Mutation-Driven Signals of ARID1A and PI3K Pathways in Ovarian Carcinomas: Alteration Is An Opportunity

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Prologue
2. ARID1A, A Member of the Mammalian Chromatin Remodeling Complex, SWI/SNF
3. ARID1A as an Epigenetic Tumor Suppressor
4. ARID1A Mutation in Ovarian Carcinomas
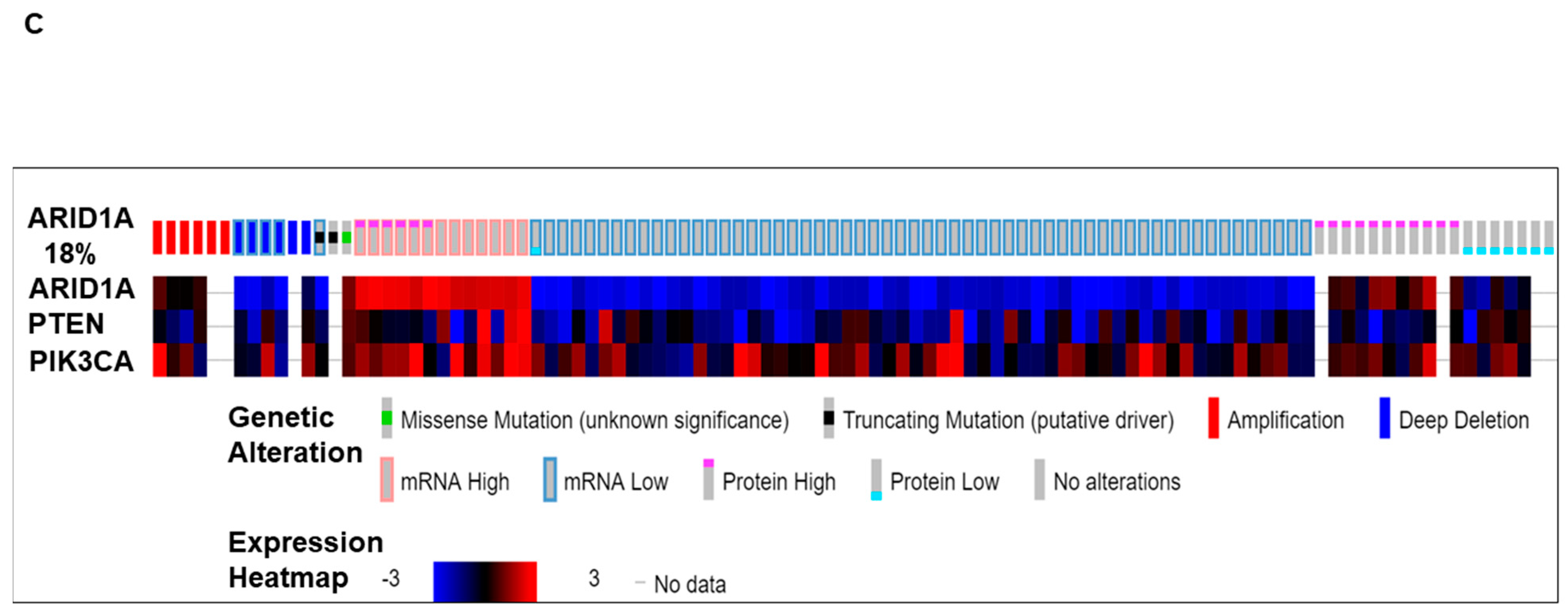
5. Alterations in the ARID1A Gene and Its Co-Occurrence with Alterations of the PI3K Pathway Genes in Ovarian Carcinomas: The Avera Experience
6. ARID1A Protein and its Role in Tumorigenic Transformation
7. Co-Alterations of ARID1A with Other Oncogenic Pathways in the Context of Mutation-driven Signaling in Tumor cells
8. Is Mutation-Driven Co-Alterations in the ARID1A-PI3K Signaling an Opportunity?
9. Epilogue
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Aalfs, J.D.; Kingston, R.E. What does ‘chromatin remodeling’ mean? Trends Biochem. Sci. 2000, 25, 548–555. [Google Scholar] [CrossRef]
- Clapier, C.R.; Cairns, B.R. The biology of chromatin remodeling complexes. Annu. Rev. Biochem. 2009, 78, 273–304. [Google Scholar] [CrossRef] [PubMed]
- Wilson, B.G.; Roberts, C.W. SWI/SNF nucleosome remodellers and cancer. Nat. Rev. Cancer 2011, 11, 481–492. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.; Imbalzano, A.N.; Khavari, P.A.; Kingston, R.E.; Green, M.R. Nucleosome disruption and enhancement of activator binding by a human SW1/SNF complex. Nature 1994, 370, 477–481. [Google Scholar] [CrossRef]
- Imbalzano, A.N.; Kwon, H.; Green, M.R.; Kingston, R.E. Facilitated binding of TATA-binding protein to nucleosomal DNA. Nature 1994, 370, 481–485. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.G.; Allis, C.D.; Chi, P. Chromatin remodeling and cancer, Part II: ATP-dependent chromatin remodeling. Trends Mol. Med. 2007, 13, 373–380. [Google Scholar] [CrossRef]
- Roberts, C.W.; Orkin, S.H. The SWI/SNF complex—Chromatin and cancer. Nat. Rev. Cancer 2004, 4, 133–142. [Google Scholar] [CrossRef]
- Megaridis, M.R.; Lu, Y.; Tevonian, E.N.; Junger, K.M.; Moy, J.M.; Bohn-Wippert, K.; Dar, R.D. Fine-tuning of noise in gene expression with nucleosome remodeling. APL Bioeng. 2018, 2, 026106. [Google Scholar] [CrossRef]
- Euskirchen, G.; Auerbach, R.K.; Snyder, M. SWI/SNF chromatin-remodeling factors: Multiscale analyses and diverse functions. J. Biol. Chem. 2012, 287, 30897–30905. [Google Scholar] [CrossRef]
- Masliah-Planchon, J.; Bieche, I.; Guinebretiere, J.M.; Bourdeaut, F.; Delattre, O. SWI/SNF chromatin remodeling and human malignancies. Annu. Rev. Pathol. 2015, 10, 145–171. [Google Scholar] [CrossRef]
- Arnaud, O.; Le Loarer, F.; Tirode, F. BAFfling pathologies: Alterations of BAF complexes in cancer. Cancer Lett. 2018, 419, 266–279. [Google Scholar] [CrossRef] [PubMed]
- Kadoch, C.; Hargreaves, D.C.; Hodges, C.; Elias, L.; Ho, L.; Ranish, J.; Crabtree, G.R. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat. Genet. 2013, 45, 592–601. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.; Li, M.; Parsons, D.W.; Zhang, X.; Wesseling, J.; Kristel, P.; Schmidt, M.K.; Markowitz, S.; Yan, H.; Bigner, D.; et al. Somatic mutations in the chromatin remodeling gene ARID1A occur in several tumor types. Hum. Mutat. 2012, 33, 100–103. [Google Scholar] [CrossRef] [PubMed]
- Takeda, T.; Banno, K.; Okawa, R.; Yanokura, M.; Iijima, M.; Irie-Kunitomi, H.; Nakamura, K.; Iida, M.; Adachi, M.; Umene, K.; et al. ARID1A gene mutation in ovarian and endometrial cancers (Review). Oncol. Rep. 2016, 35, 607–613. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.N.; Roberts, C.W. ARID1A mutations in cancer: Another epigenetic tumor suppressor? Cancer Discov. 2013, 3, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.; Wang, T.L.; Shih Ie, M.; Mao, T.L.; Nakayama, K.; Roden, R.; Glas, R.; Slamon, D.; Diaz, L.A., Jr.; Vogelstein, B.; et al. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science 2010, 330, 228–231. [Google Scholar] [CrossRef]
- Ayhan, A.; Mao, T.L.; Seckin, T.; Wu, C.H.; Guan, B.; Ogawa, H.; Futagami, M.; Mizukami, H.; Yokoyama, Y.; Kurman, R.J.; et al. Loss of ARID1A expression is an early molecular event in tumor progression from ovarian endometriotic cyst to clear cell and endometrioid carcinoma. Int. J. Gynecol. Cancer 2012, 22, 1310–1315. [Google Scholar] [CrossRef]
- Wiegand, K.C.; Shah, S.P.; Al-Agha, O.M.; Zhao, Y.; Tse, K.; Zeng, T.; Senz, J.; McConechy, M.K.; Anglesio, M.S.; Kalloger, S.E.; et al. ARID1A mutations in endometriosis-associated ovarian carcinomas. N. Engl. J. Med. 2010, 363, 1532–1543. [Google Scholar] [CrossRef]
- Lowery, W.J.; Schildkraut, J.M.; Akushevich, L.; Bentley, R.; Marks, J.R.; Huntsman, D.; Berchuck, A. Loss of ARID1A-associated protein expression is a frequent event in clear cell and endometrioid ovarian cancers. Int. J. Gynecol. Cancer 2012, 22, 9–14. [Google Scholar] [CrossRef]
- Obata, K.; Hoshiai, H. Common genetic changes between endometriosis and ovarian cancer. Gynecol. Obstet. Investig. 2000, 50 (Suppl. 1), 39–43. [Google Scholar] [CrossRef]
- Maeda, D.; Shih Ie, M. Pathogenesis and the role of ARID1A mutation in endometriosis-related ovarian neoplasms. Adv. Anat. Pathol. 2013, 20, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Heckl, M.; Schmoeckel, E.; Hertlein, L.; Rottmann, M.; Jeschke, U.; Mayr, D. The ARID1A, p53 and ss-Catenin statuses are strong prognosticators in clear cell and endometrioid carcinoma of the ovary and the endometrium. PLoS ONE 2018, 13, e0192881. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multi-dimensional Cancer Genomics Data. Cancer Discovery. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed]
- Martens, J.A.; Winston, F. Recent advances in understanding chromatin remodeling by Swi/Snf complexes. Curr. Opin. Genet. Dev. 2003, 13, 136–142. [Google Scholar] [CrossRef]
- Papillon, J.P.N.; Nakajima, K.; Adair, C.D.; Hempel, J.; Jouk, A.O.; Karki, R.G.; Mathieu, S.; Möbitz, H.; Ntaganda, R.; Smith, T.; et al. Discovery of Orally Active Inhibitors of Brahma Homolog (BRM)/SMARCA2 ATPase Activity for the Treatment of Brahma Related Gene 1 (BRG1)/SMARCA4-Mutant Cancers. J. Med. Chem. 2018, 61, 10155–10172. [Google Scholar] [CrossRef] [PubMed]
- Phelan, M.L.; Sif, S.; Narlikar, G.J.; Kingston, R.E. Reconstitution of a core chromatin remodeling complex from SWI/SNF subunits. Mol. Cell 1999, 3, 247–253. [Google Scholar] [CrossRef]
- Dinulescu, D.M.; Ince, T.A.; Quade, B.J.; Shafer, S.A.; Crowley, D.; Jacks, T. Role of K-ras and Pten in the development of mouse models of endometriosis and endometrioid ovarian cancer. Nat. Med. 2005, 11, 63–70. [Google Scholar] [CrossRef]
- Obata, K.; Morland, S.J.; Watson, R.H.; Hitchcock, A.; Chenevix-Trench, G.; Thomas, E.J.; Campbell, I.G. Frequent PTEN/MMAC mutations in endometrioid but not serous or mucinous epithelial ovarian tumors. Cancer Res. 1998, 58, 2095–2097. [Google Scholar]
- Ruderman, R.; Pavone, M.E. Ovarian cancer in endometriosis: An update on the clinical and molecular aspects. Minerva Ginecol. 2017, 69, 286–294. [Google Scholar]
- Cybulska, P.; Paula, A.D.C.; Tseng, J.; Leitao, M.M., Jr.; Bashashati, A.; Huntsman, D.G.; Nazeran, T.M.; Aghajanian, C.; Abu-Rustum, N.R.; DeLair, D.F.; et al. Molecular profiling and molecular classification of endometrioid ovarian carcinomas. Gynecol. Oncol. 2019, 154, 516–523. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhao, H.; Zhang, X.; Wood, L.D.; Anders, R.A.; Choti, M.A.; Pawlik, T.M.; Daniel, H.D.; Kannangai, R.; Offerhaus, G.J.; et al. Inactivating mutations of the chromatin remodeling gene ARID2 in hepatocellular carcinoma. Nat. Genet. 2011, 43, 828–829. [Google Scholar] [CrossRef] [PubMed]
- Hodis, E.; Watson, I.R.; Kryukov, G.V.; Arold, S.T.; Imielinski, M.; Theurillat, J.P.; Nickerson, E.; Auclair, D.; Li, L.; Place, C.; et al. A landscape of driver mutations in melanoma. Cell 2012, 150, 251–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krauthammer, M.; Kong, Y.; Ha, B.H.; Evans, P.; Bacchiocchi, A.; McCusker, J.P.; Cheng, E.; Davis, M.J.; Goh, G.; Choi, M.; et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat. Genet. 2012, 44, 1006–1014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khazanchi, R.; Ronspies, C.A.; Smith, S.C.; Starr, L.J. Patient with anomalous skin pigmentation expands the phenotype of ARID2 loss-of-function disorder, a SWI/SNF-related intellectual disability. Am. J. Med. Genet. Part A 2019, 179, 808–812. [Google Scholar] [CrossRef]
- Beausoleil, S.A.; Jedrychowski, M.; Schwartz, D.; Elias, J.E.; Villen, J.; Li, J.; Cohn, M.A.; Cantley, L.C.; Gygi, S.P. Large-scale characterization of HeLa cell nuclear phosphoproteins. Proc. Natl. Acad. Sci. USA 2004, 101, 12130–12135. [Google Scholar] [CrossRef] [Green Version]
- Samartzis, E.P.; Noske, A.; Dedes, K.J.; Fink, D.; Imesch, P. ARID1A mutations and PI3K/AKT pathway alterations in endometriosis and endometriosis-associated ovarian carcinomas. Int. J. Mol. Sci. 2013, 14, 18824–18849. [Google Scholar]
- Trizzino, M.; Barbieri, E.; Petracovici, A.; Wu, S.; Welsh, S.A.; Owens, T.A.; Licciulli, S.; Zhang, R.; Gardini, A. The Tumor Suppressor ARID1A Controls Global Transcription via Pausing of RNA Polymerase, I.I. Cell Rep. 2018, 23, 3933–3945. [Google Scholar] [CrossRef]
- Guan, B.; Wang, T.L.; Shih Ie, M. ARID1A, a factor that promotes formation of SWI/SNF-mediated chromatin remodeling, is a tumor suppressor in gynecologic cancers. Cancer Res. 2011, 71, 6718–6727. [Google Scholar] [CrossRef] [Green Version]
- Flores-Alcantar, A.; Gonzalez-Sandoval, A.; Escalante-Alcalde, D.; Lomeli, H. Dynamics of expression of ARID1A and ARID1B subunits in mouse embryos and in cells during the cell cycle. Cell Tissue Res. 2011, 345, 137–148. [Google Scholar] [CrossRef]
- Nagl, N.G., Jr.; Patsialou, A.; Haines, D.S.; Dallas, P.B.; Beck, G.R., Jr.; Moran, E. The p270 (ARID1A/SMARCF1) subunit of mammalian SWI/SNF-related complexes is essential for normal cell cycle arrest. Cancer Res. 2005, 65, 9236–9244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagl, N.G., Jr.; Wang, X.; Patsialou, A.; Van Scoy, M.; Moran, E. Distinct mammalian SWI/SNF chromatin remodeling complexes with opposing roles in cell-cycle control. EMBO J. 2007, 26, 752–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, X.; Tate, P.; Hu, P.; Tjian, R.; Skarnes, W.C.; Wang, Z. ES cell pluripotency and germ-layer formation require the SWI/SNF chromatin remodeling component BAF250a. Proc. Natl. Acad. Sci. USA 2008, 105, 6656–6661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, B.; Cheung, H.W.; Subramanian, A.; Sharifnia, T.; Okamoto, M.; Yang, X.; Hinkle, G.; Boehm, J.S.; Beroukhim, R.; Weir, B.A.; et al. Highly parallel identification of essential genes in cancer cells. Proc. Natl. Acad. Sci. USA 2008, 105, 20380–20385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, B.; Lin, J.; Rong, L.; Wu, S.; Deng, Z.; Fatkhutdinov, N.; Zundell, J.; Fukumoto, T.; Liu, Q.; Kossenkov, A.; et al. ARID1A promotes genomic stability through protecting telomere cohesion. Nat. Commun. 2019, 10, 4067. [Google Scholar]
- Shen, J.; Ju, Z.; Zhao, W.; Wang, L.; Peng, Y.; Ge, Z.; Nagel, Z.D.; Zou, J.; Wang, C.; Kapoor, P.; et al. ARID1A deficiency promotes mutability and potentiates therapeutic antitumor immunity unleashed by immune checkpoint blockade. Nat. Med. 2018, 24, 556–562. [Google Scholar] [CrossRef]
- Shen, J.; Peng, Y.; Wei, L.; Zhang, W.; Yang, L.; Lan, L.; Kapoor, P.; Ju, Z.; Mo, Q.; Shih, I.M.; et al. ARID1A Deficiency Impairs the DNA Damage Checkpoint and Sensitizes Cells to PARP Inhibitors. Cancer Discov. 2015, 5, 752–767. [Google Scholar]
- Caumanns, J.J.; Wisman, G.B.A.; Berns, K.; van der Zee, A.G.J.; de Jong, S. ARID1A mutant ovarian clear cell carcinoma: A clear target for synthetic lethal strategies. Biochim. Biophys. Acta Rev. Cancer 2018, 1870, 176–184. [Google Scholar] [CrossRef]
- FDA approves PARP inhibitor for ovarian cancer. Nat. Biotechnol. 2017, 35, 398. [CrossRef]
- McCann, K.E. Novel poly-ADP-ribose polymerase inhibitor combination strategies in ovarian cancer. Curr. Opin. Obstet. Gynecol. 2018, 30, 7–16. [Google Scholar] [CrossRef]
- Schick, S.; Rendeiro, A.F.; Runggatscher, K.; Ringler, A.; Boidol, B.; Hinkel, M.; Májek, P.; Vulliard, L.; Penz, T.; Parapatics, K.; et al. Systematic characterization of BAF mutations provides insights into intracomplex synthetic lethalities in human cancers. Nat. Genet. 2019, 51, 1399–1410. [Google Scholar] [CrossRef] [PubMed]
- Bitler, B.G.; Fatkhutdinov, N.; Zhang, R. Potential therapeutic targets in ARID1A-mutated cancers. Expert Opin. Ther. Targets 2015, 19, 1419–1422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, R.C.; Wang, T.L.; Shih Ie, M. The emerging roles of ARID1A in tumor suppression. Cancer Biol. Ther. 2014, 15, 655–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathur, R. ARID1A loss in cancer: Towards a mechanistic understanding. Pharmacol. Ther. 2018, 190, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Yu, E.J.; Ham, I.H.; Hur, H.; Kim, Y.S. AKT inhibition is an effective treatment strategy in ARID1A-deficient gastric cancer cells. OncoTargets Ther. 2017, 10, 4153–4159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Yan, H.B.; Wang, J.; Cui, S.J.; Wang, X.Q.; Jiang, Y.H.; Feng, L.; Yang, P.Y.; Liu, F. Chromatin remodeling gene AT-rich interactive domain-containing protein 1A suppresses gastric cancer cell proliferation by targeting PIK3CA and PDK1. Oncotarget 2016, 7, 46127–46141. [Google Scholar] [CrossRef]
- Samartzis, E.P.; Gutsche, K.; Dedes, K.J.; Fink, D.; Stucki, M.; Imesch, P. Loss of ARID1A expression sensitizes cancer cells to PI3K- and AKT-inhibition. Oncotarget 2014, 5, 5295–5303. [Google Scholar] [CrossRef]
- Wiegand, K.C.; Hennessy, B.T.; Leung, S.; Wang, Y.; Ju, Z.; McGahren, M.; Kalloger, S.E.; Finlayson, S.; Stemke-Hale, K.; Lu, Y.; et al. A functional proteogenomic analysis of endometrioid and clear cell carcinomas using reverse phase protein array and mutation analysis: Protein expression is histotype-specific and loss of ARID1A/BAF250a is associated with AKT phosphorylation. BMC Cancer 2014, 14, 120. [Google Scholar] [CrossRef] [Green Version]
- Miller, R.E.; Brough, R.; Bajrami, I.; Williamson, C.T.; McDade, S.; Campbell, J.; Kigozi, A.; Rafiq, R.; Pemberton, H.; Natrajan, R.; et al. Synthetic Lethal Targeting of ARID1A-Mutant Ovarian Clear Cell Tumors with Dasatinib. Mol. Cancer Ther. 2016, 15, 1472–1484. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Yang, G.; Ding, Y.; Huang, Y.; Liu, S.; Zhou, L.; Wei, W.; Wang, J.; Hu, G. Combined treatment with PI3K inhibitor BKM120 and PARP inhibitor olaparib is effective in inhibiting the gastric cancer cells with ARID1A deficiency. Oncol. Rep. 2018, 40, 479–487. [Google Scholar] [CrossRef]
- Kim, Y.B.; Ahn, J.M.; Bae, W.J.; Sung, C.O.; Lee, D. Functional loss of ARID1A is tightly associated with high PD-L1 expression in gastric cancer. Int. J. Cancer 2019, 145, 916–926. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Bueno, G.; Gamallo, C.; Perez-Gallego, L.; de Mora, J.C.; Suarez, A.; Palacios, J. Beta-Catenin expression pattern, beta-catenin gene mutations, and microsatellite instability in endometrioid ovarian carcinomas and synchronous endometrial carcinomas. Diagn. Mol. Pathol. 2001, 10, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Vasileiou, G.; Ekici, A.B.; Uebe, S.; Zweier, C.; Hoyer, J.; Engels, H.; Behrens, J.; Reis, A.; Hadjihannas, M.V. Chromatin-Remodeling-Factor ARID1B Represses Wnt/beta-Catenin Signaling. Am. J. Hum. Genet. 2015, 97, 445–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bitler, B.G.; Aird, K.M.; Garipov, A.; Li, H.; Amatangelo, M.; Kossenkov, A.V.; Schultz, D.C.; Liu, Q.; Shih, I.M.; Conejo-Garcia, J.R.; et al. Synthetic lethality by targeting EZH2 methyltransferase activity in ARID1A-mutated cancers. Nat. Med. 2015, 21, 231–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berns, K.; Caumanns, J.J.; Hijmans, E.M.; Gennissen, A.M.C.; Severson, T.M.; Evers, B.; Wisman, G.B.A.; Jan Meersma, G.; Lieftink, C.; Beijersbergen, R.L.; et al. ARID1A mutation sensitizes most ovarian clear cell carcinomas to BET inhibitors. Oncogene 2018, 37, 4611–4625. [Google Scholar] [CrossRef] [PubMed]
- Chandler, R.L.; Damrauer, J.S.; Raab, J.R.; Schisler, J.C.; Wilkerson, M.D.; Didion, J.P.; Starmer, J.; Serber, D.; Yee, D.; Xiong, J.; et al. Coexistent ARID1A-PIK3CA mutations promote ovarian clear-cell tumorigenesis through pro-tumorigenic inflammatory cytokine signalling. Nat. Commun. 2015, 6, 6118. [Google Scholar] [CrossRef] [Green Version]
- Chene, G.; Ouellet, V.; Rahimi, K.; Barres, V.; Provencher, D.; Mes-Masson, A.M. The ARID1A pathway in ovarian clear cell and endometrioid carcinoma, contiguous endometriosis, and benign endometriosis. Int. J. Gynaecol. Obstet. 2015, 130, 27–30. [Google Scholar] [CrossRef]
- Watanabe, R.; Ui, A.; Kanno, S.; Ogiwara, H.; Nagase, T.; Kohno, T.; Yasui, A. SWI/SNF factors required for cellular resistance to DNA damage include ARID1A and ARID1B and show interdependent protein stability. Cancer Res. 2014, 74, 2465–2475. [Google Scholar] [CrossRef] [Green Version]
- Davidson, J.; Shen, Z.; Gong, X.; Pollack, J.R. SWI/SNF aberrations sensitize pancreatic cancer cells to DNA crosslinking agents. Oncotarget 2018, 9, 9608–9617. [Google Scholar] [CrossRef] [Green Version]
- Park, Y.; Chui, M.H.; Suryo Rahmanto, Y.; Yu, Z.C.; Shamanna, R.A.; Bellani, M.A.; Gaillard, S.; Ayhan, A.; Viswanathan, A.; Seidman, M.M.; et al. Loss of ARID1A in Tumor Cells Renders Selective Vulnerability to Combined Ionizing Radiation and PARP Inhibitor Therapy. Clin. Cancer Res. 2019, 25, 5584–5594. [Google Scholar] [CrossRef]
- Yang, L.; Yang, G.; Ding, Y.; Dai, Y.; Xu, S.; Guo, Q.; Xie, A.; Hu, G. Inhibition of PI3K/AKT Signaling Pathway Radiosensitizes Pancreatic Cancer Cells with ARID1A Deficiency In Vitro. J. Cancer 2018, 9, 890–900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abe, A.; Minaguchi, T.; Ochi, H.; Onuki, M.; Okada, S.; Matsumoto, K.; Satoh, T.; Oki, A.; Yoshikawa, H. PIK3CA overexpression is a possible prognostic factor for favorable survival in ovarian clear cell carcinoma. Hum. Pathol. 2013, 44, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Oda, K.; Hamanishi, J.; Matsuo, K.; Hasegawa, K. Genomics to immunotherapy of ovarian clear cell carcinoma: Unique opportunities for management. Gynecol. Oncol. 2018, 151, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Y.; Kuick, R.; Tipton, C.; Wu, R.; Sessine, M.; Wang, Z.; Baker, S.J.; Fearon, E.R.; Cho, K.R. Arid1a inactivation in an Apc- and Pten-defective mouse ovarian cancer model enhances epithelial differentiation and prolongs survival. J. Pathol. 2016, 238, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Berns, K.; Sonnenblick, A.; Gennissen, A.; Brohee, S.; Hijmans, E.M.; Evers, B.; Fumagalli, D.; Desmedt, C.; Loibl, S.; Denkert, C.; et al. Loss of ARID1A Activates ANXA1, which Serves as a Predictive Biomarker for Trastuzumab Resistance. Clin. Cancer Res. 2016, 22, 5238–5248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosse, T.; ter Haar, N.T.; Seeber, L.M.; v Diest, P.J.; Hes, F.J.; Vasen, H.F.; Nout, R.A.; Creutzberg, C.L.; Morreau, H.; Smit, V.T. Loss of ARID1A expression and its relationship with PI3K-Akt pathway alterations, TP53 and microsatellite instability in endometrial cancer. Mod. Pathol. 2013, 26, 1525–1535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, M.R.; Reske, J.J.; Holladay, J.; Wilber, G.E.; Rhodes, M.; Koeman, J.; Adams, M.; Johnson, B.; Su, R.W.; Joshi, N.R.; et al. ARID1A and PI3-kinase pathway mutations in the endometrium drive epithelial transdifferentiation and collective invasion. Nat. Commun. 2019, 10, 3554. [Google Scholar] [CrossRef]
- Bitler, B.G.; Wu, S.; Park, P.H.; Hai, Y.; Aird, K.M.; Wang, Y.; Zhai, Y.; Kossenkov, A.V.; Vara-Ailor, A.; Rauscher, F.J., III; et al. ARID1A-mutated ovarian cancers depend on HDAC6 activity. Nat. Cell Biol. 2017, 19, 962–973. [Google Scholar] [CrossRef] [Green Version]
- Berger, A.H.; Knudson, A.G.; Pandolfi, P.P. A continuum model for tumour suppression. Nature 2011, 476, 163–169. [Google Scholar] [CrossRef] [Green Version]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De, P.; Dey, N. Mutation-Driven Signals of ARID1A and PI3K Pathways in Ovarian Carcinomas: Alteration Is An Opportunity. Int. J. Mol. Sci. 2019, 20, 5732. https://doi.org/10.3390/ijms20225732
De P, Dey N. Mutation-Driven Signals of ARID1A and PI3K Pathways in Ovarian Carcinomas: Alteration Is An Opportunity. International Journal of Molecular Sciences. 2019; 20(22):5732. https://doi.org/10.3390/ijms20225732
Chicago/Turabian StyleDe, Pradip, and Nandini Dey. 2019. "Mutation-Driven Signals of ARID1A and PI3K Pathways in Ovarian Carcinomas: Alteration Is An Opportunity" International Journal of Molecular Sciences 20, no. 22: 5732. https://doi.org/10.3390/ijms20225732
APA StyleDe, P., & Dey, N. (2019). Mutation-Driven Signals of ARID1A and PI3K Pathways in Ovarian Carcinomas: Alteration Is An Opportunity. International Journal of Molecular Sciences, 20(22), 5732. https://doi.org/10.3390/ijms20225732