Therapeutic Evaluation of Synthetic Peucedanocoumarin III in an Animal Model of Parkinson’s Disease



Abstract
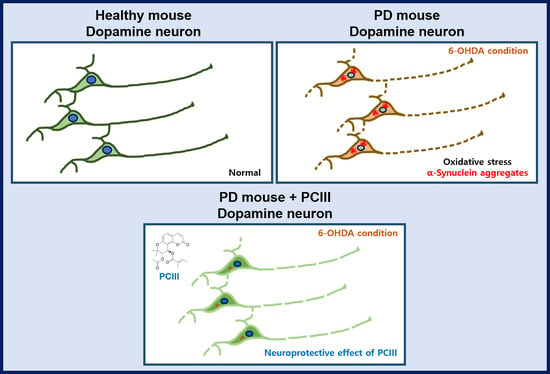
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
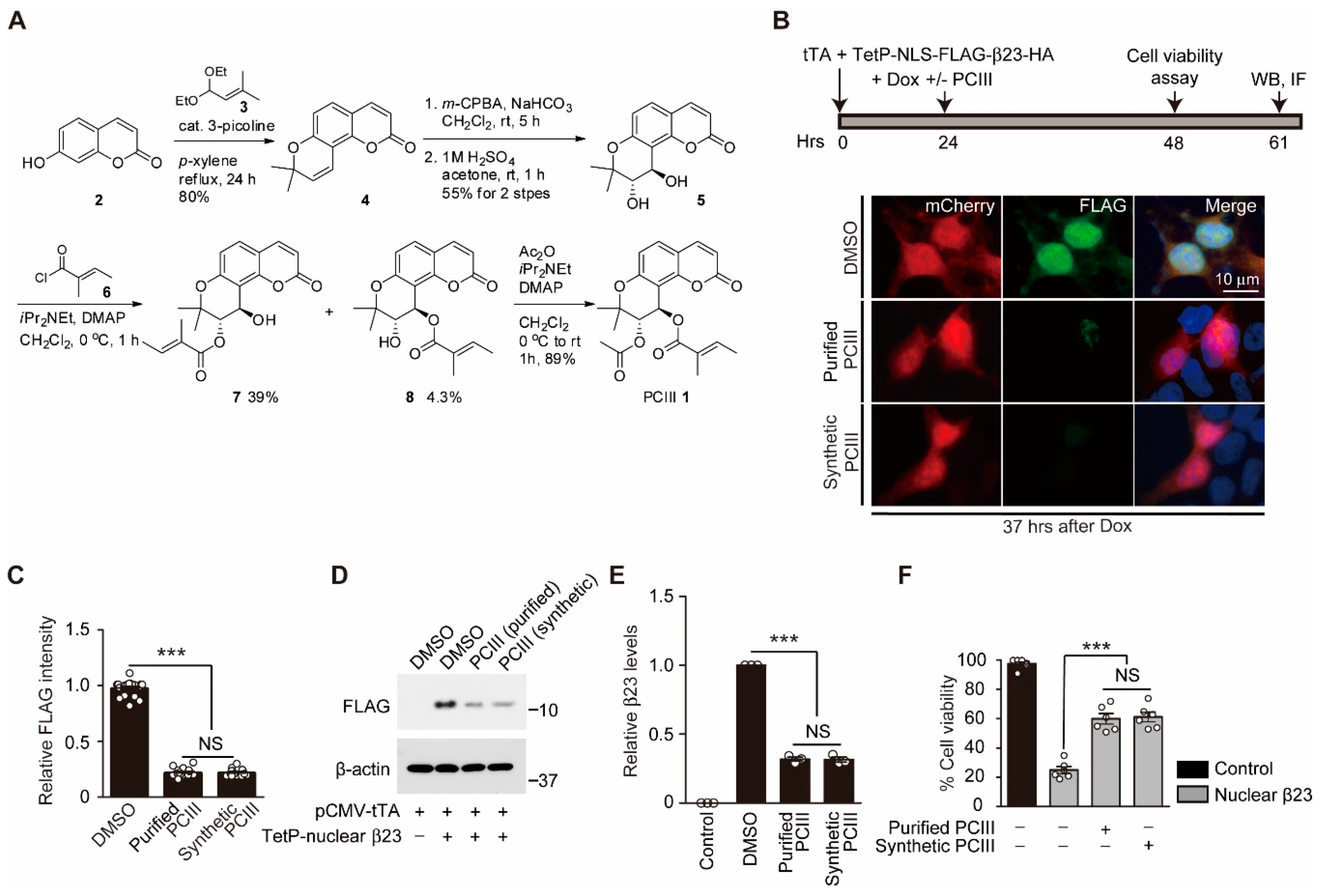
2.1. Organic Synthesis of Peucedanocoumarin III and its Evaluation
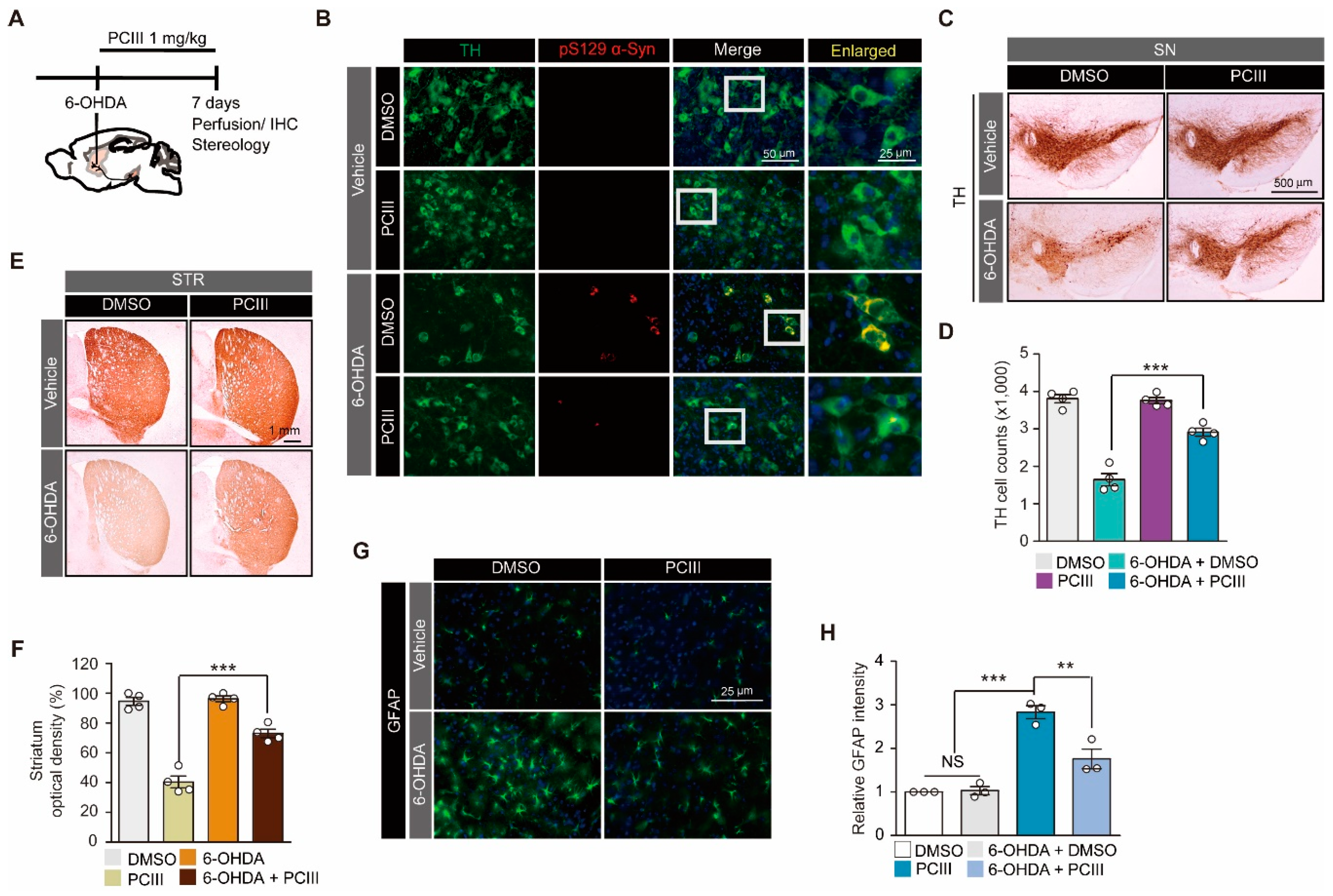
2.2. PCIII Prevents DA Cell Loss and α-synuclein Aggregation in a 6-OHDA PD Mouse Model
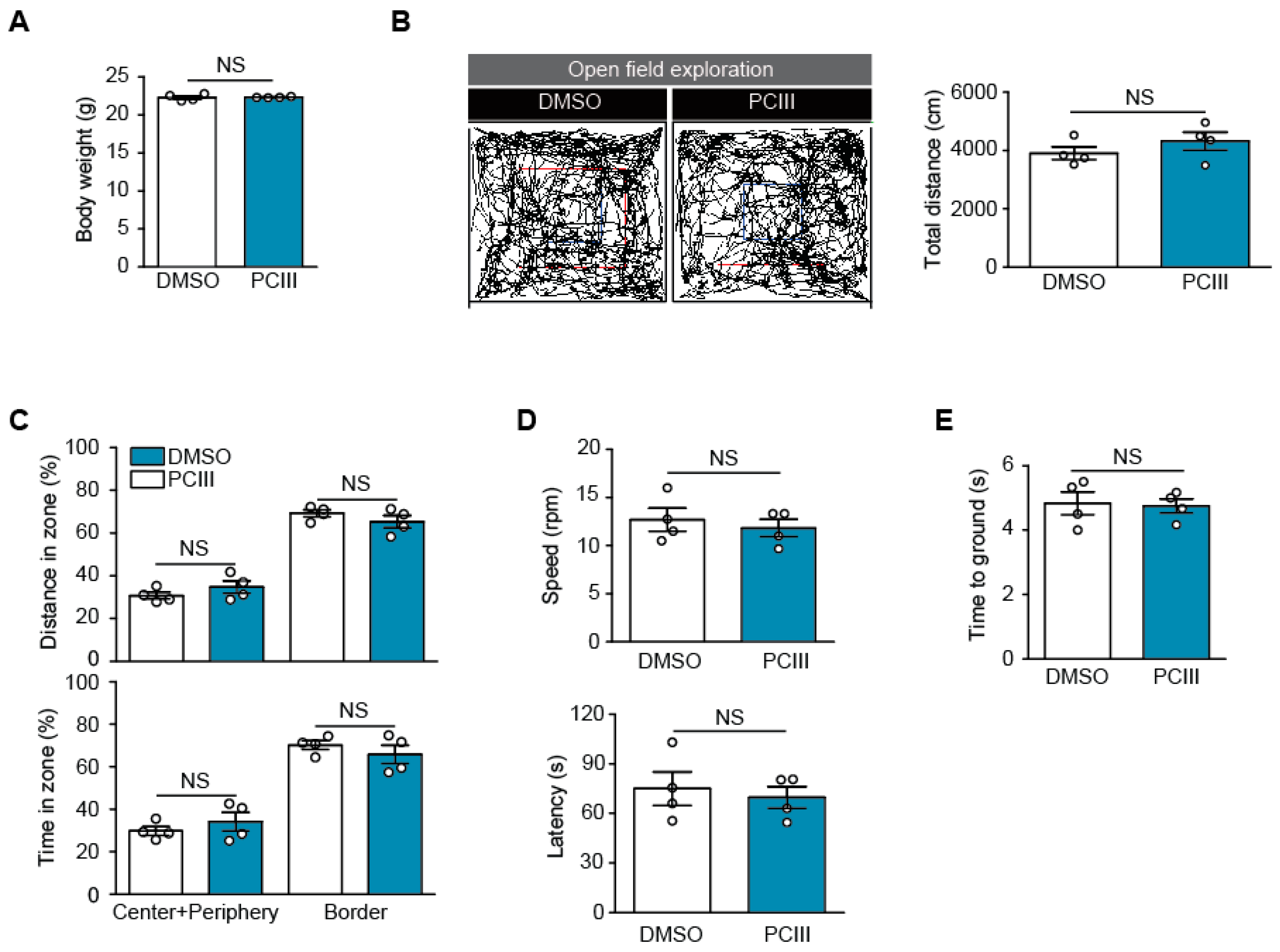
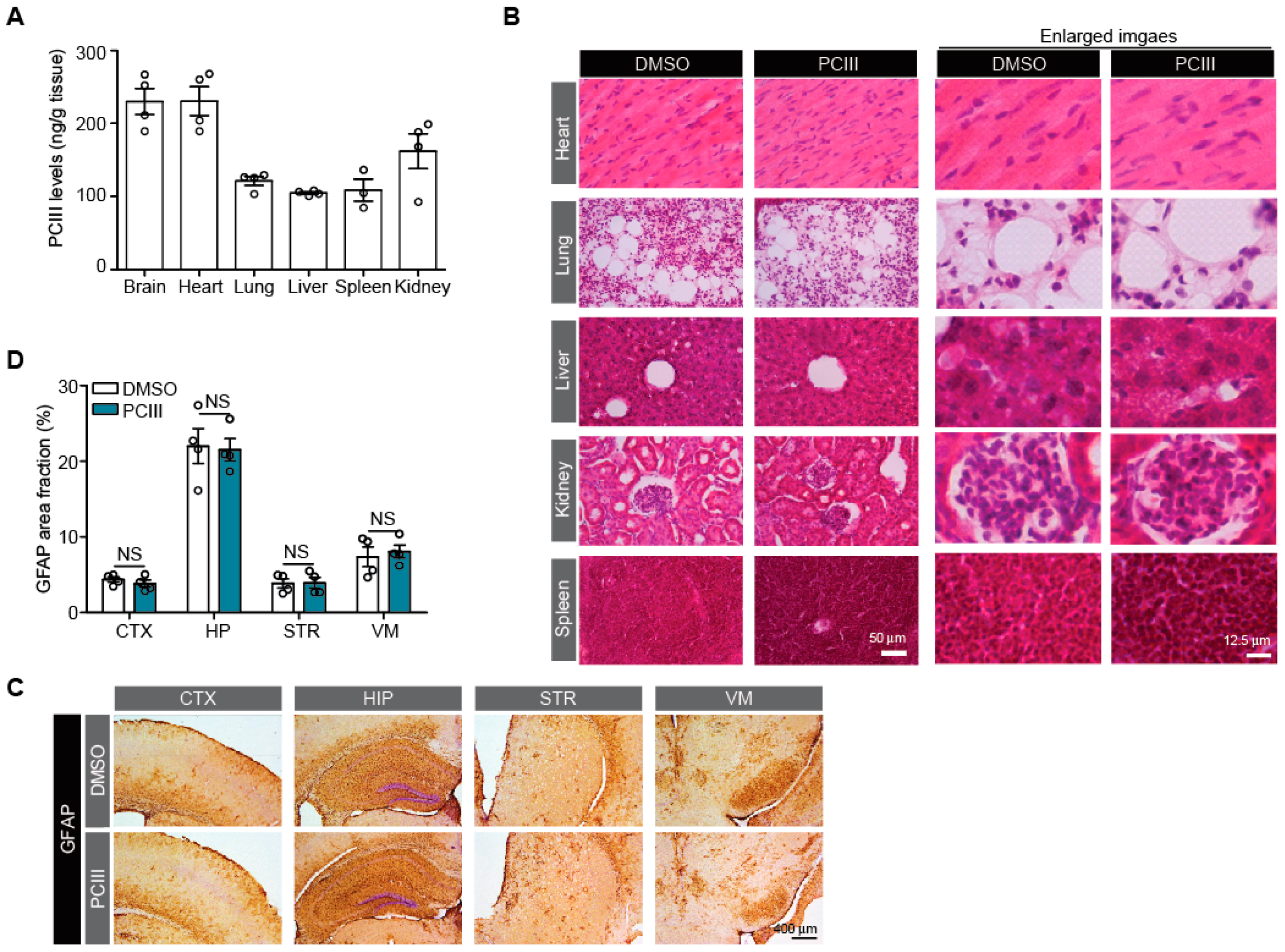
2.3. Safety Profiling of Synthetic PCIII Administration In Vivo
3. Discussion
4. Materials and Methods
4.1. Chemicals and Antibodies
4.2. Organic Synthesis of PCIII




4.3. Cell Culture and Transfection
4.4. Fluorescence Imaging
4.5. Western Blotting
4.6. Cell Viability Assay
4.7. Animal Experiments
4.8. UHPLC Bioanalytical Method of PCIII
4.9. Intrastriatal Injection of 6-OHDA
4.10. TH Stereological Cell Counting and Immunohistochemistry
4.11. Behavior Tests
4.12. Tissue Pathological Staining
4.13. Statistics
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| PD | Parkinson’s disease |
| Ac2O | Acetic anhydride |
| m-CPBA | meta-Chloroperoxybenzoic acid |
| 6-OHDA | 6-hydroxydopamine |
References
- Lang, A.E.; Lozano, A.M. Parkinson’s disease. First of two parts. N. Engl. J. Med. 1998, 339, 1044–1053. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Del Tredici, K.; Rub, U.; de Vos, R.A.; Jansen Steur, E.N.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Braak, H.; Rub, U.; Gai, W.P.; Del Tredici, K. Idiopathic Parkinson’s disease: Possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J. Neural Transm. (Vienna) 2003, 110, 517–536. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Ghebremedhin, E.; Rub, U.; Bratzke, H.; Del Tredici, K. Stages in the development of Parkinson’s disease-related pathology. Cell Tissue Res. 2004, 318, 121–134. [Google Scholar] [CrossRef]
- Hebron, M.L.; Lonskaya, I.; Moussa, C.E. Nilotinib reverses loss of dopamine neurons and improves motor behavior via autophagic degradation of alpha-synuclein in Parkinson’s disease models. Hum. Mol. Genet. 2013, 22, 3315–3328. [Google Scholar] [CrossRef]
- Karuppagounder, S.S.; Brahmachari, S.; Lee, Y.; Dawson, V.L.; Dawson, T.M.; Ko, H.S. The c-Abl inhibitor, nilotinib, protects dopaminergic neurons in a preclinical animal model of Parkinson’s disease. Sci. Rep. 2014, 4, 4874. [Google Scholar] [CrossRef]
- Yun, S.P.; Kam, T.I.; Panicker, N.; Kim, S.; Oh, Y.; Park, J.S.; Kwon, S.H.; Park, Y.J.; Karuppagounder, S.S.; Park, H.; et al. Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson’s disease. Nat. Med. 2018, 24, 931–938. [Google Scholar] [CrossRef]
- Ham, S.; Kim, H.; Hwang, S.; Kang, H.; Yun, S.P.; Kim, S.; Kim, D.; Kwon, H.S.; Lee, Y.S.; Cho, M.; et al. Cell-Based Screen Using Amyloid Mimic beta23 Expression Identifies Peucedanocoumarin III as a Novel Inhibitor of alpha-Synuclein and Huntingtin Aggregates. Mol. Cells 2019, 42, 480–494. [Google Scholar]
- Page, P.C.; Appleby, L.F.; Day, D.; Chan, Y.; Buckley, B.R.; Allin, S.M.; McKenzie, M.J. Highly enantioselective total synthesis of (-)-(3’S)-Lomatin and (+)-(3’S,4’R)-trans-khellactone. Org. Lett. 2009, 11, 1991–1993. [Google Scholar] [CrossRef]
- Efe, C.; Lykakis, I.N.; Stratakis, M. Gold nanoparticles supported on TiO2 catalyse the cycloisomerisation/oxidative dimerisation of aryl propargyl ethers. Chem. Commun. (Camb) 2011, 47, 803–805. [Google Scholar] [CrossRef]
- Reisch, J.; Voerste, A.A.W. Natural product chemistry. Part 181. Investigations on the synthesis of dihydropyrano- and dihydrofurano-coumarins by application of catalytic enantioselective cis-dihydroxylation. J. Chem. Soc. Perkin Trans. 1 1994, 3251–3256. [Google Scholar] [CrossRef]
- Huang, L.; Kashiwada, Y.; Cosentino, L.M.; Fan, S.; Lee, K.-H.J.B.; Letters, M.C. 3′, 4′-Di-o-(−)-camphanoyl-(+)-ciskhellactone and related compounds: A. new class of potent anti-HIV agents. Bioorganic Med. Chem. Lett. 1994, 4, 593–598. [Google Scholar] [CrossRef]
- Page, P.C.B.; Mace, A.; Arquier, D.; Bethell, D.; Buckley, B.R.; Willock, D.J.; Hutchings, G.J.J.C.S. Towards heterogeneous organocatalysis: Chiral iminium cations supported on porous materials for enantioselective alkene epoxidation. Catal. Sci. Technol. 2013, 3, 2330–2339. [Google Scholar] [CrossRef]
- Chen, I.-S.; Chang, C.-T.; Sheen, W.-S.; Teng, C.-M.; Tsai, I.-L.; Duh, C.-Y.; Ko, F.-N. Coumarins and antiplatelet aggregation constituents from Formosan Peucedanum japonicum. Phytochemistry 1996, 41, 525–530. [Google Scholar] [CrossRef]
- Olzscha, H.; Schermann, S.M.; Woerner, A.C.; Pinkert, S.; Hecht, M.H.; Tartaglia, G.G.; Vendruscolo, M.; Hayer-Hartl, M.; Hartl, F.U.; Vabulas, R.M. Amyloid-like aggregates sequester numerous metastable proteins with essential cellular functions. Cell 2011, 144, 67–78. [Google Scholar] [CrossRef]
- Coppola-Segovia, V.; Cavarsan, C.; Maia, F.G.; Ferraz, A.C.; Nakao, L.S.; Lima, M.M.; Zanata, S.M. ER Stress Induced by Tunicamycin Triggers alpha-Synuclein Oligomerization, Dopaminergic Neurons Death and Locomotor Impairment: A New Model of Parkinson’s Disease. Mol. Neurobiol. 2017, 54, 5798–5806. [Google Scholar] [CrossRef]
- Alvarez-Fischer, D.; Henze, C.; Strenzke, C.; Westrich, J.; Ferger, B.; Hoglinger, G.U.; Oertel, W.H.; Hartmann, A. Characterization of the striatal 6-OHDA model of Parkinson’s disease in wild type and alpha-synuclein-deleted mice. Exp. Neurol. 2008, 210, 182–193. [Google Scholar] [CrossRef]
- da Conceicao, F.S.; Ngo-Abdalla, S.; Houzel, J.C.; Rehen, S.K. Murine model for Parkinson’s disease: From 6-OH dopamine lesion to behavioral test. J. Vis. Exp. 2010, 15, 1376. [Google Scholar] [CrossRef]
- Ganapathy, K.; Datta, I.; Sowmithra, S.; Joshi, P.; Bhonde, R. Influence of 6-Hydroxydopamine Toxicity on alpha-Synuclein Phosphorylation, Resting Vesicle Expression, and Vesicular Dopamine Release. J. Cell Biochem. 2016, 117, 2719–2736. [Google Scholar] [CrossRef]
- Lang, A.E.; Lozano, A.M. Parkinson’s disease. Second of two parts. N. Engl. J. Med. 1998, 339, 1130–1143. [Google Scholar] [CrossRef]
- Jeong, Y.S.; Balla, A.; Chun, K.H.; Chung, S.J.; Maeng, H.J. Physiologically-Based Pharmacokinetic Modeling for Drug-Drug Interactions of Procainamide and N-Acetylprocainamide with Cimetidine, an Inhibitor of rOCT2 and rMATE1, in Rats. Pharmaceutics 2019, 11, 180. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Park, J.; Leem, H.; Cho, M.; Yoon, J.H.; Maeng, H.J.; Lee, Y. Rhododendrin-Induced RNF146 Expression via Estrogen Receptor beta Activation is Cytoprotective Against 6-OHDA-Induced Oxidative Stress. Int. J. Mol. Sci. 2019, 20, 1772. [Google Scholar] [CrossRef] [PubMed]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ham, S.; Kim, H.; Yoon, J.-H.; Kim, H.; Song, B.R.; Choi, J.-Y.; Lee, Y.-S.; Paek, S.-M.; Maeng, H.-J.; Lee, Y. Therapeutic Evaluation of Synthetic Peucedanocoumarin III in an Animal Model of Parkinson’s Disease. Int. J. Mol. Sci. 2019, 20, 5481. https://doi.org/10.3390/ijms20215481
Ham S, Kim H, Yoon J-H, Kim H, Song BR, Choi J-Y, Lee Y-S, Paek S-M, Maeng H-J, Lee Y. Therapeutic Evaluation of Synthetic Peucedanocoumarin III in an Animal Model of Parkinson’s Disease. International Journal of Molecular Sciences. 2019; 20(21):5481. https://doi.org/10.3390/ijms20215481
Chicago/Turabian StyleHam, Sangwoo, Heejeong Kim, Jin-Ha Yoon, Hyojung Kim, Bo Reum Song, Jeong-Yun Choi, Yun-Song Lee, Seung-Mann Paek, Han-Joo Maeng, and Yunjong Lee. 2019. "Therapeutic Evaluation of Synthetic Peucedanocoumarin III in an Animal Model of Parkinson’s Disease" International Journal of Molecular Sciences 20, no. 21: 5481. https://doi.org/10.3390/ijms20215481
APA StyleHam, S., Kim, H., Yoon, J. -H., Kim, H., Song, B. R., Choi, J. -Y., Lee, Y. -S., Paek, S. -M., Maeng, H. -J., & Lee, Y. (2019). Therapeutic Evaluation of Synthetic Peucedanocoumarin III in an Animal Model of Parkinson’s Disease. International Journal of Molecular Sciences, 20(21), 5481. https://doi.org/10.3390/ijms20215481