.; Rao, N.V.S. Natural occurrence of 3-aryl-4-hydroxycoumarins. Part II. Phytochemical examination of Derris scandens (Roxb.) Benth. J. Chem. Soc. C Org. 1969, 3, 374. [Google Scholar] [CrossRef]
- Jin, Y.; Chen, Q.; Shi, X.; Lu, Z.; Cheng, C.; Lai, Y.; Zheng, Q.; Pan, J. Activity of triptolide against human mast cells harboring the kinase domain mutant KIT. Cancer Sci. 2009, 100, 1335–1343. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Jing, D.; Chen, R.; Haroon, U.R.; Jiang, J.; Liu, X.; Wang, L.; Wang, P. Design, synthesis and evaluation of novel sophoridinic imine derivatives containing conjugated planar structure as potent anticancer agents. Bioorg. Med. Chem. 2018, 26, 4136–4144. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | HL-60 | A-549 | SMMC-7721 | HepG2 | Hela | LO2 | BEAS-2B |
|---|---|---|---|---|---|---|---|
| 2a | >100 | 98.42 ± 4.18 | 40.24 ± 1.38 | >100 | >100 | >100 | >100 |
| 2b | >100 | >100 | >100 | >100 | >100 | >100 | >100 |
| 2c | >100 | >100 | >100 | >100 | >100 | >100 | >100 |
| 2d | 21.04 ± 0.43 | 53.33 ± 1.37 | 36.79 ± 1.06 | 40.12 ± 1.36 | 37.03 ± 0.97 | >100 | >100 |
| 2e | 80.19 ± 3.72 | 91.63 ± 2.21 | 68.18 ± 3.92 | 58.99 ± 3.11 | >100 | >100 | >100 |
| 2f | >100 | >100 | 66.18 ± 5.70 | 87.27 ± 2.14 | 65.07 ± 1.27 | >100 | >100 |
| 2g | 16.63 ± 0.12 | 40.26 ± 0.62 | 30.47 ± 1.59 | 55.87± 0.42 | 27.45 ± 0.38 | 20.79 ± 0.97 | >100 |
| 2h | >100 | >100 | 92.01 ± 6.10 | >100 | >100 | >100 | >100 |
| 2i | 16.38 ± 0.27 | 37.06 ± 0.61 | 31.58 ± 1.25 | 44.23 ± 0.89 | 67.45 ± 1.24 | >100 | >100 |
| 2j | 87.44 ± 7.12 | >100 | 78.15 ± 4.35 | >100 | >100 | >100 | >100 |
| RA | >100 | >100 | >100 | >100 | >100 | >100 | >100 |
| CPT | 12.25 ± 1.06 | 15.34 ± 1.22 | 18.9 ± 4.12 | 1.3 ± 0.62 | 0.5 ± 0.08 | 0.030 ± 0.002 | 2.159 ± 0.47 |
| cisplatin | 14.23 ± 0.56 | 25.40 ± 1.06 | 1.32 ± 0.97 | 10.59 ± 1.40 | 5.83 ± 0.14 | <0.008 | 57.66 ± 3.10 |
| Compound | Amino Acid | Type | Hydrophobic Residues | Total Score a |
|---|---|---|---|---|
| 2a | DA113 | π–π | LYS374, GLU356, DC112, TGP11, DT10, TYR426, ALA351, ASN352, MET428, LYS425 | 8.4481 |
| DC112 | π–π | |||
| DA113 | Hydrogen Bond | |||
| 2b | TGP11 | π–π | ASP533, THR718, TGP11, DT10, GLU356, ASN352, DC112, DG12 | 6.9191 |
| DA113 | π–π | |||
| ARG364 | Hydrogen Bond | |||
| DA113 | Hydrogen Bond | |||
| 2c | TGP11 | π–π | ASP533, TGP11, GLU356, DC112, LYS532, HIS632, DT10, ASN722, ASN352, DA113 | 8.9089 |
| DA113 | π–π | |||
| DC112 | π–π | |||
| DT10 | π–π | |||
| THR718 | Hydrogen Bond | |||
| ARG364 | Hydrogen Bond | |||
| 2d | TGP11 | π–π | ASN722, DT10, TGP11, ALN351, ASN352, GLU356, DA113, ASP533, PTR723, ARG488, LYS532, HIS632 | 8.7600 |
| DA113 | π–π | |||
| DT10 | π–π | |||
| ARG364 | Hydrogen Bond | |||
| THR718 | Hydrogen Bond | |||
| 2e | TGP11 | π–π | ASN722, DT10, TGP11, ALN351, ASN352, GLU356, DA113, ASP533, PTR723, ARG488, LYS532, HIS632 | 8.6166 |
| DA113 | π–π | |||
| DC112 | π–π | |||
| ARG364 | Hydrogen Bond | |||
| DA113 | Hydrogen Bond | |||
| 2f | TGP11 | π–π | DC112, TGP11, THR718, DT10, TRP416, GLU356, LYS354, ASN352 | 8.5897 |
| DA113 | π–π | |||
| DT10 | π–π | |||
| DA113 | Hydrogen Bond | |||
| ARP364 | Hydrogen Bond | |||
| 2g | DC112 | π–π | ILE427, TGP11, DT10, DA113, ARG364, DC112, PRO357, GLU356, LYS374, GLU418, TYR426 | 10.0229 |
| TGP11 | π–π | |||
| DA113 | π–π | |||
| MET428 | Hydrogen Bond | |||
| LYS425 | Hydrogen Bond | |||
| 2h | DT10 | π–π | DC112, TGP11, THR718, DT10, LYS436, PRO431, ASN352, TRP416, LYS354, GLU356 | 9.2983 |
| TGP11 | π–π | |||
| DA113 | π–π | |||
| DA113 | Hydrogen Bond | |||
| ARG364 | Hydrogen Bond | |||
| 2i | DA113 | π–π | DT10, TGP11, THR718, DC112, LYS354, GLU356, TRP416, ASN352 | 8.8944 |
| TGP11 | π–π | |||
| DT10 | π–π | |||
| DA113 | Hydrogen Bond | |||
| ARG364 | Hydrogen Bond | |||
| 2j | TGP11 | π–π | DC112, TYR426, LYS425, ASN352, THR718, DT10, TGP11 | 8.4305 |
| DA113 | π–π | |||
| ARG364 | Hydrogen Bond | |||
| DA113 | Hydrogen Bond | |||
| RA | TGP11 | π–π | ARG364, ASP533, TGP11, DA113, ALA351, ASN352, LEU429, MET428 | 7.8183 |
| DA113 | π–π | |||
| DT10 | Hydrogen Bond | |||
| CPT | TGP11 | π–π | DC112, DA113, TGP11, DT10, DG12 | 10.3269 |
| DA113 | π–π | |||
| DC112 | π–π | |||
| ASP533 | Hydrogen Bond | |||
| TGP11 | π–π |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, R.; Huo, L.; Jaiswal, Y.; Wei, J.; Li, D.; Zhong, J.; Williams, L.; Xia, X.; Liang, Y. Synthesis and Evaluation of Anticancer Activity of New 4-Acyloxy Derivatives of Robustic Acid. Int. J. Mol. Sci. 2019, 20, 5336. https://doi.org/10.3390/ijms20215336
Chen R, Huo L, Jaiswal Y, Wei J, Li D, Zhong J, Williams L, Xia X, Liang Y. Synthesis and Evaluation of Anticancer Activity of New 4-Acyloxy Derivatives of Robustic Acid. International Journal of Molecular Sciences. 2019; 20(21):5336. https://doi.org/10.3390/ijms20215336
Chicago/Turabian StyleChen, Rui, Lini Huo, Yogini Jaiswal, Jianhua Wei, Dianpeng Li, Jing Zhong, Leonard Williams, Xing Xia, and Yan Liang. 2019. "Synthesis and Evaluation of Anticancer Activity of New 4-Acyloxy Derivatives of Robustic Acid" International Journal of Molecular Sciences 20, no. 21: 5336. https://doi.org/10.3390/ijms20215336
APA StyleChen, R., Huo, L., Jaiswal, Y., Wei, J., Li, D., Zhong, J., Williams, L., Xia, X., & Liang, Y. (2019). Synthesis and Evaluation of Anticancer Activity of New 4-Acyloxy Derivatives of Robustic Acid. International Journal of Molecular Sciences, 20(21), 5336. https://doi.org/10.3390/ijms20215336