Effect of Kinase Inhibiting RNase Attenuator (KIRA) Compounds on the Formation of Face-to-Face Dimers of Inositol-Requiring Enzyme 1: Insights from Computational Modeling

 ,
,  and
and
Abstract
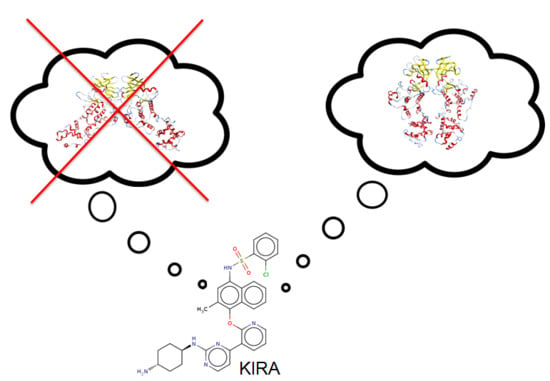
:
1. Introduction
2. Methods
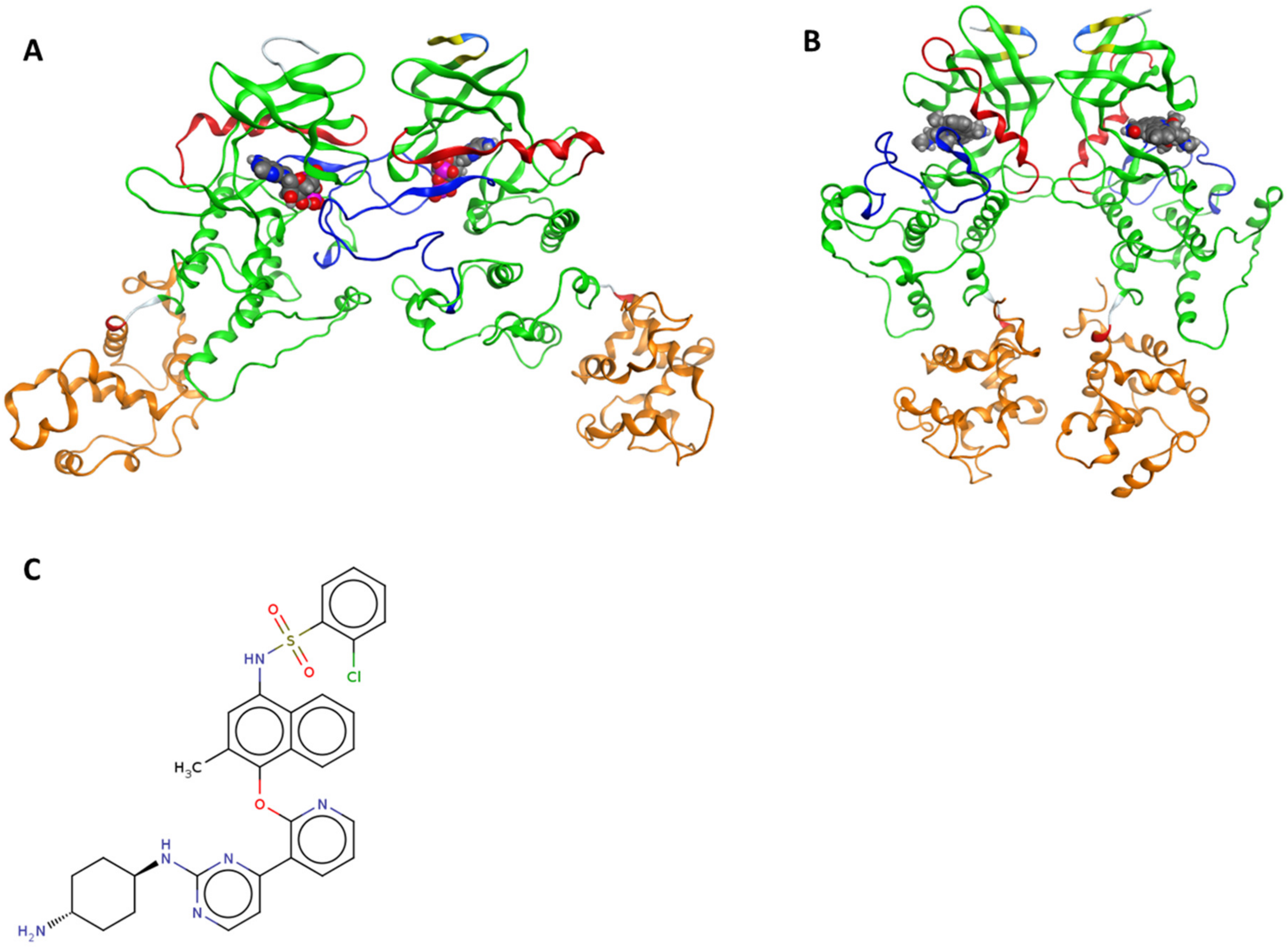
2.1. Selection and Preparation of IRE1 Crystal Structure
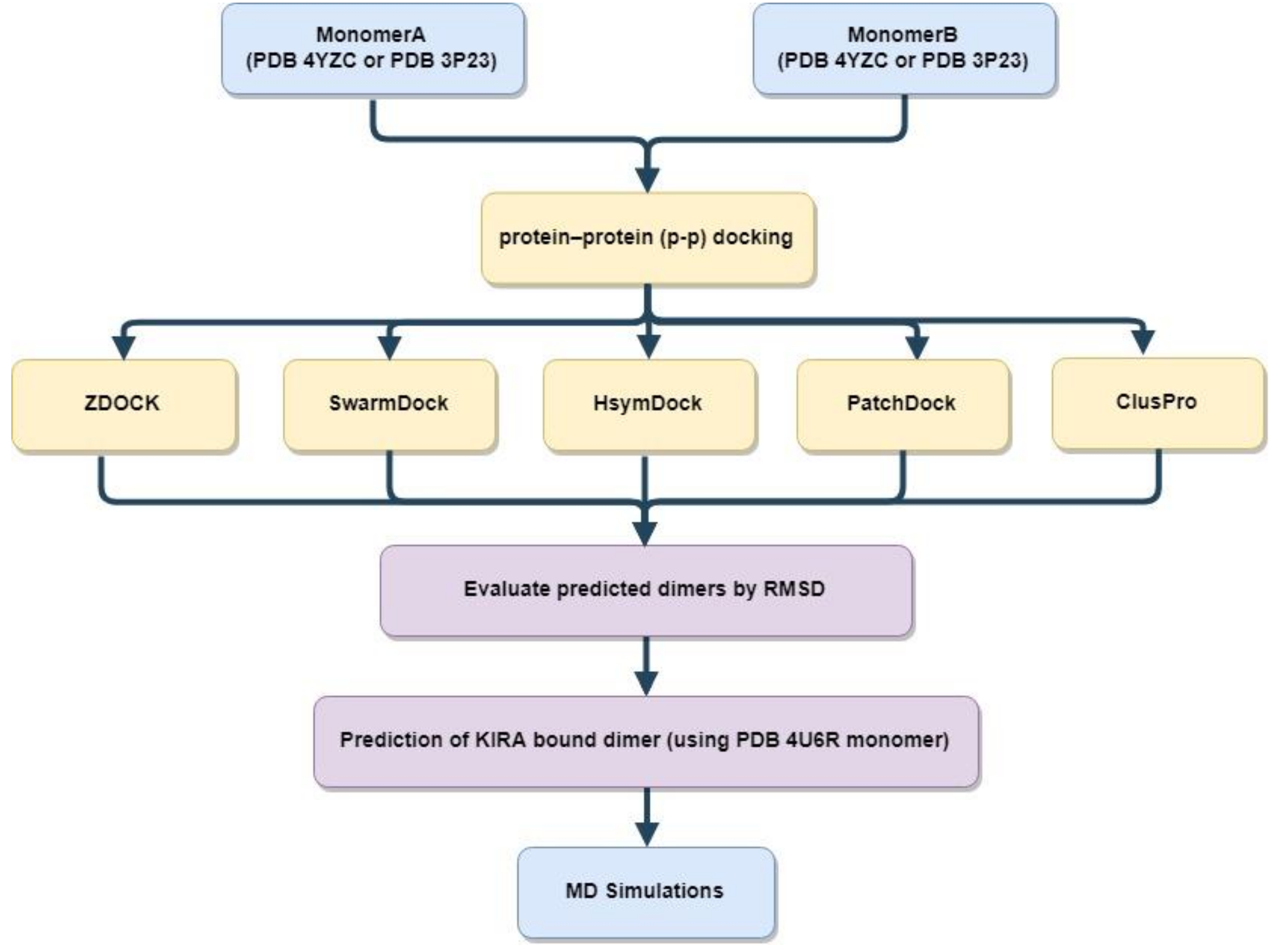
2.2. Protein-Protein Docking
2.3. KIRA Preparation for Docking Studies
2.4. Molecular Docking of KIRA
2.5. Molecular Dynamics Simulations
- Systems preparation: systems included the experimental IRE1 dimer structures (PDB 4YZC, 3P23), predicted dimers (from KIRA-bound monomer) (Section 2.2.), and KIRA-docked dimer forms (Section 2.4.). The systems were prepared separately as discussed in Section 2.1.
- Molecular dynamics simulation protocol: MD simulations were performed using the GROMACS 5.1 package [28] with the AMBER14SB force field for the protein [29]. The systems were explicitly solvated using cubic water boxes with cell borders placed at least 10 Å away from the protein or ligand atoms using TIP3P water [30] under periodic boundary conditions. The rational for the choice of the 10 Å cutoff distances was to place the protein or ligand atoms at a distance longer than the non-bonded interactions cut-off (i.e., 8 Å). The systems were first neutralized and Na+/Cl– counter ions were added to give a physiological salt concentration of 0.154 M. All simulation runs consisted of energy minimization until the force was less than 1000 kJ mol−1 nm−1, 200 ps under NVT conditions subjected to position-restrained equilibration on the heavy atoms of IRE1, snf 200 ps equilibration and 300 ns of classical molecular dynamics simulation under NPT conditions. The simulations were run in triplicate (referred to as Replica 1, 2, and 3). In all simulations, the temperature was kept at 300 K by the velocity rescaling thermostat [31] with a coupling constant of 0.1 ps and pressure at 1.01325 bar using the Parrinello–Rahman barostat [32] with a coupling time of 5.0 ps, excluding NVT pre-simulation steps. Constraints were applied on all bonds using the LINCS algorithm [33]. The leap-frog algorithm [34] was employed in the simulations with integration timesteps of 2 fs.
2.6. Data Availability
- a source PDB (.pdb) file
- leap.log—commands used to create the. prmtop and. inpcrd files
- two AMBER parameter/topology (.prmtop) and an AMBER coordinate (.inpcrd) file
- .mdp file used for performing all the minimisation, relaxation, equilibration, and production run steps
- Executable script (i.e., job009) that was used to perform the production run
- trajectory (.xtc) files for each independent MD simulation
3. Results and Discussion
3.1. Protein–Ligand Docking Analysis
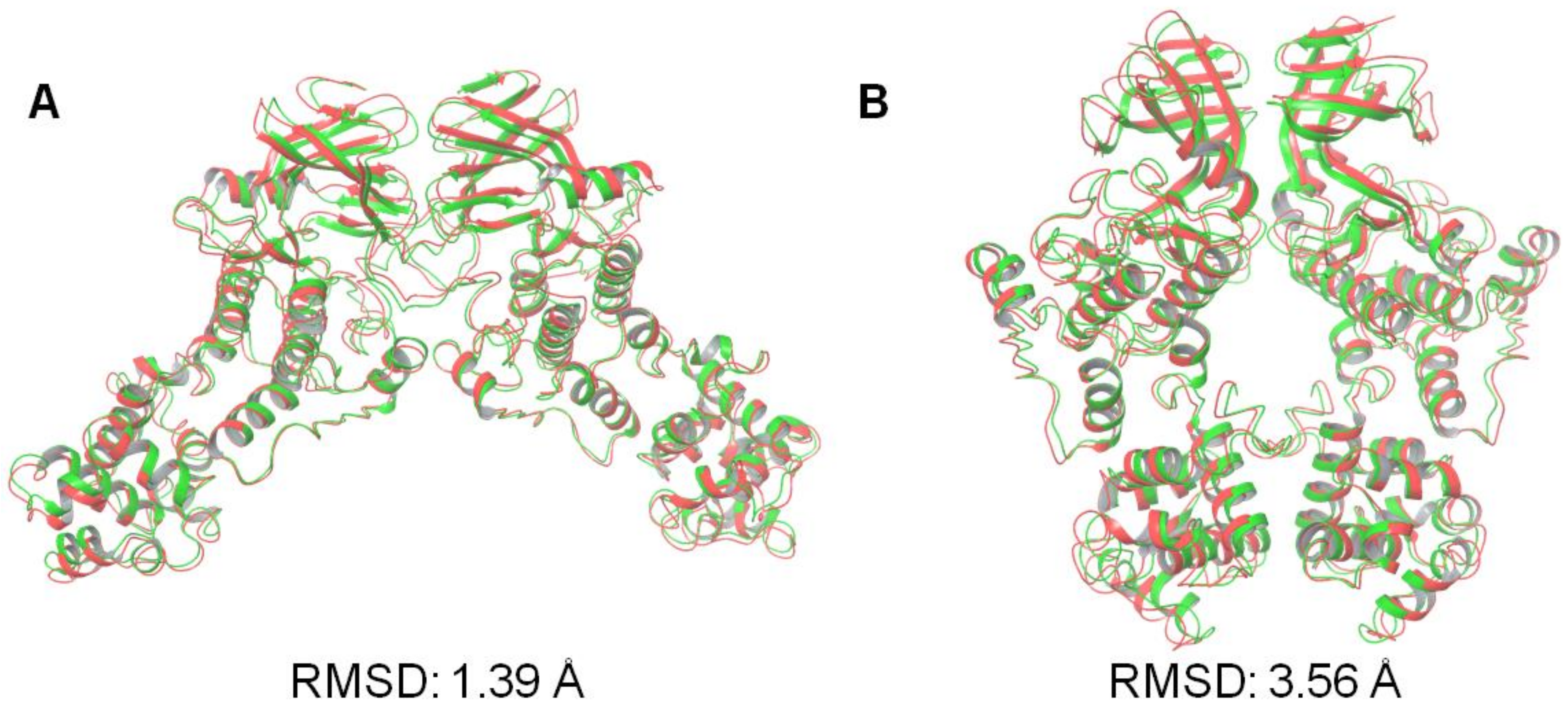
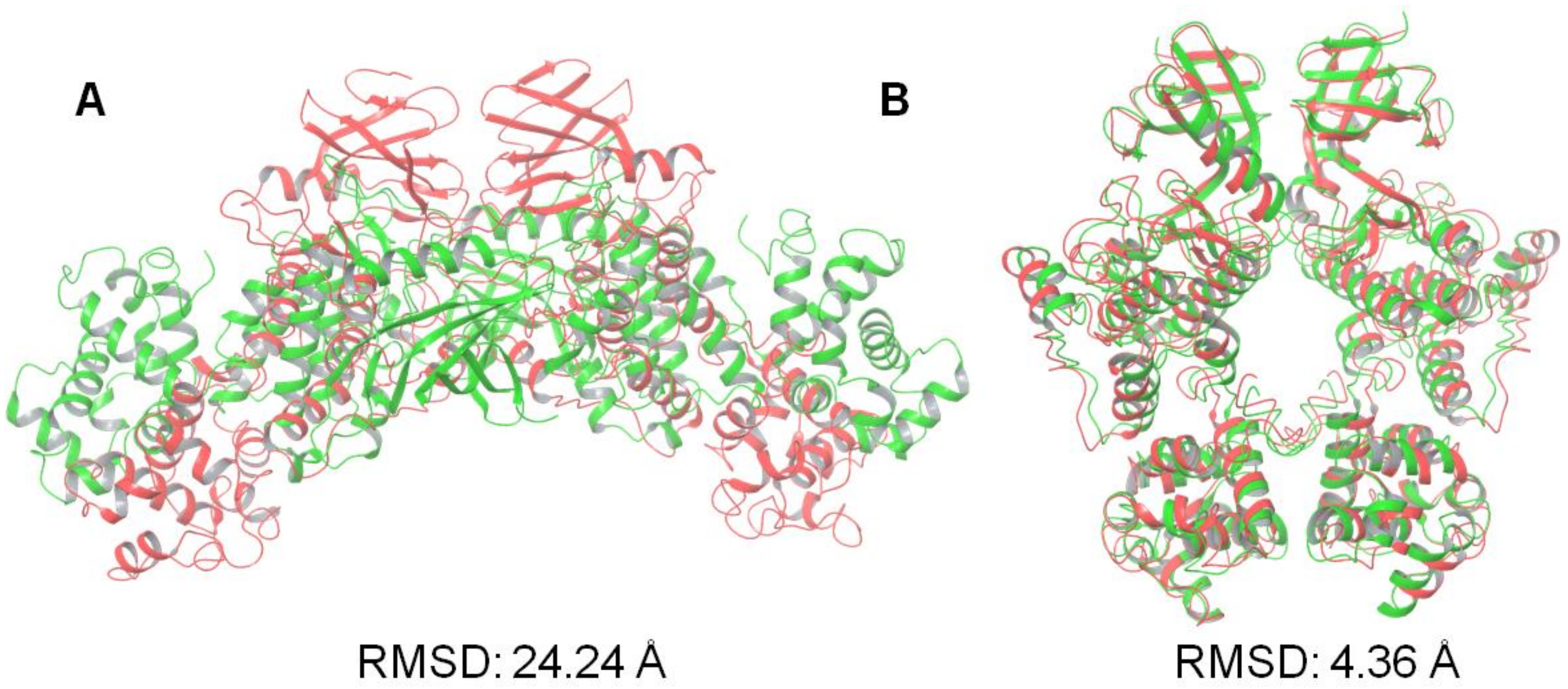
3.2. Protein–Protein Docking Analysis
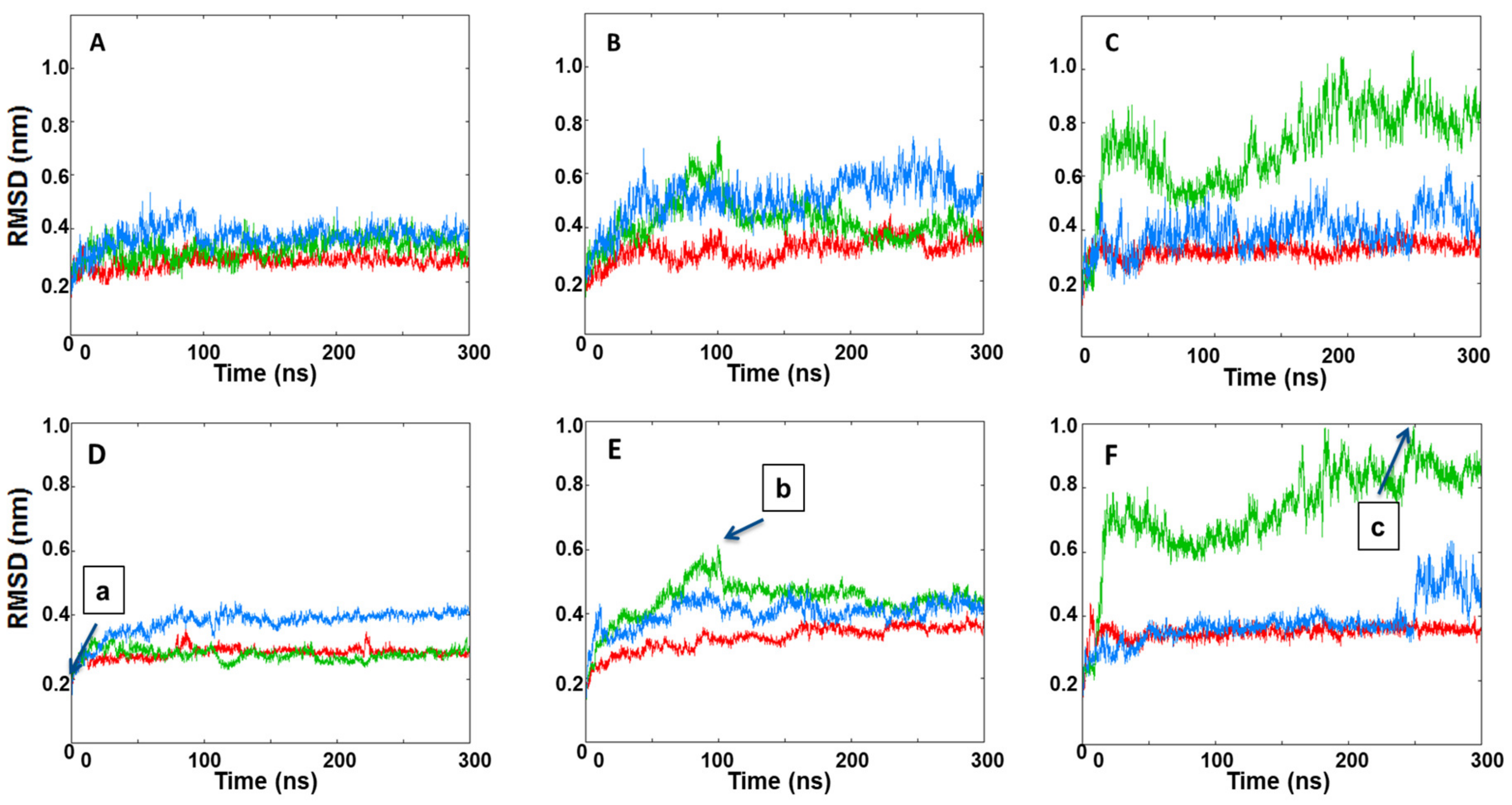
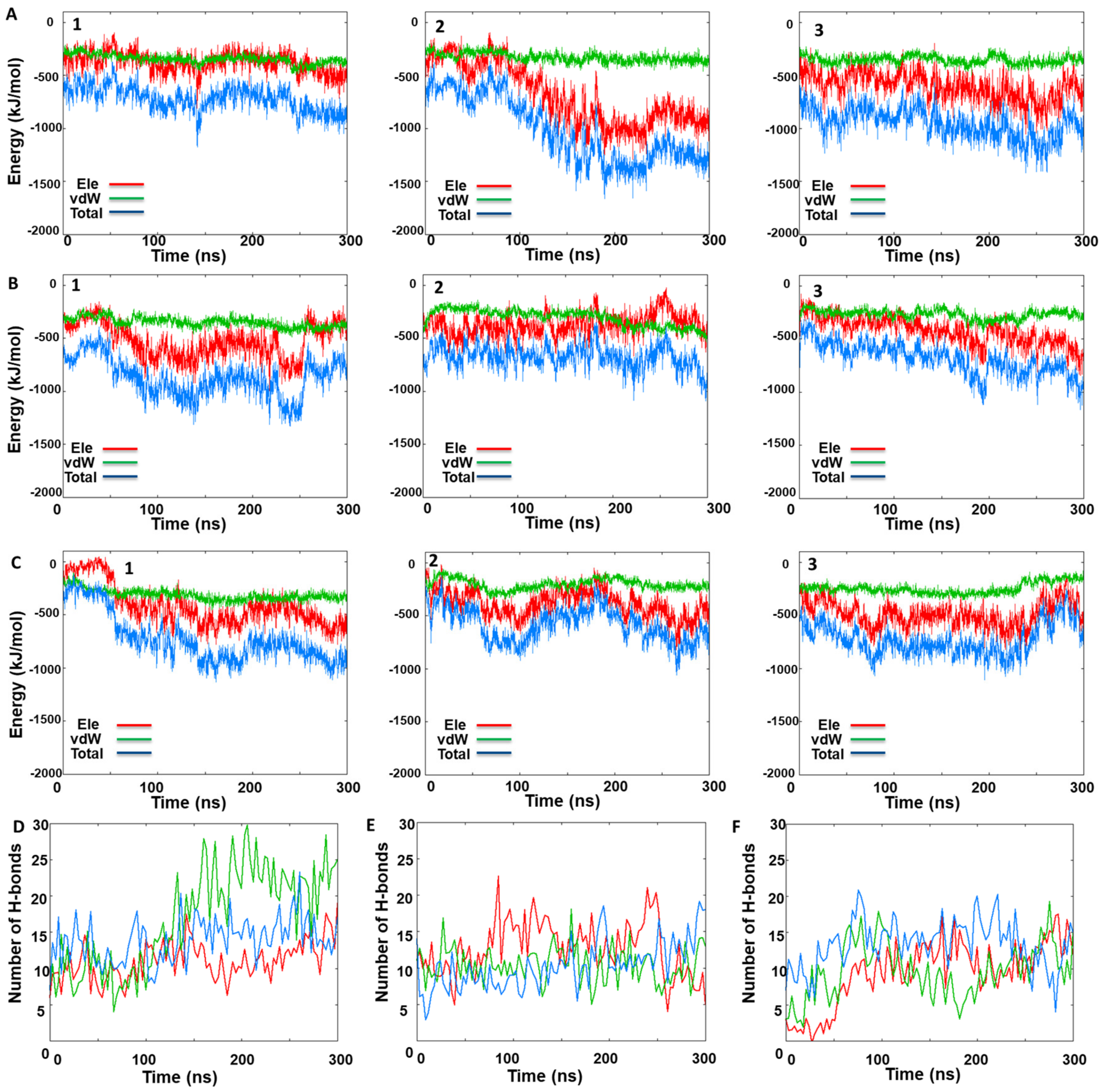
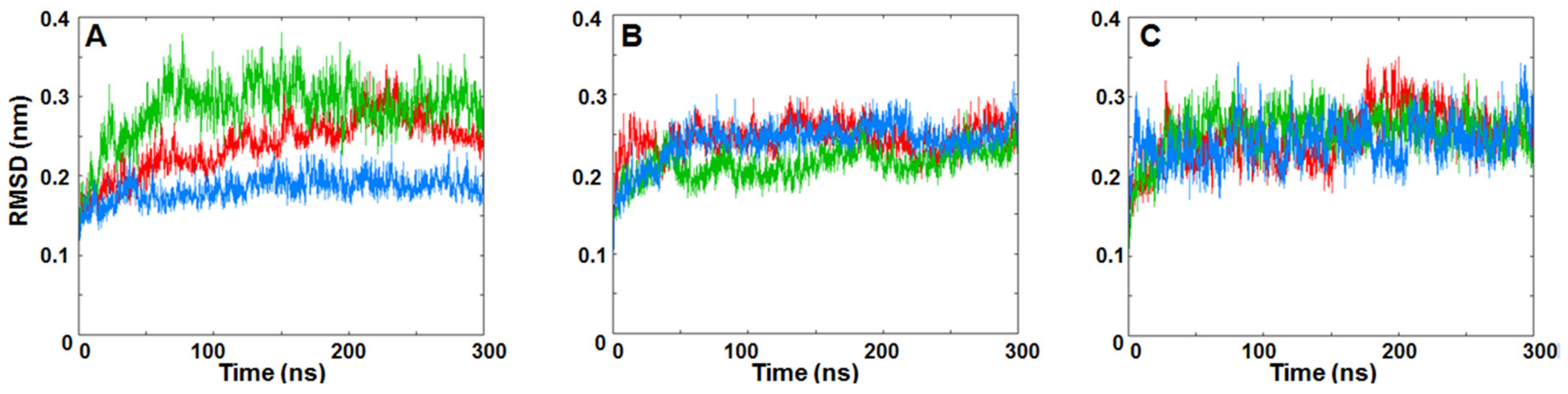
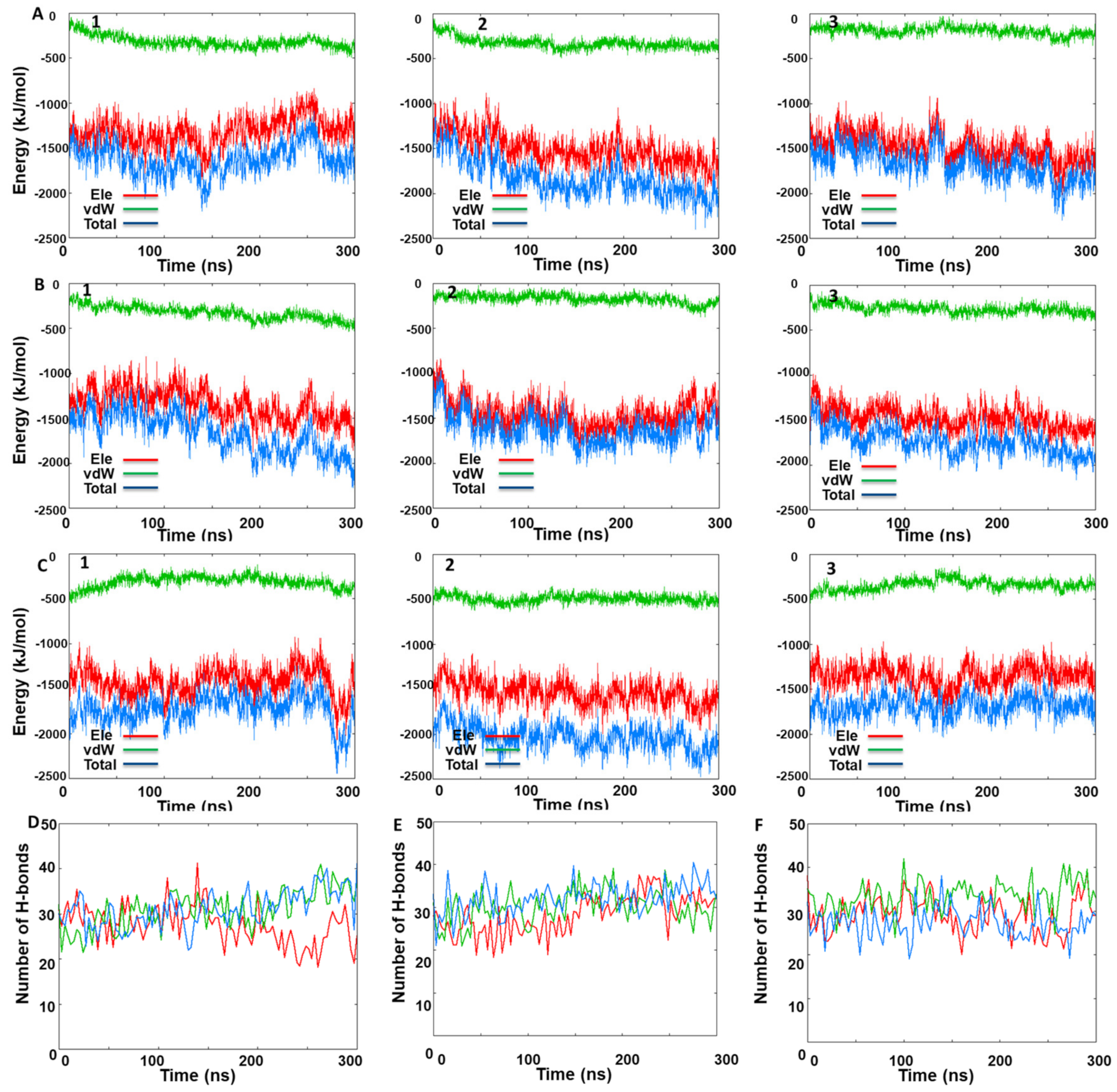
3.3. MD Simulations Analysis: Influence of KIRA on the Face-to-Face Dimer
3.4. MD Simulations Analysis: Influence of KIRA on the Back-to-Back Dimer
4. Conclusions and Perspective
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Almanza, A.; Carlesso, A.; Chintha, C.; Creedican, S.; Doultsinos, D.; Leuzzi, B.; Luís, A.; McCarthy, N.; Montibeller, L.; More, S.; et al. Endoplasmic reticulum stress signalling–from basic mechanisms to clinical applications. FEBS J. 2019, 286, 241–278. [Google Scholar] [CrossRef] [PubMed]
- Adams, C.J.; Kopp, M.C.; Larburu, N.; Nowak, P.R.; Ali, M.M.U. Structure and Molecular Mechanism of ER Stress Signaling by the Unfolded Protein Response Signal Activator IRE1. Front. Mol. Biosci. 2019, 6, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Thamsen, M.; Ghosh, R.; Auyeung, V.C.; Brumwell, A.; Chapman, H.A.; Backes, B.J.; Perara, G.; Maly, D.J.; Sheppard, D.; Papa, F.R. Small molecule inhibition of IRE1α kinase/ RNase has anti-fibrotic effects in the lung. PLoS ONE 2019, 14, e0209824. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.A.; Groenendyk, J.; Michalak, M. Endoplasmic reticulum stress associated responses in cancer. Biochim. Biophys. Acta Mol. Cell Res. 2014, 1843, 2143–2149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maly, D.J.; Papa, F.R. Druggable sensors of the unfolded protein response. Nat. Chem. Biol. 2014, 10, 892–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanches, M.; Duffy, N.M.; Talukdar, M.; Thevakumaran, N.; Chiovitti, D.; Canny, M.D.; Lee, K.; Kurinov, I.; Uehling, D.; Al-Awar, R.; et al. Structure and mechanism of action of the hydroxy–aryl–aldehyde class of IRE1 endoribonuclease inhibitors. Nat. Commun. 2014, 5, 4202. [Google Scholar] [CrossRef]
- Feldman, H.C.; Tong, M.; Wang, L.; Meza-Acevedo, R.; Gobillot, T.A.; Lebedev, I.; Gliedt, M.J.; Hari, S.B.; Mitra, A.K.; Backes, B.J.; et al. Structural and Functional Analysis of the Allosteric Inhibition of IRE1α with ATP-Competitive Ligands. ACS Chem. Biol. 2016, 11, 2195–2205. [Google Scholar] [CrossRef]
- Harrington, P.E.; Biswas, K.; Malwitz, D.; Tasker, A.S.; Mohr, C.; Andrews, K.L.; Dellamaggiore, K.; Kendall, R.; Beckmann, H.; Jaeckel, P.; et al. Unfolded protein response in cancer: IRE1α inhibition by selective kinase ligands does not impair tumor cell viability. ACS Med. Chem. Lett. 2015, 6, 68–72. [Google Scholar] [CrossRef]
- Ghosh, R.; Wang, L.; Wang, E.S.; Perera, B.G.K.; Igbaria, A.; Morita, S.; Prado, K.; Thamsen, M.; Caswell, D.; Macias, H.; et al. Allosteric Inhibition of the IRE1α RNase Preserves Cell Viability and Function during Endoplasmic Reticulum Stress. Cell 2014, 158, 534–548. [Google Scholar] [CrossRef]
- Morita, S.; Villalta, S.A.; Feldman, H.C.; Register, A.C.; Rosenthal, W.; Hoffmann-Petersen, I.T.; Mehdizadeh, M.; Ghosh, R.; Wang, L.; Colon-Negron, K.; et al. Erratum: Targeting ABL-IRE1α Signaling Spares ER-Stressed Pancreatic β Cells to Reverse Autoimmune Diabetes. Cell Metab. 2017, 25, 883–897. [Google Scholar] [CrossRef] [PubMed]
- Porter, K.A.; Desta, I.; Kozakov, D.; Vajda, S. What method to use for protein–protein docking? Curr. Opin. Struct. Biol. 2019, 55, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Salmaso, V.; Moro, S. Bridging molecular docking to molecular dynamics in exploring ligand-protein recognition process: An overview. Front. Pharmacol. 2018, 9, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Madhavi Sastry, G.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Jacobson, M.P.; Pincus, D.L.; Rapp, C.S.; Day, T.J.F.; Honig, B.; Shaw, D.E.; Friesner, R.A. A Hierarchical Approach to All-Atom Protein Loop Prediction. Proteins Struct. Funct. Genet. 2004, 55, 351–367. [Google Scholar] [CrossRef]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. ACS Publ. 2016, 12, 281–296. [Google Scholar] [CrossRef]
- Torchala, M.; Moal, I.H.; Chaleil, R.A.G.; Fernandez-Recio, J.; Bates, P.A. SwarmDock: A server for flexible protein–protein docking. Bioinformatics 2013, 29, 807–809. [Google Scholar] [CrossRef]
- Pierce, B.G.; Wiehe, K.; Hwang, H.; Kim, B.H.; Vreven, T.; Weng, Z. ZDOCK server: Interactive docking prediction of protein–protein complexes and symmetric multimers. Bioinformatics 2014, 30, 1771–1773. [Google Scholar] [CrossRef]
- Yan, Y.; Tao, H.; Huang, S.Y. HSYMDOCK: A docking web server for predicting the structure of protein homo-oligomers with Cn or Dn symmetry. Nucleic Acids Res. 2018, 46, W423–W431. [Google Scholar] [CrossRef]
- Schneidman-Duhovny, D.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. PatchDock and SymmDock: Servers for rigid and symmetric docking. Nucleic Acids Res. 2005, 33, 363–367. [Google Scholar] [CrossRef]
- Beglov, D.; Padhorny, D.; Hall, D.R.; Yueh, C.; Porter, K.A.; Kozakov, D.; Vajda, S.; Xia, B. The ClusPro web server for protein–protein docking. Nat. Protoc. 2017, 12, 255–278. [Google Scholar]
- Schrödinger Release 2015-4: LigPrep: Maestro; Schrödinger LLC: New York, NY, USA, 2015.
- Maestro Schrödinger Release 2015-4: Maestro; Schrödinger LLC: New York, NY, USA, 2015.
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein-Ligand Complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general Amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Jakalian, A.; Jack, D.B.; Bayly, C.I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem. 2002, 23, 1623–1641. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09; Revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Hess, B.; Kutzner, C.; Van Der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Jagsi, R.; Jiang, J.; Momoh, A.O.; Alderman, A.; Giordano, S.H.; Buchholz, T.A.; Pierce, L.J.; Kronowitz, S.J.; Smith, B.D. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB; 2017; Volume 263, pp. 219–227. [Google Scholar]
- Mark, P.; Nilsson, L. Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K. J. Phys. Chem. A 2001, 105, 9954–9960. [Google Scholar] [CrossRef]
- Bussi, G.; Zykova-Timan, T.; Parrinello, M. Isothermal-isobaric molecular dynamics using stochastic velocity rescaling. J. Chem. Phys. 2009, 130, 074101. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A Linear Constraint Solver for Molecular Simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar]
- Van Gunsteren, W.F.; Berendsen, H.J.C. A Leap-Frog Algorithm for Stochastic Dynamics. Mol. Simul. 1988, 1, 173–185. [Google Scholar] [CrossRef]
- Wang, Q.; Pechersky, Y.; Sagawa, S.; Pan, A.C.; Shaw, D.E. Structural mechanism for Bruton’s tyrosine kinase activation at the cell membrane. Proc. Natl. Acad. Sci. USA 2019, 116, 9390–9399. [Google Scholar] [CrossRef]
- Pan, A.C.; Jacobson, D.; Yatsenko, K.; Sritharan, D.; Weinreich, T.M.; Shaw, D.E. Atomic-level characterization of protein–protein association. Proc. Natl. Acad. Sci. USA 2019, 116, 4244–4249. [Google Scholar] [CrossRef] [PubMed]
- Carlesso, A.; Chintha, C.; Gorman, A.M.; Samali, A.; Eriksson, L.A. Binding Analysis of the Inositol-Requiring Enzyme 1 Kinase Domain. ACS Omega 2018, 3, 13313–13322. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Face-to-Face Dimer (PDB Code: 3P23) | Back-to-Back Dimer (PDB Code: 4YZC) | |
|---|---|---|
| SwarmDock | 1.39 | 3.56 |
| ZDOCK | 12.48 | 3.32 |
| HsymDock | 3.12 | 13.25 |
| PatchDock | 24.33 | 29.49 |
| ClusPro | 3.58 | 31.01 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carlesso, A.; Chintha, C.; Gorman, A.M.; Samali, A.; Eriksson, L.A. Effect of Kinase Inhibiting RNase Attenuator (KIRA) Compounds on the Formation of Face-to-Face Dimers of Inositol-Requiring Enzyme 1: Insights from Computational Modeling. Int. J. Mol. Sci. 2019, 20, 5538. https://doi.org/10.3390/ijms20225538
Carlesso A, Chintha C, Gorman AM, Samali A, Eriksson LA. Effect of Kinase Inhibiting RNase Attenuator (KIRA) Compounds on the Formation of Face-to-Face Dimers of Inositol-Requiring Enzyme 1: Insights from Computational Modeling. International Journal of Molecular Sciences. 2019; 20(22):5538. https://doi.org/10.3390/ijms20225538
Chicago/Turabian StyleCarlesso, Antonio, Chetan Chintha, Adrienne M. Gorman, Afshin Samali, and Leif A. Eriksson. 2019. "Effect of Kinase Inhibiting RNase Attenuator (KIRA) Compounds on the Formation of Face-to-Face Dimers of Inositol-Requiring Enzyme 1: Insights from Computational Modeling" International Journal of Molecular Sciences 20, no. 22: 5538. https://doi.org/10.3390/ijms20225538
APA StyleCarlesso, A., Chintha, C., Gorman, A. M., Samali, A., & Eriksson, L. A. (2019). Effect of Kinase Inhibiting RNase Attenuator (KIRA) Compounds on the Formation of Face-to-Face Dimers of Inositol-Requiring Enzyme 1: Insights from Computational Modeling. International Journal of Molecular Sciences, 20(22), 5538. https://doi.org/10.3390/ijms20225538