The Alteration of CTNNBIP1 in Lung Cancer

Abstract
:1. Introduction
2. Results
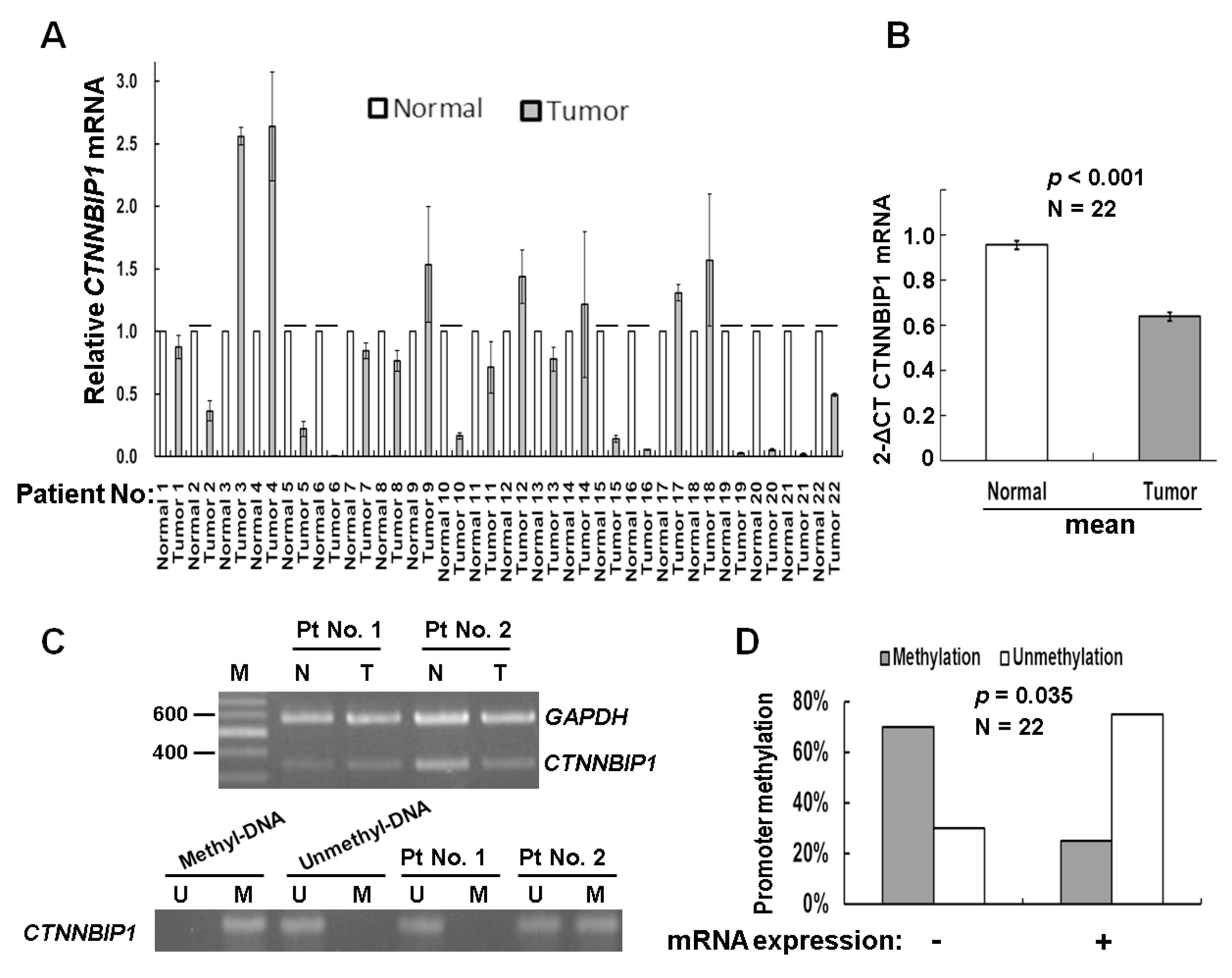
2.1. Analysis of the Factors Affecting CTNNBIP1 Gene Expression in Lung Cancer Patients
2.2. CTNNBIP1 is Reactivated by 5-aza-dC in Lung Cancer Cells
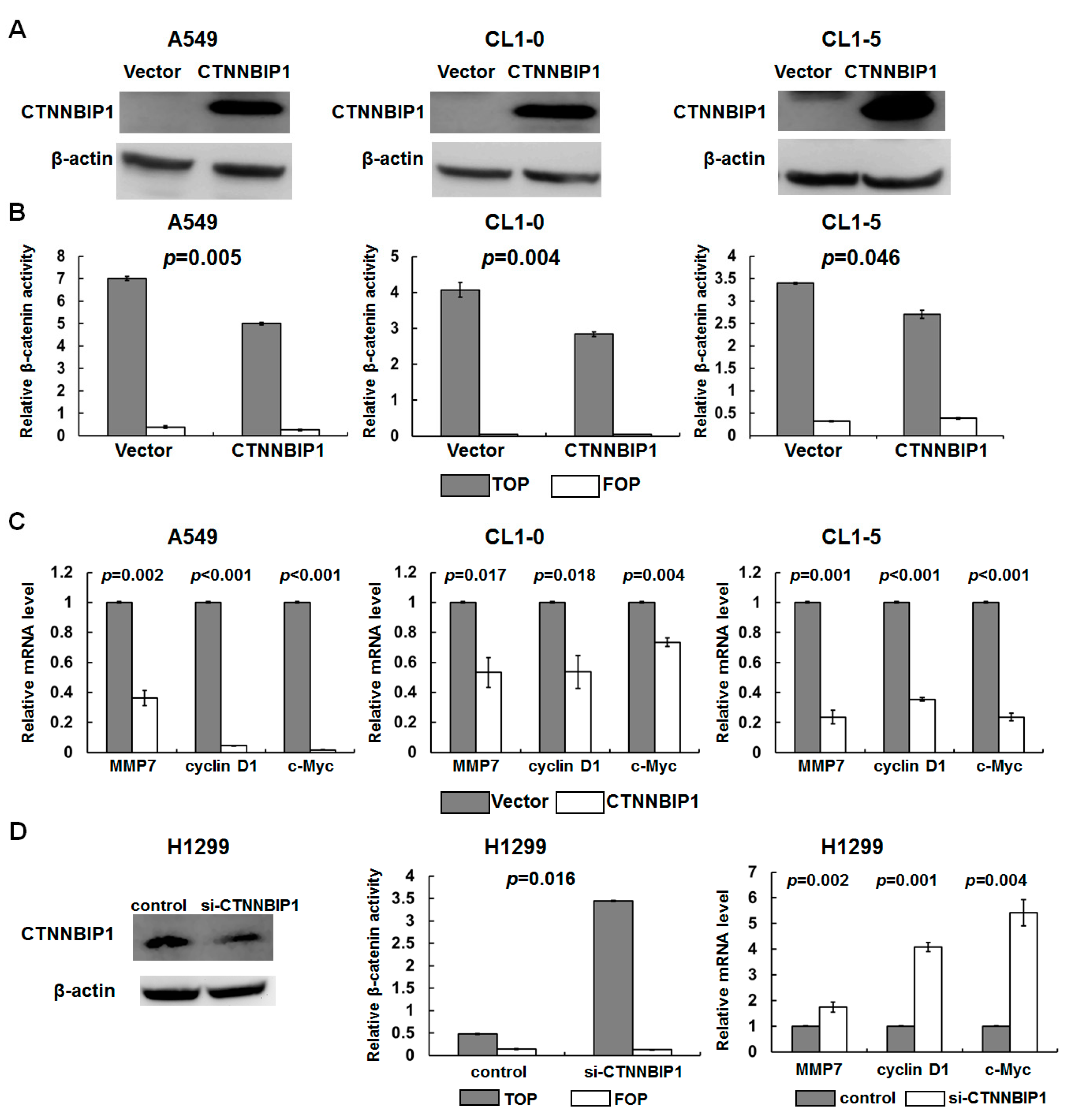
2.3. Ectopical Expression and Knockdown of CTNNBIP1 Influences the Transactivation of β-catenin in Lung Cancer Cell Lines
2.4. Ectopical Expression and Knockdown of CTNNBIP1 Influences the Migration of Lung Cancer Cells
2.5. Correlation between the Expression of CTNNBIP1 and the Clinical Characteristics of Lung Cancer Patients
3. Discussion
4. Materials and Methods
4.1. Subjects
4.2. Cell Culture
4.3. RT-PCR Analysis
4.4. RT-qPCR Assays
4.5. Western Blot Analysis
4.6. Methylation-Specific PCR (MSP) Assay
4.7. 5-aza-2′-Deoxycytidine (5-aza-dC) Treatment
4.8. Knockdown and Ectopic Expression
4.9. Luciferase Reporter Assay
4.10. Wound Healing Assay
4.11. Transwell Migration Assay
4.12. Adhesion Assay
4.13. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Wood, S.L.; Pernemalm, M.; Crosbie, P.A.; Whetton, A.D. Molecular histology of lung cancer: From targets to treatments. Cancer Treat. Rev. 2015, 41, 361–375. [Google Scholar] [CrossRef]
- Tseng, R.C.; Chang, J.W.; Hsien, F.J.; Chang, Y.H.; Hsiao, C.F.; Chen, J.T.; Chen, C.Y.; Jou, Y.S.; Wang, Y.C. Genomewide loss of heterozygosity and its clinical associations in non small cell lung cancer. Int. J. Cancer 2005, 117, 241–247. [Google Scholar] [CrossRef]
- Polakis, P. Wnt signaling and cancer. Genes Dev. 2000, 14, 1837–1851. [Google Scholar] [CrossRef]
- Chen, R.H.; Ding, W.V.; McCormick, F. Wnt signaling to beta-catenin involves two interactive components. Glycogen synthase kinase-3beta inhibition and activation of protein kinase C. J. Biol. Chem. 2000, 275, 17894–17899. [Google Scholar] [CrossRef]
- He, T.C.; Sparks, A.B.; Rago, C.; Hermeking, H.; Zawel, L.; da Costa, L.T.; Morin, P.J.; Vogelstein, B.; Kinzler, K.W. Identification of c-MYC as a target of the APC pathway. Science 1998, 281, 1509–1512. [Google Scholar] [CrossRef]
- Shtutman, M.; Zhurinsky, J.; Simcha, I.; Albanese, C.; D’Amico, M.; Pestell, R.; Ben-Ze’ev, A. The cyclin D1 gene is a target of the beta-catenin/LEF-1 pathway. Proc. Natl. Acad. Sci. USA 1999, 96, 5522–5527. [Google Scholar] [CrossRef]
- Li, Y.J.; Wei, Z.M.; Meng, Y.X.; Ji, X.R. Beta-catenin up-regulates the expression of cyclinD1, c-myc and MMP-7 in human pancreatic cancer: Relationships with carcinogenesis and metastasis. World J. Gastroenterol. 2005, 11, 2117–2123. [Google Scholar] [CrossRef] [PubMed]
- Tago, K.; Nakamura, T.; Nishita, M.; Hyodo, J.; Nagai, S.; Murata, Y.; Adachi, S.; Ohwada, S.; Morishita, Y.; Shibuya, H.; et al. Inhibition of Wnt signaling by ICAT, a novel beta-catenin-interacting protein. Genes Dev. 2000, 14, 1741–1749. [Google Scholar] [PubMed]
- Satoh, K.; Kasai, M.; Ishidao, T.; Tago, K.; Ohwada, S.; Hasegawa, Y.; Senda, T.; Takada, S.; Nada, S.; Nakamura, T.; et al. Anteriorization of neural fate by inhibitor of beta-catenin and T cell factor (ICAT), a negative regulator of Wnt signaling. Proc. Natl. Acad. Sci. USA 2004, 101, 8017–8021. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.Y.; Pan, H.F.; Leng, R.X.; Ye, D.Q. Long noncoding RNAs: Novel insights into gastric cancer. Cancer Lett. 2015, 356, 357–366. [Google Scholar] [CrossRef]
- Reifenberger, J.; Knobbe, C.B.; Wolter, M.; Blaschke, B.; Schulte, K.W.; Pietsch, T.; Ruzicka, T.; Reifenberger, G. Molecular genetic analysis of malignant melanomas for aberrations of the WNT signaling pathway genes CTNNB1, APC, ICAT and BTRC. Int. J. Cancer 2002, 100, 549–556. [Google Scholar] [CrossRef] [PubMed]
- Sekiya, T.; Nakamura, T.; Kazuki, Y.; Oshimura, M.; Kohu, K.; Tago, K.; Ohwada, S.; Akiyama, T. Overexpression of Icat induces G(2) arrest and cell death in tumor cell mutants for adenomatous polyposis coli, beta-catenin, or Axin. Cancer Res. 2002, 62, 3322–3326. [Google Scholar] [PubMed]
- Zhang, K.; Zhu, S.; Liu, Y.; Dong, X.; Shi, Z.; Zhang, A.; Liu, C.; Chen, L.; Wei, J.; Pu, P.; et al. ICAT inhibits glioblastoma cell proliferation by suppressing Wnt/beta-catenin activity. Cancer Lett. 2015, 357, 404–411. [Google Scholar] [CrossRef] [PubMed]
- Hommura, F.; Furuuchi, K.; Yamazaki, K.; Ogura, S.; Kinoshita, I.; Shimizu, M.; Moriuchi, T.; Katoh, H.; Nishimura, M.; Dosaka-Akita, H. Increased expression of beta-catenin predicts better prognosis in nonsmall cell lung carcinomas. Cancer 2002, 94, 752–758. [Google Scholar] [CrossRef]
- Chiu, C.G.; Chan, S.K.; Fang, Z.A.; Masoudi, H.; Wood-Baker, R.; Jones, S.J.; Gilks, B.; Laskin, J.; Wiseman, S.M. Beta-catenin expression is prognostic of improved non-small cell lung cancer survival. Am. J. Surg. 2012, 203, 654–659. [Google Scholar] [CrossRef] [PubMed]
- Baylin, S.; Bestor, T.H. Altered methylation patterns in cancer cell genomes: Cause or consequence? Cancer Cell 2002, 1, 299–305. [Google Scholar] [CrossRef]
- Bjaanaes, M.M.; Fleischer, T.; Halvorsen, A.R.; Daunay, A.; Busato, F.; Solberg, S.; Jorgensen, L.; Kure, E.; Edvardsen, H.; Borresen-Dale, A.L.; et al. Genome-wide DNA methylation analyses in lung adenocarcinomas: Association with EGFR, KRAS and TP53 mutation status, gene expression and prognosis. Mol. Oncol. 2016, 10, 330–343. [Google Scholar] [CrossRef]
- Zeng, Z.S.; Shu, W.P.; Cohen, A.M.; Guillem, J.G. Matrix metalloproteinase-7 expression in colorectal cancer liver metastases: Evidence for involvement of MMP-7 activation in human cancer metastases. Clin. Cancer Res. 2002, 8, 144–148. [Google Scholar]
- Okayama, H.; Kohno, T.; Ishii, Y.; Shimada, Y.; Shiraishi, K.; Iwakawa, R.; Furuta, K.; Tsuta, K.; Shibata, T.; Yamamoto, S.; et al. Identification of genes upregulated in ALK-positive and EGFR/KRAS/ALK-negative lung adenocarcinomas. Cancer Res. 2012, 72, 100–111. [Google Scholar] [CrossRef]
- Klutstein, M.; Nejman, D.; Greenfield, R.; Cedar, H. DNA Methylation in Cancer and Aging. Cancer Res. 2016, 76, 3446–3450. [Google Scholar] [CrossRef]
- Xie, W.; Baylin, S.B.; Easwaran, H. DNA methylation in senescence, aging and cancer. Oncoscience 2019, 6, 291–293. [Google Scholar] [PubMed]
- Cruickshanks, H.A.; McBryan, T.; Nelson, D.M.; Vanderkraats, N.D.; Shah, P.P.; van Tuyn, J.; Singh Rai, T.; Brock, C.; Donahue, G.; Dunican, D.S.; et al. Senescent cells harbour features of the cancer epigenome. Nat. Cell Biol. 2013, 15, 1495–1506. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Kagiampakis, I.; Pan, L.; Zhang, Y.W.; Murphy, L.; Tao, Y.; Kong, X.; Kang, B.; Xia, L.; Carvalho, F.L.F.; et al. DNA Methylation Patterns Separate Senescence from Transformation Potential and Indicate Cancer Risk. Cancer Cell 2018, 33, 309–321. [Google Scholar] [CrossRef] [PubMed]
- Sadikovic, B.; Al-Romaih, K.; Squire, J.A.; Zielenska, M. Cause and consequences of genetic and epigenetic alterations in human cancer. Curr. Genom. 2008, 9, 394–408. [Google Scholar] [CrossRef]
- Dehan, E.; Ben-Dor, A.; Liao, W.; Lipson, D.; Frimer, H.; Rienstein, S.; Simansky, D.; Krupsky, M.; Yaron, P.; Friedman, E.; et al. Chromosomal aberrations and gene expression profiles in non-small cell lung cancer. Lung Cancer 2007, 56, 175–184. [Google Scholar] [CrossRef]
- Mukherjee, N.; Dasgupta, H.; Bhattacharya, R.; Pal, D.; Roy, R.; Islam, S.; Alam, N.; Biswas, J.; Roy, A.; Roychoudhury, S.; et al. Frequent inactivation of MCC/CTNNBIP1 and overexpression of phospho-beta-catenin(Y654) are associated with breast carcinoma: Clinical and prognostic significance. Biochim. Biophys. Acta 2016, 1862, 1472–1484. [Google Scholar] [CrossRef]
- Domingues, M.J.; Rambow, F.; Job, B.; Papon, L.; Liu, W.; Larue, L.; Bonaventure, J. Beta-catenin inhibitor ICAT modulates the invasive motility of melanoma cells. Cancer Res. 2014, 74, 1983–1995. [Google Scholar] [CrossRef]
- Director’s Challenge Consortium for the Molecular Classification of Lung Adenocarcinoma; Shedden, K.; Taylor, J.M.; Enkemann, S.A.; Tsao, M.S.; Yeatman, T.J.; Gerald, W.L.; Eschrich, S.; Jurisica, I.; Giordano, T.J.; et al. Gene expression-based survival prediction in lung adenocarcinoma: A multi-site, blinded validation study. Nat. Med. 2008, 14, 822–827. [Google Scholar]
- Qi, W.; Chen, J.; Cheng, X.; Huang, J.; Xiang, T.; Li, Q.; Long, H.; Zhu, B. Targeting the Wnt-Regulatory Protein CTNNBIP1 by microRNA-214 Enhances the Stemness and Self-Renewal of Cancer Stem-Like Cells in Lung Adenocarcinomas. Stem Cells 2015, 33, 3423–3436. [Google Scholar] [CrossRef]
- Xu, J.; Chen, Y.; Huo, D.; Khramtsov, A.; Khramtsova, G.; Zhang, C.; Goss, K.H.; Olopade, O.I. β-catenin regulates c-Myc and CDKN1A expression in breast cancer cells. Mol. Carcinog. 2016, 55, 431–439. [Google Scholar] [CrossRef]
- Wang, J.S.; Wang, W.J.; Wang, T.; Zhang, Y. Expression of ICAT and Wnt signaling-related proteins in the monocytic differentiation of HL-60 cells induced by a new steroidal drug NSC67657. Chin. J. Oncol. 2016, 38, 246–251. [Google Scholar]
- Yuan, B.Z.; Jefferson, A.M.; Popescu, N.C.; Reynolds, S.H. Aberrant gene expression in human non small cell lung carcinoma cells exposed to demethylating agent 5-aza-2’-deoxycytidine. Neoplasia 2004, 6, 412–419. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.W.; Yang, P.C.; Yang, S.C.; Shyu, Y.C.; Hendrix, M.J.; Wu, R.; Wu, C.W. Selection of invasive and metastatic subpopulations from a human lung adenocarcinoma cell line. Am. J. Respir. Cell Mol. Biol. 1997, 17, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.C.; Lin, R.K.; Tan, Y.H.; Chen, J.T.; Chen, C.Y.; Wang, Y.C. Wild-type p53 overexpression and its correlation with MDM2 and p14ARF alterations: An alternative pathway to non-small-cell lung cancer. J. Clin. Oncol. 2005, 23, 154–164. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | CTNNBIP1 Expression | ||||
|---|---|---|---|---|---|
| Total | + | − (%) | p-Value b | ||
| Overall | 204 | 162 | 42 (20.6) | ||
| Sex | Female | 109 | 92 | 17 (15.6) | 0.059 |
| Male | 95 | 70 | 25 (26.3) | ||
| Smoker | No | 105 | 91 | 14 (13.3) | 0.008 |
| Yes | 99 | 71 | 28 (28.3) | ||
| Tumor stage | IA | 109 | 95 | 14 (12.8) | 0.006 |
| IB | 53 | 40 | 13 (24.5) | ||
| II | 42 | 27 | 15 (35.7) | ||
| MMP7 | high | 40 | 24 | 16 (40.0) | 0.001 |
| low | 164 | 138 | 26 (15.9) | ||
| Cyclin D1 | high | 34 | 26 | 8 (23.5) | 0.642 |
| low | 170 | 136 | 34 (20.0) | ||
| c-MYC | high | 61 | 49 | 12 (19.7) | 0.833 |
| low | 143 | 113 | 30 (21.0) | ||
| Characteristics | Univariate Analysis | Multivariate Analysis | ||
|---|---|---|---|---|
| HR (95%CI) | p Value * | HR (95%CI) | p Value b | |
| CTNNBIP1 | ||||
| Expression | 1.00 | 1.00 | ||
| Low | 2.40 (1.36–4.24) | 0.002 | 1.85 (1.02–3.37) | 0.043 |
| Sex | ||||
| Female | 1.00 | 1.00 | ||
| Male | 1.40 (0.82–2.38) | 0.220 | 1.18 (0.57–2.41) | 0.659 |
| Smokier | ||||
| No | 1.00 | 1.00 | ||
| Yes | 1.43 (0.84–2.44) | 0.190 | 1.07 (0.52–2.22) | 0.853 |
| Tumor stage | ||||
| I | 1.00 | 1.00 | ||
| II | 3.44 (1.98–6.00) | <0.001 | 2.91 (1.63–5.18) | <0.001 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, J.-M.; Tsai, A.C.-D.; Huang, W.-R.; Tseng, R.-C. The Alteration of CTNNBIP1 in Lung Cancer. Int. J. Mol. Sci. 2019, 20, 5684. https://doi.org/10.3390/ijms20225684
Chang J-M, Tsai AC-D, Huang W-R, Tseng R-C. The Alteration of CTNNBIP1 in Lung Cancer. International Journal of Molecular Sciences. 2019; 20(22):5684. https://doi.org/10.3390/ijms20225684
Chicago/Turabian StyleChang, Jia-Ming, Alexander Charng-Dar Tsai, Way-Ren Huang, and Ruo-Chia Tseng. 2019. "The Alteration of CTNNBIP1 in Lung Cancer" International Journal of Molecular Sciences 20, no. 22: 5684. https://doi.org/10.3390/ijms20225684
APA StyleChang, J. -M., Tsai, A. C. -D., Huang, W. -R., & Tseng, R. -C. (2019). The Alteration of CTNNBIP1 in Lung Cancer. International Journal of Molecular Sciences, 20(22), 5684. https://doi.org/10.3390/ijms20225684