Beyond the Cell Surface: Targeting Intracellular Negative Regulators to Enhance T cell Anti-Tumor Activity

 ,
, 
Abstract
:1. Introduction
2. E3 Ubiquitin Ligases that Negatively Regulate T Cells
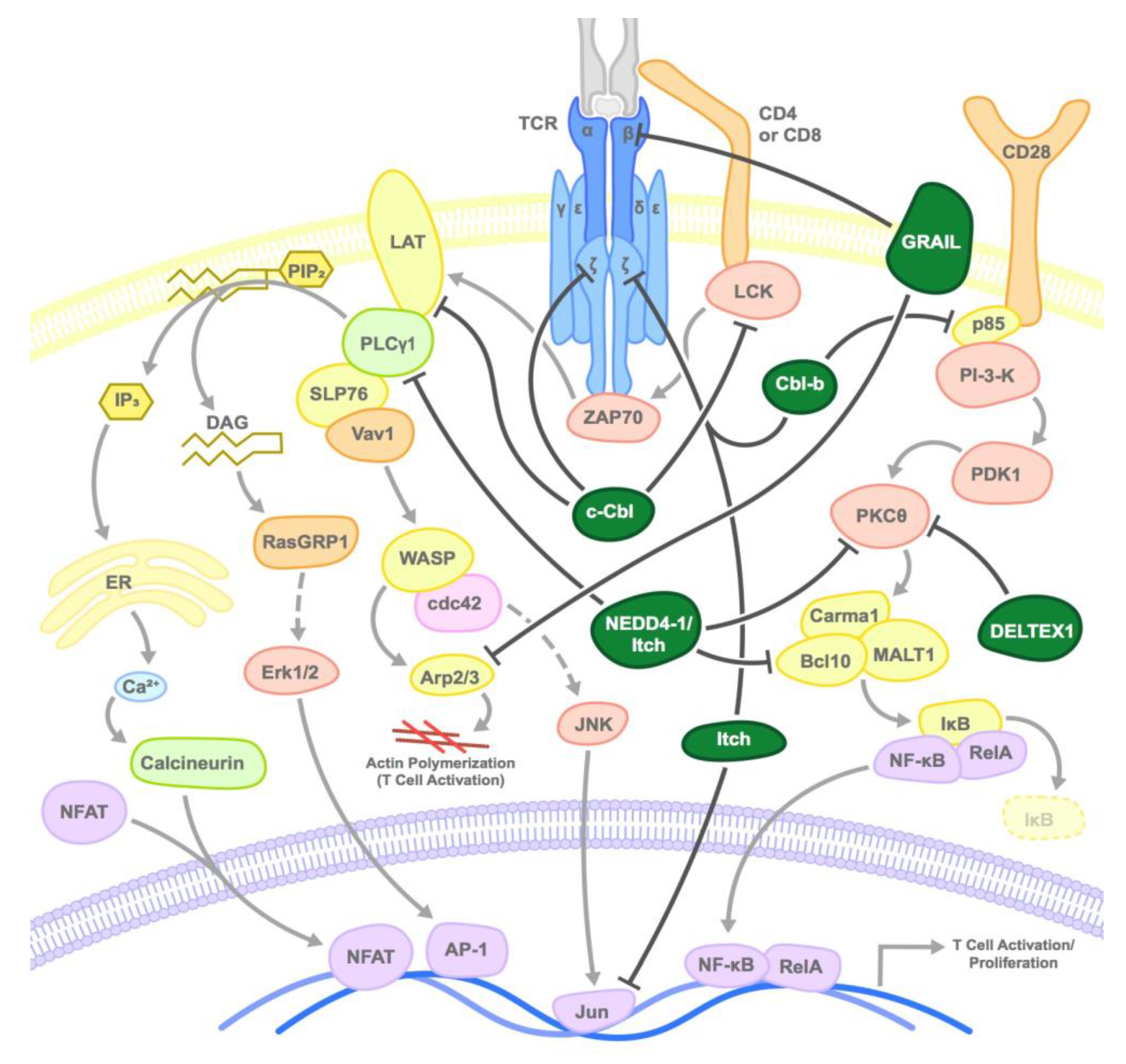
2.1. CBL Family
2.2. GRAIL
2.3. NEDD4 Family
2.4. Deltex1
2.5. Other E3 Ligases
3. Kinases and Phosphatases that Negatively Regulate T Cells
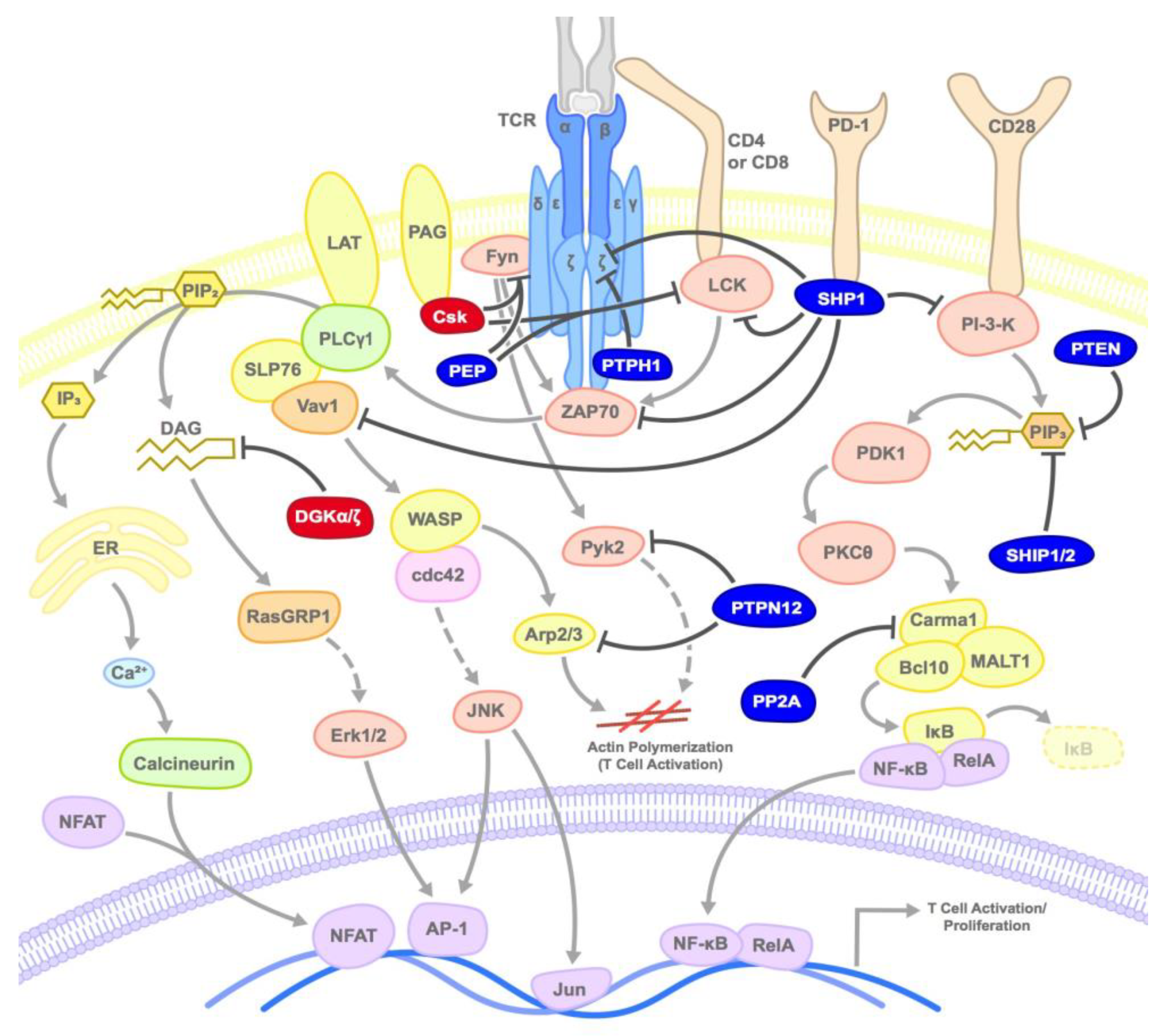
3.1. SHP1/2
3.2. PTEN
3.3. SHIP
3.4. PEP/PTPN22
3.5. PTP-PEST/PTPN12
3.6. PTPH1
3.7. PP2A
3.8. Csk
3.9. DGKs
4. Autoimmune Considerations
5. Conclusions
Funding
Conflicts of Interest
References
- Burton, J.C.; Grimsey, N.J. Ubiquitination as a Key Regulator of Endosomal Signaling by GPCRs. Front. Cell Dev. Biol. 2019, 7, 43. [Google Scholar] [CrossRef]
- Kar, G.; Keskin, O.; Nussinov, R.; Gursoy, A. Human proteome-scale structural modeling of E2-E3 interactions exploiting interface motifs. J. Proteome Res. 2012, 11, 1196–1207. [Google Scholar] [CrossRef]
- Loureiro, J.; Ploegh, H.L. Antigen presentation and the ubiquitin-proteasome system in host-pathogen interactions. Adv. Immunol. 2006, 92, 225–305. [Google Scholar]
- Kanarek, N.; Ben-Neriah, Y. Regulation of NF-kappaB by ubiquitination and degradation of the IkappaBs. Immunol. Rev. 2012, 246, 77–94. [Google Scholar] [CrossRef] [PubMed]
- Keane, M.M.; Ettenberg, S.A.; Nau, M.M.; Banerjee, P.; Cuello, M.; Penninger, J.; Lipkowitz, S. cbl-3: A new mammalian cbl family protein. Oncogene 1999, 18, 3365–3375. [Google Scholar] [CrossRef] [PubMed]
- Keane, M.M.; Rivero-Lezcano, O.M.; Mitchell, J.A.; Robbins, K.C.; Lipkowitz, S. Cloning and characterization of cbl-b: A SH3 binding protein with homology to the c-cbl proto-oncogene. Oncogene 1995, 10, 2367–2377. [Google Scholar] [PubMed]
- Kim, M.; Tezuka, T.; Suziki, Y.; Sugano, S.; Hirai, M.; Yamamoto, T. Molecular cloning and characterization of a novel cbl-family gene, cbl-c. Gene 1999, 239, 145–154. [Google Scholar] [CrossRef]
- Nau, M.M.; Lipkowitz, S. Comparative genomic organization of the cbl genes. Gene 2003, 308, 103–113. [Google Scholar] [CrossRef]
- Liu, Y.C. Ubiquitin ligases and the immune response. Annu. Rev. Immunol. 2004, 22, 81–127. [Google Scholar] [CrossRef]
- Thien, C.B.; Langdon, W.Y. c-Cbl and Cbl-b ubiquitin ligases: Substrate diversity and the negative regulation of signalling responses. Biochem. J. 2005, 391, 153–166. [Google Scholar] [CrossRef]
- Huang, F.; Gu, H. Negative regulation of lymphocyte development and function by the Cbl family of proteins. Immunol. Rev. 2008, 224, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Langdon, W.Y.; Hyland, C.D.; Grumont, R.J.; Morse, H.C., 3rd. The c-cbl proto-oncogene is preferentially expressed in thymus and testis tissue and encodes a nuclear protein. J. Virol. 1989, 63, 5420–5424. [Google Scholar] [PubMed]
- Wang, H.Y.; Altman, Y.; Fang, D.; Elly, C.; Dai, Y.; Shao, Y.; Liu, Y.C. Cbl promotes ubiquitination of the T cell receptor zeta through an adaptor function of Zap-70. J. Biol. Chem. 2001, 276, 26004–26011. [Google Scholar] [CrossRef] [PubMed]
- Naramura, M.; Kole, H.K.; Hu, R.J.; Gu, H. Altered thymic positive selection and intracellular signals in Cbl-deficient mice. Proc. Natl. Acad. Sci. USA 1998, 95, 15547–15552. [Google Scholar] [CrossRef] [PubMed]
- Hicke, L. A new ticket for entry into budding vesicles-ubiquitin. Cell 2001, 106, 527–530. [Google Scholar] [CrossRef]
- Liu, Y.C.; Gu, H. Cbl and Cbl-b in T-cell regulation. Trends Immunol. 2002, 23, 140–143. [Google Scholar] [CrossRef]
- Rao, N.; Miyake, S.; Reddi, A.L.; Douillard, P.; Ghosh, A.K.; Dodge, I.L.; Zhou, P.; Fernandes, N.D.; Band, H. Negative regulation of Lck by Cbl ubiquitin ligase. Proc. Natl. Acad. Sci. USA 2002, 99, 3794–3799. [Google Scholar] [CrossRef]
- Balagopalan, L.; Barr, V.A.; Sommers, C.L.; Barda-Saad, M.; Goyal, A.; Isakowitz, M.S.; Samelson, L.E. c-Cbl-mediated regulation of LAT-nucleated signaling complexes. Mol. Cell. Biol. 2007, 27, 8622–8636. [Google Scholar] [CrossRef]
- Boussiotis, V.A.; Freeman, G.J.; Berezovskaya, A.; Barber, D.L.; Nadler, L.M. Maintenance of human T cell anergy: Blocking of IL-2 gene transcription by activated Rap1. Science 1997, 278, 124–128. [Google Scholar] [CrossRef]
- Shao, Y.; Elly, C.; Liu, Y.C. Negative regulation of Rap1 activation by the Cbl E3 ubiquitin ligase. EMBO Rep. 2003, 4, 425–431. [Google Scholar] [CrossRef]
- Zhang, W.; Shao, Y.; Fang, D.; Huang, J.; Jeon, M.S.; Liu, Y.C. Negative regulation of T cell antigen receptor-mediated Crk-L-C3G signaling and cell adhesion by Cbl-b. J. Biol. Chem. 2003, 278, 23978–23983. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Zhou, H.; Langdon, W.Y.; Zhang, J. E3 ubiquitin ligase Cbl-b in innate and adaptive immunity. Cell Cycle 2014, 13, 1875–1884. [Google Scholar] [PubMed]
- Bachmaier, K.; Krawczyk, C.; Kozieradzki, I.; Kong, Y.Y.; Sasaki, T.; Oliveira-dos-Santos, A.; Mariathasan, S.; Bouchard, D.; Wakeham, A.; Itie, A.; et al. Negative regulation of lymphocyte activation and autoimmunity by the molecular adaptor Cbl-b. Nature 2000, 403, 211–216. [Google Scholar] [CrossRef] [PubMed]
- Chiang, Y.J.; Kole, H.K.; Brown, K.; Naramura, M.; Fukuhara, S.; Hu, R.J.; Jang, I.K.; Gutkind, J.S.; Shevach, E.; Gu, H. Cbl-b regulates the CD28 dependence of T-cell activation. Nature 2000, 403, 216–220. [Google Scholar] [CrossRef]
- Krawczyk, C.; Bachmaier, K.; Sasaki, T.; Jones, R.G.; Snapper, S.B.; Bouchard, D.; Kozieradzki, I.; Ohashi, P.S.; Alt, F.W.; Penninger, J.M. Cbl-b is a negative regulator of receptor clustering and raft aggregation in T cells. Immunity 2000, 13, 463–473. [Google Scholar] [CrossRef]
- Michel, F.; Mangino, G.; Attal-Bonnefoy, G.; Tuosto, L.; Alcover, A.; Roumier, A.; Olive, D.; Acuto, O. CD28 utilizes Vav-1 to enhance TCR-proximal signaling and NF-AT activation. J. Immunol. 2000, 165, 3820–3829. [Google Scholar] [CrossRef]
- Salojin, K.V.; Zhang, J.; Delovitch, T.L. TCR and CD28 are coupled via ZAP-70 to the activation of the Vav/Rac-1-/PAK-1/p38 MAPK signaling pathway. J. Immunol. 1999, 163, 844–853. [Google Scholar]
- Gruber, T.; Hermann-Kleiter, N.; Hinterleitner, R.; Fresser, F.; Schneider, R.; Gastl, G.; Penninger, J.M.; Baier, G. PKC-theta modulates the strength of T cell responses by targeting Cbl-b for ubiquitination and degradation. Sci. Signal. 2009, 2, ra30. [Google Scholar] [CrossRef]
- Zhang, J.; Bardos, T.; Li, D.; Gal, I.; Vermes, C.; Xu, J.; Mikecz, K.; Finnegan, A.; Lipkowitz, S.; Glant, T.T. Cutting edge: Regulation of T cell activation threshold by CD28 costimulation through targeting Cbl-b for ubiquitination. J. Immunol. 2002, 169, 2236–2240. [Google Scholar] [CrossRef]
- Yang, B.; Gay, D.L.; MacLeod, M.K.; Cao, X.; Hala, T.; Sweezer, E.M.; Kappler, J.; Marrack, P.; Oliver, P.M. Nedd4 augments the adaptive immune response by promoting ubiquitin-mediated degradation of Cbl-b in activated T cells. Nat. Immunol. 2008, 9, 1356–1363. [Google Scholar] [CrossRef]
- Li, D.; Gal, I.; Vermes, C.; Alegre, M.L.; Chong, A.S.; Chen, L.; Shao, Q.; Adarichev, V.; Xu, X.; Koreny, T.; et al. Cutting edge: Cbl-b: One of the key molecules tuning CD28- and CTLA-4-mediated T cell costimulation. J. Immunol. 2004, 173, 7135–7139. [Google Scholar] [CrossRef]
- Nanjappa, S.G.; Mudalagiriyappa, S.; Fites, J.S.; Suresh, M.; Klein, B.S. CBLB Constrains Inactivated Vaccine-Induced CD8(+) T Cell Responses and Immunity against Lethal Fungal Pneumonia. J. Immunol. 2018, 201, 1717–1726. [Google Scholar] [CrossRef]
- Tang, R.; Langdon, W.Y.; Zhang, J. Regulation of immune responses by E3 ubiquitin ligase Cbl-b. Cell Immunol. 2018, 340, 103878. [Google Scholar] [CrossRef] [PubMed]
- Chiang, J.Y.; Jang, I.K.; Hodes, R.; Gu, H. Ablation of Cbl-b provides protection against transplanted and spontaneous tumors. J. Clin. Investig. 2007, 117, 1029–1036. [Google Scholar] [CrossRef] [Green Version]
- Loeser, S.; Loser, K.; Bijker, M.S.; Rangachari, M.; van der Burg, S.H.; Wada, T.; Beissert, S.; Melief, C.J.; Penninger, J.M. Spontaneous tumor rejection by cbl-b-deficient CD8+ T cells. J. Exp. Med. 2007, 204, 879–891. [Google Scholar] [CrossRef] [Green Version]
- Peer, S.; Baier, G.; Gruber, T. Cblb-deficient T cells are less susceptible to PD-L1-mediated inhibition. Oncotarget 2017, 8, 41841–41853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Triozzi, P.; Kooshki, M.; Alistar, A.; Bitting, R.; Neal, A.; Lametschwandtner, G.; Loibner, H. Phase I clinical trial of adoptive cellular immunotherapy with APN401 in patients with solid tumors. J. Immunother. Cancer 2015, 3, 175. [Google Scholar] [CrossRef] [Green Version]
- Riling, C.; Sokirniy, I.; Cunnion, B.; Todd, E.; Mattern, M.; Wu, J.; Kambayashi, T.; Kumar, S. Abstract A206: Small-molecule Cbl-b inhibitors as novel intracellular checkpoint inhibitors for cancer immunotherapy. Mol. Targets Cancer Ther. 2018, 17. [Google Scholar] [CrossRef]
- Wu, Y.H.; Wu, W.S.; Lin, L.C.; Liu, C.S.; Ho, S.Y.; Wang, B.J.; Huang, B.M.; Yeh, Y.L.; Chiu, H.W.; Yang, W.L.; et al. Bortezomib enhances radiosensitivity in oral cancer through inducing autophagy-mediated TRAF6 oncoprotein degradation. J. Exp. Clin. Cancer Res. 2018, 37, 91. [Google Scholar] [CrossRef] [Green Version]
- Kararoudi, M.N.; Elmas, E.; Lamb, M.; Chakravarti, N.; Trikha, P.; Lee, D.A. Disruption of SOCS3 Promotes the Anti-Cancer Efficacy of Primary NK Cells. Blood 2018, 132. [Google Scholar] [CrossRef]
- Anandasabapathy, N.; Ford, G.S.; Bloom, D.; Holness, C.; Paragas, V.; Seroogy, C.; Skrenta, H.; Hollenhorst, M.; Fathman, C.G.; Soares, L. GRAIL: An E3 ubiquitin ligase that inhibits cytokine gene transcription is expressed in anergic CD4+ T cells. Immunity 2003, 18, 535–547. [Google Scholar] [CrossRef] [Green Version]
- Seroogy, C.M.; Soares, L.; Ranheim, E.A.; Su, L.; Holness, C.; Bloom, D.; Fathman, C.G. The gene related to anergy in lymphocytes, an E3 ubiquitin ligase, is necessary for anergy induction in CD4 T cells. J. Immunol. 2004, 173, 79–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kriegel, M.A.; Rathinam, C.; Flavell, R.A. E3 ubiquitin ligase GRAIL controls primary T cell activation and oral tolerance. Proc. Natl. Acad. Sci. USA 2009, 106, 16770–16775. [Google Scholar] [CrossRef] [Green Version]
- Nurieva, R.I.; Zheng, S.; Jin, W.; Chung, Y.; Zhang, Y.; Martinez, G.J.; Reynolds, J.M.; Wang, S.L.; Lin, X.; Sun, S.C.; et al. The E3 ubiquitin ligase GRAIL regulates T cell tolerance and regulatory T cell function by mediating T cell receptor-CD3 degradation. Immunity 2010, 32, 670–680. [Google Scholar] [CrossRef] [Green Version]
- Czyzyk, J.; Chen, H.C.; Bottomly, K.; Flavell, R.A. p21 Ras/impedes mitogenic signal propagation regulates cytokine production and migration in CD4 T cells. J. Biol. Chem. 2008, 283, 23004–23015. [Google Scholar] [CrossRef] [Green Version]
- Lineberry, N.; Su, L.; Soares, L.; Fathman, C.G. The single subunit transmembrane E3 ligase gene related to anergy in lymphocytes (GRAIL) captures and then ubiquitinates transmembrane proteins across the cell membrane. J. Biol. Chem. 2008, 283, 28497–28505. [Google Scholar] [CrossRef] [Green Version]
- Lineberry, N.B.; Su, L.L.; Lin, J.T.; Coffey, G.P.; Seroogy, C.M.; Fathman, C.G. Cutting edge: The transmembrane E3 ligase GRAIL ubiquitinates the costimulatory molecule CD40 ligand during the induction of T cell anergy. J. Immunol. 2008, 181, 1622–1626. [Google Scholar] [CrossRef] [Green Version]
- Su, L.L.; Iwai, H.; Lin, J.T.; Fathman, C.G. The transmembrane E3 ligase GRAIL ubiquitinates and degrades CD83 on CD4 T cells. J. Immunol. 2009, 183, 438–444. [Google Scholar] [CrossRef] [Green Version]
- Grewal, I.S.; Flavell, R.A. CD40 and CD154 in cell-mediated immunity. Annu. Rev. Immunol. 1998, 16, 111–135. [Google Scholar] [CrossRef]
- Reinwald, S.; Wiethe, C.; Westendorf, A.M.; Breloer, M.; Probst-Kepper, M.; Fleischer, B.; Steinkasserer, A.; Buer, J.; Hansen, W. CD83 expression in CD4+ T cells modulates inflammation and autoimmunity. J. Immunol. 2008, 180, 5890–5897. [Google Scholar] [CrossRef] [Green Version]
- Ichikawa, D.; Mizuno, M.; Yamamura, T.; Miyake, S. GRAIL (gene related to anergy in lymphocytes) regulates cytoskeletal reorganization through ubiquitination and degradation of Arp2/3 subunit 5 and coronin 1A. J. Biol. Chem. 2011, 286, 43465–43474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, L.; Lineberry, N.; Huh, Y.; Soares, L.; Fathman, C.G. A novel E3 ubiquitin ligase substrate screen identifies Rho guanine dissociation inhibitor as a substrate of gene related to anergy in lymphocytes. J. Immunol. 2006, 177, 7559–7566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haymaker, C.; Yang, Y.; Wang, J.; Zou, Q.; Sahoo, A.; Alekseev, A.; Singh, D.; Ritthipichai, K.; Hailemichael, Y.; Hoang, O.N.; et al. Absence of Grail promotes CD8(+) T cell anti-tumour activity. Nat. Commun. 2017, 8, 239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, X.; Levy-Cohen, G.; Blank, M. Molecular functions of NEDD4 E3 ubiquitin ligases in cancer. Biochim. Biophys. Acta 2015, 1856, 91–106. [Google Scholar] [CrossRef] [PubMed]
- Scharschmidt, E.; Wegener, E.; Heissmeyer, V.; Rao, A.; Krappmann, D. Degradation of Bcl10 induced by T-cell activation negatively regulates NF-kappa B signaling. Mol. Cell. Biol. 2004, 24, 3860–3873. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Matsumoto, R.; You, Y.; Che, T.; Lin, X.Y.; Gaffen, S.L.; Lin, X. CD3/CD28 costimulation-induced NF-kappaB activation is mediated by recruitment of protein kinase C-theta, Bcl10, and IkappaB kinase beta to the immunological synapse through CARMA1. Mol. Cell. Biol. 2004, 24, 164–171. [Google Scholar] [CrossRef] [Green Version]
- Hustad, C.M.; Perry, W.L.; Siracusa, L.D.; Rasberry, C.; Cobb, L.; Cattanach, B.M.; Kovatch, R.; Copeland, N.G.; Jenkins, N.A. Molecular genetic characterization of six recessive viable alleles of the mouse agouti locus. Genetics 1995, 140, 255–265. [Google Scholar]
- Perry, W.L.; Hustad, C.M.; Swing, D.A.; O’Sullivan, T.N.; Jenkins, N.A.; Copeland, N.G. The itchy locus encodes a novel ubiquitin protein ligase that is disrupted in a18H mice. Nat. Genet. 1998, 18, 143–146. [Google Scholar] [CrossRef]
- Ho, K.C.; Zhou, Z.; She, Y.M.; Chun, A.; Cyr, T.D.; Yang, X. Itch E3 ubiquitin ligase regulates large tumor suppressor 1 stability [corrected]. Proc. Natl. Acad. Sci. USA 2011, 108, 4870–4875. [Google Scholar] [CrossRef] [Green Version]
- Infante, P.; Faedda, R.; Bernardi, F.; Bufalieri, F.; Lospinoso Severini, L.; Alfonsi, R.; Mazza, D.; Siler, M.; Coni, S.; Po, A.; et al. Itch/beta-arrestin2-dependent non-proteolytic ubiquitylation of SuFu controls Hedgehog signalling and medulloblastoma tumorigenesis. Nat. Commun. 2018, 9, 976. [Google Scholar] [CrossRef]
- Salah, Z.; Melino, G.; Aqeilan, R.I. Negative regulation of the Hippo pathway by E3 ubiquitin ligase ITCH is sufficient to promote tumorigenicity. Cancer Res. 2011, 71, 2010–2020. [Google Scholar] [CrossRef] [PubMed]
- Fang, D.; Elly, C.; Gao, B.; Fang, N.; Altman, Y.; Joazeiro, C.; Hunter, T.; Copeland, N.; Jenkins, N.; Liu, Y.C. Dysregulation of T lymphocyte function in itchy mice: A role for Itch in TH2 differentiation. Nat. Immunol. 2002, 3, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Labuda, T.; Xia, Y.; Gallagher, E.; Fang, D.; Liu, Y.C.; Karin, M. Jun turnover is controlled through JNK-dependent phosphorylation of the E3 ligase Itch. Science 2004, 306, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Venuprasad, K.; Elly, C.; Gao, M.; Salek-Ardakani, S.; Harada, Y.; Luo, J.L.; Yang, C.; Croft, M.; Inoue, K.; Karin, M.; et al. Convergence of Itch-induced ubiquitination with MEKK1-JNK signaling in Th2 tolerance and airway inflammation. J. Clin. Investig. 2006, 116, 1117–1126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kathania, M.; Khare, P.; Zeng, M.; Cantarel, B.; Zhang, H.; Ueno, H.; Venuprasad, K. Itch inhibits IL-17-mediated colon inflammation and tumorigenesis by ROR-gammat ubiquitination. Nat. Immunol. 2016, 17, 997–1004. [Google Scholar] [CrossRef]
- Bai, Y.; Yang, C.; Hu, K.; Elly, C.; Liu, Y.C. Itch E3 ligase-mediated regulation of TGF-beta signaling by modulating smad2 phosphorylation. Mol. Cell 2004, 15, 825–831. [Google Scholar] [CrossRef]
- Heissmeyer, V.; Rao, A. E3 ligases in T cell anergy—Turning immune responses into tolerance. Sci. STKE 2004, 2004, pe29. [Google Scholar] [CrossRef]
- Aki, D.; Li, H.; Zhang, W.; Zheng, M.; Elly, C.; Lee, J.H.; Zou, W.; Liu, Y.C. The E3 ligases Itch and WWP2 cooperate to limit TH2 differentiation by enhancing signaling through the TCR. Nat. Immunol. 2018, 19, 766–775. [Google Scholar] [CrossRef]
- Huang, H.; Jeon, M.S.; Liao, L.; Yang, C.; Elly, C.; Yates, J.R., 3rd; Liu, Y.C. K33-linked polyubiquitination of T cell receptor-zeta regulates proteolysis-independent T cell signaling. Immunity 2010, 33, 60–70. [Google Scholar] [CrossRef] [Green Version]
- Rossi, M.; Rotblat, B.; Ansell, K.; Amelio, I.; Caraglia, M.; Misso, G.; Bernassola, F.; Cavasotto, C.N.; Knight, R.A.; Ciechanover, A.; et al. High throughput screening for inhibitors of the HECT ubiquitin E3 ligase ITCH identifies antidepressant drugs as regulators of autophagy. Cell Death Dis. 2014, 5, e1203. [Google Scholar] [CrossRef] [Green Version]
- Sampath, D.; Calin, G.A.; Puduvalli, V.K.; Gopisetty, G.; Taccioli, C.; Liu, C.G.; Ewald, B.; Liu, C.; Keating, M.J.; Plunkett, W. Specific activation of microRNA106b enables the p73 apoptotic response in chronic lymphocytic leukemia by targeting the ubiquitin ligase Itch for degradation. Blood 2009, 113, 3744–3753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleine-Eggebrecht, N.; Staufner, C.; Kathemann, S.; Elgizouli, M.; Kopajtich, R.; Prokisch, H.; Lainka, E. Mutation in ITCH Gene Can Cause Syndromic Multisystem Autoimmune Disease With Acute Liver Failure. Pediatrics 2019, 143, e20181554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deftos, M.L.; He, Y.W.; Ojala, E.W.; Bevan, M.J. Correlating notch signaling with thymocyte maturation. Immunity 1998, 9, 777–786. [Google Scholar] [CrossRef] [Green Version]
- Hsiao, H.W.; Liu, W.H.; Wang, C.J.; Lo, Y.H.; Wu, Y.H.; Jiang, S.T.; Lai, M.Z. Deltex1 is a target of the transcription factor NFAT that promotes T cell anergy. Immunity 2009, 31, 72–83. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.H.; Lai, M.Z. Deltex regulates T-cell activation by targeted degradation of active MEKK1. Mol. Cell. Biol. 2005, 25, 1367–1378. [Google Scholar] [CrossRef] [Green Version]
- Hsu, T.S.; Hsiao, H.W.; Wu, P.J.; Liu, W.H.; Lai, M.Z. Deltex1 promotes protein kinase Ctheta degradation and sustains Casitas B-lineage lymphoma expression. J. Immunol. 2014, 193, 1672–1680. [Google Scholar] [CrossRef] [Green Version]
- King, C.G.; Kobayashi, T.; Cejas, P.J.; Kim, T.; Yoon, K.; Kim, G.K.; Chiffoleau, E.; Hickman, S.P.; Walsh, P.T.; Turka, L.A.; et al. TRAF6 is a T cell-intrinsic negative regulator required for the maintenance of immune homeostasis. Nat. Med. 2006, 12, 1088–1092. [Google Scholar] [CrossRef]
- Wang, C.; Deng, L.; Hong, M.; Akkaraju, G.R.; Inoue, J.; Chen, Z.J. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature 2001, 412, 346–351. [Google Scholar] [CrossRef]
- Brenke, J.K.; Popowicz, G.M.; Schorpp, K.; Rothenaigner, I.; Roesner, M.; Meininger, I.; Kalinski, C.; Ringelstetter, L.; R’Kyek, O.; Jurjens, G.; et al. Targeting TRAF6 E3 ligase activity with a small-molecule inhibitor combats autoimmunity. J. Biol. Chem. 2018, 293, 13191–13203. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Lozano, G. Molecular pathways: Targeting Mdm2 and Mdm4 in cancer therapy. Clin. Cancer Res. 2013, 19, 34–41. [Google Scholar] [CrossRef] [Green Version]
- Zou, Q.; Jin, J.; Hu, H.; Li, H.S.; Romano, S.; Xiao, Y.; Nakaya, M.; Zhou, X.; Cheng, X.; Yang, P.; et al. USP15 stabilizes MDM2 to mediate cancer-cell survival and inhibit antitumor T cell responses. Nat. Immunol. 2014, 15, 562–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tisato, V.; Voltan, R.; Gonelli, A.; Secchiero, P.; Zauli, G. MDM2/X inhibitors under clinical evaluation: Perspectives for the management of hematological malignancies and pediatric cancer. J. Hematol. Oncol. 2017, 10, 133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, M.; Jin, W.; Chang, J.H.; Xiao, Y.; Brittain, G.C.; Yu, J.; Zhou, X.; Wang, Y.H.; Cheng, X.; Li, P.; et al. The ubiquitin ligase Peli1 negatively regulates T cell activation and prevents autoimmunity. Nat. Immunol. 2011, 12, 1002–1009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Huang, X.; Hao, S.; Wang, Y.; Liu, M.; Xu, J.; Zhang, X.; Yu, T.; Gan, S.; Dai, D.; et al. Peli1 negatively regulates noncanonical NF-kappaB signaling to restrain systemic lupus erythematosus. Nat. Commun. 2018, 9, 1136. [Google Scholar] [CrossRef] [Green Version]
- Bonnevier, J.L.; Zhang, R.; Mueller, D.L. E3 ubiquitin ligases and their control of T cell autoreactivity. Arthritis Res. Ther. 2005, 7, 233–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnston, J.A. Are SOCS suppressors, regulators, and degraders? J. Leukoc. Biol. 2004, 75, 743–748. [Google Scholar] [CrossRef]
- Petroski, M.D.; Deshaies, R.J. Function and regulation of cullin-RING ubiquitin ligases. Nat. Rev. Mol. Cell. Biol. 2005, 6, 9–20. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.R.; Mahdi, R.M.; Ebong, S.; Vistica, B.P.; Gery, I.; Egwuagu, C.E. Suppressor of cytokine signaling 3 regulates proliferation and activation of T-helper cells. J. Biol. Chem. 2003, 278, 29752–29759. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.B.; Son, M.; Park, M.; Shin, J.; Yun, Y. SOCS-6 negatively regulates T cell activation through targeting p56lck to proteasomal degradation. J. Biol. Chem. 2010, 285, 7271–7280. [Google Scholar] [CrossRef] [Green Version]
- Guittard, G.; Dios-Esponera, A.; Palmer, D.C.; Akpan, I.; Barr, V.A.; Manna, A.; Restifo, N.P.; Samelson, L.E. The Cish SH2 domain is essential for PLC-gamma1 regulation in TCR stimulated CD8(+) T cells. Sci. Rep. 2018, 8, 5336. [Google Scholar] [CrossRef] [Green Version]
- Lorenz, U. SHP-1 and SHP-2 in T cells: Two phosphatases functioning at many levels. Immunol. Rev. 2009, 228, 342–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pei, D.; Lorenz, U.; Klingmuller, U.; Neel, B.G.; Walsh, C.T. Intramolecular regulation of protein tyrosine phosphatase SH-PTP1: A new function for Src homology 2 domains. Biochemistry 1994, 33, 15483–15493. [Google Scholar] [CrossRef] [PubMed]
- Townley, R.; Shen, S.H.; Banville, D.; Ramachandran, C. Inhibition of the activity of protein tyrosine phosphate 1C by its SH2 domains. Biochemistry 1993, 32, 13414–13418. [Google Scholar] [CrossRef] [PubMed]
- Binstadt, B.A.; Billadeau, D.D.; Jevremovic, D.; Williams, B.L.; Fang, N.; Yi, T.; Koretzky, G.A.; Abraham, R.T.; Leibson, P.J. SLP-76 is a direct substrate of SHP-1 recruited to killer cell inhibitory receptors. J. Biol. Chem. 1998, 273, 27518–27523. [Google Scholar] [CrossRef] [Green Version]
- Brockdorff, J.; Williams, S.; Couture, C.; Mustelin, T. Dephosphorylation of ZAP-70 and inhibition of T cell activation by activated SHP1. Eur. J. Immunol. 1999, 29, 2539–2550. [Google Scholar] [CrossRef]
- Cuevas, B.; Lu, Y.; Watt, S.; Kumar, R.; Zhang, J.; Siminovitch, K.A.; Mills, G.B. SHP-1 regulates Lck-induced phosphatidylinositol 3-kinase phosphorylation and activity. J. Biol. Chem. 1999, 274, 27583–27589. [Google Scholar] [CrossRef] [Green Version]
- Kon-Kozlowski, M.; Pani, G.; Pawson, T.; Siminovitch, K.A. The tyrosine phosphatase PTP1C associates with Vav, Grb2, and mSos1 in hematopoietic cells. J. Biol. Chem. 1996, 271, 3856–3862. [Google Scholar] [CrossRef] [Green Version]
- Plas, D.R.; Johnson, R.; Pingel, J.T.; Matthews, R.J.; Dalton, M.; Roy, G.; Chan, A.C.; Thomas, M.L. Direct regulation of ZAP-70 by SHP-1 in T cell antigen receptor signaling. Science 1996, 272, 1173–1176. [Google Scholar] [CrossRef]
- Sozio, M.S.; Mathis, M.A.; Young, J.A.; Walchli, S.; Pitcher, L.A.; Wrage, P.C.; Bartok, B.; Campbell, A.; Watts, J.D.; Aebersold, R.; et al. PTPH1 is a predominant protein-tyrosine phosphatase capable of interacting with and dephosphorylating the T cell receptor zeta subunit. J. Biol. Chem. 2004, 279, 7760–7769. [Google Scholar] [CrossRef] [Green Version]
- Stefanova, I.; Hemmer, B.; Vergelli, M.; Martin, R.; Biddison, W.E.; Germain, R.N. TCR ligand discrimination is enforced by competing ERK positive and SHP-1 negative feedback pathways. Nat. Immunol. 2003, 4, 248–254. [Google Scholar] [CrossRef]
- Zhao, Q.; Weiss, A. Enhancement of lymphocyte responsiveness by a gain-of-function mutation of ZAP-70. Mol. Cell. Biol. 1996, 16, 6765–6774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, Y.; Qiao, G.; Tang, J.; Tang, R.; Guo, H.; Warwar, S.; Langdon, W.Y.; Tao, L.; Zhang, J. Protein Tyrosine Phosphatase SHP-1 Modulates T Cell Responses by Controlling Cbl-b Degradation. J. Immunol. 2015, 195, 4218–4227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dempke, W.C.M.; Uciechowski, P.; Fenchel, K.; Chevassut, T. Targeting SHP-1, 2 and SHIP Pathways: A Novel Strategy for Cancer Treatment? Oncology 2018, 95, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Watson, H.A.; Wehenkel, S.; Matthews, J.; Ager, A. SHP-1: The next checkpoint target for cancer immunotherapy? Biochem. Soc. Trans. 2016, 44, 356–362. [Google Scholar] [CrossRef] [Green Version]
- Sathish, J.G.; Dolton, G.; Leroy, F.G.; Matthews, R.J. Loss of Src homology region 2 domain-containing protein tyrosine phosphatase-1 increases CD8+ T cell-APC conjugate formation and is associated with enhanced in vivo CTL function. J. Immunol. 2007, 178, 330–337. [Google Scholar] [CrossRef] [Green Version]
- Sathish, J.G.; Johnson, K.G.; LeRoy, F.G.; Fuller, K.J.; Hallett, M.B.; Brennan, P.; Borysiewicz, L.K.; Sims, M.J.; Matthews, R.J. Requirement for CD28 co-stimulation is lower in SHP-1-deficient T cells. Eur. J. Immunol. 2001, 31, 3649–3658. [Google Scholar] [CrossRef]
- Carter, J.D.; Neel, B.G.; Lorenz, U. The tyrosine phosphatase SHP-1 influences thymocyte selection by setting TCR signaling thresholds. Int. Immunol. 1999, 11, 1999–2014. [Google Scholar] [CrossRef] [Green Version]
- Lorenz, U.; Bergemann, A.D.; Steinberg, H.N.; Flanagan, J.G.; Li, X.; Galli, S.J.; Neel, B.G. Genetic analysis reveals cell type-specific regulation of receptor tyrosine kinase c-Kit by the protein tyrosine phosphatase SHP1. J. Exp. Med. 1996, 184, 1111–1126. [Google Scholar] [CrossRef] [Green Version]
- Shultz, L.D.; Schweitzer, P.A.; Rajan, T.V.; Yi, T.; Ihle, J.N.; Matthews, R.J.; Thomas, M.L.; Beier, D.R. Mutations at the murine motheaten locus are within the hematopoietic cell protein-tyrosine phosphatase (Hcph) gene. Cell 1993, 73, 1445–1454. [Google Scholar] [CrossRef]
- Tsui, H.W.; Siminovitch, K.A.; de Souza, L.; Tsui, F.W. Motheaten and viable motheaten mice have mutations in the haematopoietic cell phosphatase gene. Nat. Genet. 1993, 4, 124–129. [Google Scholar] [CrossRef]
- Fawcett, V.C.; Lorenz, U. Localization of Src homology 2 domain-containing phosphatase 1 (SHP-1) to lipid rafts in T lymphocytes: Functional implications and a role for the SHP-1 carboxyl terminus. J. Immunol. 2005, 174, 2849–2859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sankarshanan, M.; Ma, Z.; Iype, T.; Lorenz, U. Identification of a novel lipid raft-targeting motif in Src homology 2-containing phosphatase 1. J. Immunol. 2007, 179, 483–490. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.V.; Ke, Y.; Zhang, E.E.; Feng, G.S. Conditional deletion of Shp2 tyrosine phosphatase in thymocytes suppresses both pre-TCR and TCR signals. J. Immunol. 2006, 177, 5990–5996. [Google Scholar] [CrossRef]
- Huang, R.Y.; Eppolito, C.; Lele, S.; Shrikant, P.; Matsuzaki, J.; Odunsi, K. LAG3 and PD1 co-inhibitory molecules collaborate to limit CD8+ T cell signaling and dampen antitumor immunity in a murine ovarian cancer model. Oncotarget 2015, 6, 27359–27377. [Google Scholar] [CrossRef]
- Hui, E.; Cheung, J.; Zhu, J.; Su, X.; Taylor, M.J.; Wallweber, H.A.; Sasmal, D.K.; Huang, J.; Kim, J.M.; Mellman, I.; et al. T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science 2017, 355, 1428–1433. [Google Scholar] [CrossRef]
- Latchman, Y.; Wood, C.R.; Chernova, T.; Chaudhary, D.; Borde, M.; Chernova, I.; Iwai, Y.; Long, A.J.; Brown, J.A.; Nunes, R.; et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat. Immunol. 2001, 2, 261–268. [Google Scholar] [CrossRef]
- Sheppard, K.A.; Fitz, L.J.; Lee, J.M.; Benander, C.; George, J.A.; Wooters, J.; Qiu, Y.; Jussif, J.M.; Carter, L.L.; Wood, C.R.; et al. PD-1 inhibits T-cell receptor induced phosphorylation of the ZAP70/CD3zeta signalosome and downstream signaling to PKCtheta. FEBS Lett. 2004, 574, 37–41. [Google Scholar] [CrossRef] [Green Version]
- Chemnitz, J.M.; Parry, R.V.; Nichols, K.E.; June, C.H.; Riley, J.L. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J. Immunol. 2004, 173, 945–954. [Google Scholar] [CrossRef]
- Hebeisen, M.; Baitsch, L.; Presotto, D.; Baumgaertner, P.; Romero, P.; Michielin, O.; Speiser, D.E.; Rufer, N. SHP-1 phosphatase activity counteracts increased T cell receptor affinity. J. Clin. Investig. 2013, 123, 1044–1056. [Google Scholar] [CrossRef]
- Yokosuka, T.; Takamatsu, M.; Kobayashi-Imanishi, W.; Hashimoto-Tane, A.; Azuma, M.; Saito, T. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J. Exp. Med. 2012, 209, 1201–1217. [Google Scholar] [CrossRef] [Green Version]
- Bardhan, K.; Patsoukis, N.; Sari, D.; Anagnostou, T.; Chatterjee, P.; Freeman, G.J.; Li, L.; Boussiotis, V.A. PD-1 Inhibits TCR Proximal Signaling By Sequestering SHP-2 Phosphatase and Facilitating Csk-Mediated Inhibitory Phosphorylation of Lck. Blood 2015, 126, 283. [Google Scholar] [CrossRef]
- Chen, W.; Wang, J.; Jia, L.; Liu, J.; Tian, Y. Attenuation of the programmed cell death-1 pathway increases the M1 polarization of macrophages induced by zymosan. Cell Death Dis. 2016, 7, e2115. [Google Scholar] [CrossRef]
- Kundu, S.; Fan, K.; Cao, M.; Lindner, D.J.; Zhao, Z.J.; Borden, E.; Yi, T. Novel SHP-1 inhibitors tyrosine phosphatase inhibitor-1 and analogs with preclinical anti-tumor activities as tolerated oral agents. J. Immunol. 2010, 184, 6529–6536. [Google Scholar] [CrossRef] [Green Version]
- Russo, R.; Argentino, G.; Maresca, I.D.; Sannino, G.; Memoli, A.; Memoli, B. The use of Cinacalcet in chronic kidney disease: A case report. G. Ital. Nefrol. 2015, 32, gin/32.4.9. [Google Scholar]
- Xie, J.; Si, X.; Gu, S.; Wang, M.; Shen, J.; Li, H.; Shen, J.; Li, D.; Fang, Y.; Liu, C.; et al. Allosteric Inhibitors of SHP2 with Therapeutic Potential for Cancer Treatment. J. Med. Chem. 2017, 60, 10205–10219. [Google Scholar] [CrossRef]
- Zeng, L.F.; Zhang, R.Y.; Yu, Z.H.; Li, S.; Wu, L.; Gunawan, A.M.; Lane, B.S.; Mali, R.S.; Li, X.; Chan, R.J.; et al. Therapeutic potential of targeting the oncogenic SHP2 phosphatase. J. Med. Chem. 2014, 57, 6594–6609. [Google Scholar] [CrossRef] [Green Version]
- Chio, C.M.; Lim, C.S.; Bishop, A.C. Targeting a cryptic allosteric site for selective inhibition of the oncogenic protein tyrosine phosphatase Shp2. Biochemistry 2015, 54, 497–504. [Google Scholar] [CrossRef] [Green Version]
- Fodor, M.; Price, E.; Wang, P.; Lu, H.; Argintaru, A.; Chen, Z.; Glick, M.; Hao, H.X.; Kato, M.; Koenig, R.; et al. Dual Allosteric Inhibition of SHP2 Phosphatase. ACS Chem. Biol. 2018, 13, 647–656. [Google Scholar] [CrossRef]
- Zhao, M.; Guo, W.; Wu, Y.; Yang, C.; Zhong, L.; Deng, G.; Zhu, Y.; Liu, W.; Gu, Y.; Lu, Y.; et al. SHP2 inhibition triggers anti-tumor immunity and synergizes with PD-1 blockade. Acta Pharm. Sin. B 2019, 9, 304–315. [Google Scholar] [CrossRef]
- Rota, G.; Niogret, C.; Dang, A.T.; Barros, C.R.; Fonta, N.P.; Alfei, F.; Morgado, L.; Zehn, D.; Birchmeier, W.; Vivier, E.; et al. Shp-2 is dispensable for establishing T cell exhaustion and for PD-1 signaling in vivo. Cell Rep. 2018, 23, 39–49. [Google Scholar] [CrossRef] [Green Version]
- Niogret, C.; Miah, S.M.S.; Rota, G.; Fonta, N.P.; Wang, H.; Held, W.; Birchmeier, W.; Sexl, V.; Yang, W.; Vivier, E.; et al. Shp-2 is critical for ERK and metabolic engagement downstream of IL-15 receptor in NK cells. Nat. Commun. 2019, 10, 1444. [Google Scholar] [CrossRef] [PubMed]
- Stromnes, I.M.; Fowler, C.; Casamina, C.C.; Georgopolos, C.M.; McAfee, M.S.; Schmitt, T.M.; Tan, X.; Kim, T.D.; Choi, I.; Blattman, J.N.; et al. Abrogation of SRC homology region 2 domain-containing phosphatase 1 in tumor-specific T cells improves efficacy of adoptive immunotherapy by enhancing the effector function and accumulation of short-lived effector T cells in vivo. J. Immunol. 2012, 189, 1812–1825. [Google Scholar] [CrossRef] [PubMed]
- Baldan, V.; Ghongane, P.; Kokalaki, E.; Lim, W.C.; Onuoha, S.; Cordoba, S.P.; Thomas, S.; Pule, M. A Dominant Negative SHP-2 Which Abrogates PD-1 Signalling Pathways and Restores Function of Cytotoxic CAR T Cells. Blood 2017, 130, 3190. [Google Scholar]
- Chen, C.Y.; Chen, J.; He, L.; Stiles, B.L. PTEN: Tumor Suppressor and Metabolic Regulator. Front. Endocrinol. (Lausanne) 2018, 9, 338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.R.; Chen, M.; Pandolfi, P.P. The functions and regulation of the PTEN tumour suppressor: New modes and prospects. Nat. Rev. Mol. Cell. Biol. 2018, 19, 547–562. [Google Scholar] [CrossRef] [PubMed]
- Buckler, J.L.; Walsh, P.T.; Porrett, P.M.; Choi, Y.; Turka, L.A. Cutting edge: T cell requirement for CD28 costimulation is due to negative regulation of TCR signals by PTEN. J. Immunol. 2006, 177, 4262–4266. [Google Scholar] [CrossRef] [Green Version]
- Di Cristofano, A.; Kotsi, P.; Peng, Y.F.; Cordon-Cardo, C.; Elkon, K.B.; Pandolfi, P.P. Impaired Fas response and autoimmunity in Pten+/− mice. Science 1999, 285, 2122–2125. [Google Scholar] [CrossRef]
- Hagenbeek, T.J.; Spits, H. T-cell lymphomas in T-cell-specific Pten-deficient mice originate in the thymus. Leukemia 2008, 22, 608–619. [Google Scholar] [CrossRef]
- Xue, L.; Nolla, H.; Suzuki, A.; Mak, T.W.; Winoto, A. Normal development is an integral part of tumorigenesis in T cell-specific PTEN-deficient mice. Proc. Natl. Acad. Sci. USA 2008, 105, 2022–2027. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Karnell, J.L.; Yin, B.; Zhang, R.; Zhang, J.; Li, P.; Choi, Y.; Maltzman, J.S.; Pear, W.S.; Bassing, C.H.; et al. Distinct roles for PTEN in prevention of T cell lymphoma and autoimmunity in mice. J. Clin. Investig. 2010, 120, 2497–2507. [Google Scholar] [CrossRef]
- Harris, S.J.; Parry, R.V.; Westwick, J.; Ward, S.G. Phosphoinositide lipid phosphatases: Natural regulators of phosphoinositide 3-kinase signaling in T lymphocytes. J. Biol. Chem. 2008, 283, 2465–2469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parry, R.V.; Harris, S.J.; Ward, S.G. Fine tuning T lymphocytes: A role for the lipid phosphatase SHIP-1. Biochim. Biophys. Acta 2010, 1804, 592–597. [Google Scholar] [CrossRef]
- Kerr, W.G. Inhibitor and activator: Dual functions for SHIP in immunity and cancer. Ann. N Y Acad. Sci. 2011, 1217, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamilton, M.J.; Ho, V.W.; Kuroda, E.; Ruschmann, J.; Antignano, F.; Lam, V.; Krystal, G. Role of SHIP in cancer. Exp. Hematol. 2011, 39, 2–13. [Google Scholar] [CrossRef] [PubMed]
- Pauls, S.D.; Marshall, A.J. Regulation of immune cell signaling by SHIP1: A phosphatase, scaffold protein, and potential therapeutic target. Eur. J. Immunol. 2017, 47, 932–945. [Google Scholar] [CrossRef] [PubMed]
- Aman, M.J.; Lamkin, T.D.; Okada, H.; Kurosaki, T.; Ravichandran, K.S. The inositol phosphatase SHIP inhibits Akt/PKB activation in B cells. J. Biol. Chem. 1998, 273, 33922–33928. [Google Scholar] [CrossRef] [Green Version]
- Helou, Y.A.; Petrashen, A.P.; Salomon, A.R. Vav1 Regulates T-Cell Activation through a Feedback Mechanism and Crosstalk between the T-Cell Receptor and CD28. J. Proteome Res. 2015, 14, 2963–2975. [Google Scholar] [CrossRef] [Green Version]
- Tamir, I.; Stolpa, J.C.; Helgason, C.D.; Nakamura, K.; Bruhns, P.; Daeron, M.; Cambier, J.C. The RasGAP-binding protein p62dok is a mediator of inhibitory FcgammaRIIB signals in B cells. Immunity 2000, 12, 347–358. [Google Scholar] [CrossRef] [Green Version]
- Tridandapani, S.; Kelley, T.; Cooney, D.; Pradhan, M.; Coggeshall, K.M. Negative signaling in B cells: SHIP Grbs Shc. Immunol. Today 1997, 18, 424–427. [Google Scholar] [CrossRef]
- Tridandapani, S.; Kelley, T.; Pradhan, M.; Cooney, D.; Justement, L.B.; Coggeshall, K.M. Recruitment and phosphorylation of SH2-containing inositol phosphatase and Shc to the B-cell Fc gamma immunoreceptor tyrosine-based inhibition motif peptide motif. Mol. Cell. Biol. 1997, 17, 4305–4311. [Google Scholar] [CrossRef] [Green Version]
- Conde, C.; Rambout, X.; Lebrun, M.; Lecat, A.; Di Valentin, E.; Dequiedt, F.; Piette, J.; Gloire, G.; Legrand, S. The inositol phosphatase SHIP-1 inhibits NOD2-induced NF-kappaB activation by disturbing the interaction of XIAP with RIP2. PLoS ONE 2012, 7, e41005. [Google Scholar] [CrossRef] [PubMed]
- Tarasenko, T.; Kole, H.K.; Chi, A.W.; Mentink-Kane, M.M.; Wynn, T.A.; Bolland, S. T cell-specific deletion of the inositol phosphatase SHIP reveals its role in regulating Th1/Th2 and cytotoxic responses. Proc. Natl. Acad. Sci. USA 2007, 104, 11382–11387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobranowski, P.; Sly, L.M. SHIP negatively regulates type II immune responses in mast cells and macrophages. J. Leukoc. Biol. 2018. [Google Scholar] [CrossRef]
- Saz-Leal, P.; Del Fresno, C.; Brandi, P.; Martinez-Cano, S.; Dungan, O.M.; Chisholm, J.D.; Kerr, W.G.; Sancho, D. Targeting SHIP-1 in Myeloid Cells Enhances Trained Immunity and Boosts Response to Infection. Cell Rep. 2018, 25, 1118–1126. [Google Scholar] [CrossRef] [Green Version]
- Brooks, R.; Fuhler, G.M.; Iyer, S.; Smith, M.J.; Park, M.Y.; Paraiso, K.H.; Engelman, R.W.; Kerr, W.G. SHIP1 inhibition increases immunoregulatory capacity and triggers apoptosis of hematopoietic cancer cells. J. Immunol. 2010, 184, 3582–3589. [Google Scholar] [CrossRef] [Green Version]
- Fuhler, G.M.; Brooks, R.; Toms, B.; Iyer, S.; Gengo, E.A.; Park, M.Y.; Gumbleton, M.; Viernes, D.R.; Chisholm, J.D.; Kerr, W.G. Therapeutic potential of SH2 domain-containing inositol-5′-phosphatase 1 (SHIP1) and SHIP2 inhibition in cancer. Mol. Med. 2012, 18, 65–75. [Google Scholar] [CrossRef]
- Kennah, M.; Yau, T.Y.; Nodwell, M.; Krystal, G.; Andersen, R.J.; Ong, C.J.; Mui, A.L. Activation of SHIP via a small molecule agonist kills multiple myeloma cells. Exp. Hematol. 2009, 37, 1274–1283. [Google Scholar] [CrossRef]
- Mustelin, T.; Tasken, K. Positive and negative regulation of T-cell activation through kinases and phosphatases. Biochem. J. 2003, 371, 15–27. [Google Scholar] [CrossRef] [Green Version]
- Cloutier, J.F.; Veillette, A. Association of inhibitory tyrosine protein kinase p50csk with protein tyrosine phosphatase PEP in T cells and other hemopoietic cells. EMBO J. 1996, 15, 4909–4918. [Google Scholar] [CrossRef]
- Cloutier, J.F.; Veillette, A. Cooperative inhibition of T-cell antigen receptor signaling by a complex between a kinase and a phosphatase. J. Exp. Med. 1999, 189, 111–121. [Google Scholar] [CrossRef] [Green Version]
- Gjorloff-Wingren, A.; Saxena, M.; Williams, S.; Hammi, D.; Mustelin, T. Characterization of TCR-induced receptor-proximal signaling events negatively regulated by the protein tyrosine phosphatase PEP. Eur. J. Immunol. 1999, 29, 3845–3854. [Google Scholar] [CrossRef]
- Bray, C.; Wright, D.; Haupt, S.; Thomas, S.; Stauss, H.; Zamoyska, R. Crispr/Cas Mediated Deletion of PTPN22 in Jurkat T Cells Enhances TCR Signaling and Production of IL-2. Front. Immunol. 2018, 9, 2595. [Google Scholar] [CrossRef] [PubMed]
- Brownlie, R.J.; Zamoyska, R.; Salmond, R.J. Regulation of autoimmune and anti-tumour T-cell responses by PTPN22. Immunology 2018, 154, 377–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Katrekar, A.; Honigberg, L.A.; Smith, A.M.; Conn, M.T.; Tang, J.; Jeffery, D.; Mortara, K.; Sampang, J.; Williams, S.R.; et al. Identification of substrates of human protein-tyrosine phosphatase PTPN22. J. Biol. Chem. 2006, 281, 11002–11010. [Google Scholar] [CrossRef] [Green Version]
- Bottini, N.; Musumeci, L.; Alonso, A.; Rahmouni, S.; Nika, K.; Rostamkhani, M.; MacMurray, J.; Meloni, G.F.; Lucarelli, P.; Pellecchia, M.; et al. A functional variant of lymphoid tyrosine phosphatase is associated with type I diabetes. Nat. Genet. 2004, 36, 337–338. [Google Scholar] [CrossRef]
- Gregersen, P.K.; Lee, H.S.; Batliwalla, F.; Begovich, A.B. PTPN22: Setting thresholds for autoimmunity. Semin. Immunol. 2006, 18, 214–223. [Google Scholar] [CrossRef]
- Zheng, J.; Ibrahim, S.; Petersen, F.; Yu, X. Meta-analysis reveals an association of PTPN22 C1858T with autoimmune diseases, which depends on the localization of the affected tissue. Genes Immun. 2012, 13, 641–652. [Google Scholar] [CrossRef]
- Zheng, J.; Petersen, F.; Yu, X. The role of PTPN22 in autoimmunity: Learning from mice. Autoimmun. Rev. 2014, 13, 266–271. [Google Scholar] [CrossRef]
- Zhang, J.; Zahir, N.; Jiang, Q.; Miliotis, H.; Heyraud, S.; Meng, X.; Dong, B.; Xie, G.; Qiu, F.; Hao, Z.; et al. The autoimmune disease-associated PTPN22 variant promotes calpain-mediated Lyp/Pep degradation associated with lymphocyte and dendritic cell hyperresponsiveness. Nat. Genet. 2011, 43, 902–907. [Google Scholar] [CrossRef]
- Brownlie, R.J.; Garcia, C.; Ravasz, M.; Zehn, D.; Salmond, R.J.; Zamoyska, R. Resistance to TGFbeta suppression and improved anti-tumor responses in CD8(+) T cells lacking PTPN22. Nat. Commun. 2017, 8, 1343. [Google Scholar] [CrossRef]
- Li, K.; Hou, X.; Li, R.; Bi, W.; Yang, F.; Chen, X.; Xiao, P.; Liu, T.; Lu, T.; Zhou, Y.; et al. Identification and structure-function analyses of an allosteric inhibitor of the tyrosine phosphatase PTPN22. J. Biol. Chem. 2019, 294, 8653–8663. [Google Scholar] [CrossRef] [PubMed]
- Vang, T.; Liu, W.H.; Delacroix, L.; Wu, S.; Vasile, S.; Dahl, R.; Yang, L.; Musumeci, L.; Francis, D.; Landskron, J.; et al. LYP inhibits T-cell activation when dissociated from CSK. Nat. Chem. Biol. 2012, 8, 437–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, S.; Bottini, M.; Rickert, R.C.; Mustelin, T.; Tautz, L. In silico screening for PTPN22 inhibitors: Active hits from an inactive phosphatase conformation. ChemMedChem 2009, 4, 440–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.; Sun, J.P.; He, Y.; Guo, X.; Liu, S.; Zhou, B.; Hudmon, A.; Zhang, Z.Y. Structure, inhibitor, and regulatory mechanism of Lyp, a lymphoid-specific tyrosine phosphatase implicated in autoimmune diseases. Proc. Natl. Acad. Sci. USA 2007, 104, 19767–19772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Q.; Co, D.; Sommercorn, J.; Tonks, N.K. Cloning and expression of PTP-PEST. A novel, human, nontransmembrane protein tyrosine phosphatase. J. Biol. Chem. 1993, 268, 17650. [Google Scholar] [PubMed]
- Davidson, D.; Cloutier, J.F.; Gregorieff, A.; Veillette, A. Inhibitory tyrosine protein kinase p50csk is associated with protein-tyrosine phosphatase PTP-PEST in hemopoietic and non-hemopoietic cells. J. Biol. Chem. 1997, 272, 23455–23462. [Google Scholar] [CrossRef] [Green Version]
- Charest, A.; Wagner, J.; Kwan, M.; Tremblay, M.L. Coupling of the murine protein tyrosine phosphatase PEST to the epidermal growth factor (EGF) receptor through a Src homology 3 (SH3) domain-mediated association with Grb2. Oncogene 1997, 14, 1643–1651. [Google Scholar] [CrossRef] [Green Version]
- Cote, J.F.; Charest, A.; Wagner, J.; Tremblay, M.L. Combination of gene targeting and substrate trapping to identify substrates of protein tyrosine phosphatases using PTP-PEST as a model. Biochemistry 1998, 37, 13128–13137. [Google Scholar] [CrossRef]
- Davidson, D.; Shi, X.; Zhong, M.C.; Rhee, I.; Veillette, A. The phosphatase PTP-PEST promotes secondary T cell responses by dephosphorylating the protein tyrosine kinase Pyk2. Immunity 2010, 33, 167–180. [Google Scholar] [CrossRef] [Green Version]
- Davidson, D.; Veillette, A. PTP-PEST, a scaffold protein tyrosine phosphatase, negatively regulates lymphocyte activation by targeting a unique set of substrates. EMBO J. 2001, 20, 3414–3426. [Google Scholar] [CrossRef]
- Garton, A.J.; Flint, A.J.; Tonks, N.K. Identification of p130(cas) as a substrate for the cytosolic protein tyrosine phosphatase PTP-PEST. Mol. Cell. Biol. 1996, 16, 6408–6418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cote, J.F.; Chung, P.L.; Theberge, J.F.; Halle, M.; Spencer, S.; Lasky, L.A.; Tremblay, M.L. PSTPIP is a substrate of PTP-PEST and serves as a scaffold guiding PTP-PEST toward a specific dephosphorylation of WASP. J. Biol. Chem. 2002, 277, 2973–2986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.; Rhee, I. Important roles of protein tyrosine phosphatase PTPN12 in tumor progression. Pharmacol. Res. 2019, 144, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Nair, A.; Chung, H.C.; Sun, T.; Tyagi, S.; Dobrolecki, L.E.; Dominguez-Vidana, R.; Kurley, S.J.; Orellana, M.; Renwick, A.; Henke, D.M.; et al. Combinatorial inhibition of PTPN12-regulated receptors leads to a broadly effective therapeutic strategy in triple-negative breast cancer. Nat. Med. 2018, 24, 505–511. [Google Scholar] [CrossRef]
- Shen, N.; Li, L.; Xu, W.; Tian, J.; Yang, Y.; Zhu, Y.; Gong, Y.; Ke, J.; Gong, J.; Chang, J.; et al. A missense variant in PTPN12 associated with the risk of colorectal cancer by modifying Ras/MEK/ERK signaling. Cancer Epidemiol. 2019, 59, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Aceto, N.; Meerbrey, K.L.; Kessler, J.D.; Zhou, C.; Migliaccio, I.; Nguyen, D.X.; Pavlova, N.N.; Botero, M.; Huang, J.; et al. Activation of multiple proto-oncogenic tyrosine kinases in breast cancer via loss of the PTPN12 phosphatase. Cell 2011, 144, 703–718. [Google Scholar] [CrossRef] [Green Version]
- Han, S.; Williams, S.; Mustelin, T. Cytoskeletal protein tyrosine phosphatase PTPH1 reduces T cell antigen receptor signaling. Eur. J. Immunol. 2000, 30, 1318–1325. [Google Scholar] [CrossRef]
- Bauler, T.J.; Hughes, E.D.; Arimura, Y.; Mustelin, T.; Saunders, T.L.; King, P.D. Normal TCR signal transduction in mice that lack catalytically active PTPN3 protein tyrosine phosphatase. J. Immunol. 2007, 178, 3680–3687. [Google Scholar] [CrossRef] [Green Version]
- Patrignani, C.; Lafont, D.T.; Muzio, V.; Greco, B.; Hooft van Huijsduijnen, R.; Zaratin, P.F. Characterization of protein tyrosine phosphatase H1 knockout mice in animal models of local and systemic inflammation. J. Inflamm. 2010, 7, 16. [Google Scholar] [CrossRef] [Green Version]
- Janssens, V.; Goris, J. Protein phosphatase 2A: A highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem. J. 2001, 353, 417–439. [Google Scholar] [CrossRef]
- Breuer, R.; Becker, M.S.; Brechmann, M.; Mock, T.; Arnold, R.; Krammer, P.H. The protein phosphatase 2A regulatory subunit B56gamma mediates suppression of T cell receptor (TCR)-induced nuclear factor-kappaB (NF-kappaB) activity. J. Biol. Chem. 2014, 289, 14996–15004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eitelhuber, A.C.; Warth, S.; Schimmack, G.; Duwel, M.; Hadian, K.; Demski, K.; Beisker, W.; Shinohara, H.; Kurosaki, T.; Heissmeyer, V.; et al. Dephosphorylation of Carma1 by PP2A negatively regulates T-cell activation. EMBO J. 2011, 30, 594–605. [Google Scholar] [CrossRef] [PubMed]
- Baroja, M.L.; Vijayakrishnan, L.; Bettelli, E.; Darlington, P.J.; Chau, T.A.; Ling, V.; Collins, M.; Carreno, B.M.; Madrenas, J.; Kuchroo, V.K. Inhibition of CTLA-4 function by the regulatory subunit of serine/threonine phosphatase 2A. J. Immunol. 2002, 168, 5070–5078. [Google Scholar] [CrossRef] [PubMed]
- Lauritsen, J.P.; Menne, C.; Kastrup, J.; Dietrich, J.; Geisler, C. Protein phosphatase 2A isotypes regulate cell surface expression of the T cell receptor. Exp. Clin. Immunogenet. 2001, 18, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Bergman, M.; Mustelin, T.; Oetken, C.; Partanen, J.; Flint, N.A.; Amrein, K.E.; Autero, M.; Burn, P.; Alitalo, K. The human p50csk tyrosine kinase phosphorylates p56lck at Tyr-505 and down regulates its catalytic activity. EMBO J. 1992, 11, 2919–2924. [Google Scholar] [CrossRef]
- Chapman, N.M.; Connolly, S.F.; Reinl, E.L.; Houtman, J.C. Focal adhesion kinase negatively regulates Lck function downstream of the T cell antigen receptor. J. Immunol. 2013, 191, 6208–6221. [Google Scholar] [CrossRef] [Green Version]
- Davidson, D.; Zhong, M.C.; Pandolfi, P.P.; Bolland, S.; Xavier, R.J.; Seed, B.; Li, X.; Gu, H.; Veillette, A. The Csk-Associated Adaptor PAG Inhibits Effector T Cell Activation in Cooperation with Phosphatase PTPN22 and Dok Adaptors. Cell Rep. 2016, 17, 2776–2788. [Google Scholar] [CrossRef] [Green Version]
- Solheim, S.A.; Petsalaki, E.; Stokka, A.J.; Russell, R.B.; Tasken, K.; Berge, T. Interactions between the Fyn SH3-domain and adaptor protein Cbp/PAG derived ligands, effects on kinase activity and affinity. FEBS J. 2008, 275, 4863–4874. [Google Scholar] [CrossRef]
- Solheim, S.A.; Torgersen, K.M.; Tasken, K.; Berge, T. Regulation of FynT function by dual domain docking on PAG/Cbp. J. Biol. Chem. 2008, 283, 2773–2783. [Google Scholar] [CrossRef] [Green Version]
- Brdicka, T.; Pavlistova, D.; Leo, A.; Bruyns, E.; Korinek, V.; Angelisova, P.; Scherer, J.; Shevchenko, A.; Hilgert, I.; Cerny, J.; et al. Phosphoprotein associated with glycosphingolipid-enriched microdomains (PAG), a novel ubiquitously expressed transmembrane adaptor protein, binds the protein tyrosine kinase csk and is involved in regulation of T cell activation. J. Exp. Med. 2000, 191, 1591–1604. [Google Scholar] [CrossRef]
- Davidson, D.; Bakinowski, M.; Thomas, M.L.; Horejsi, V.; Veillette, A. Phosphorylation-dependent regulation of T-cell activation by PAG/Cbp, a lipid raft-associated transmembrane adaptor. Mol. Cell. Biol. 2003, 23, 2017–2028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torgersen, K.M.; Vang, T.; Abrahamsen, H.; Yaqub, S.; Horejsi, V.; Schraven, B.; Rolstad, B.; Mustelin, T.; Tasken, K. Release from tonic inhibition of T cell activation through transient displacement of C-terminal Src kinase (Csk) from lipid rafts. J. Biol. Chem. 2001, 276, 29313–29318. [Google Scholar] [CrossRef] [Green Version]
- Wallis, A.M.; Bishop, G.A. TRAF3 regulation of inhibitory signaling pathways in B and T lymphocytes by kinase and phosphatase localization. J. Leukoc. Biol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Wallis, A.M.; Wallace, E.C.; Hostager, B.S.; Yi, Z.; Houtman, J.C.D.; Bishop, G.A. TRAF3 enhances TCR signaling by regulating the inhibitors Csk and PTPN22. Sci. Rep. 2017, 7, 2081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chow, L.M.; Fournel, M.; Davidson, D.; Veillette, A. Negative regulation of T-cell receptor signalling by tyrosine protein kinase p50csk. Nature 1993, 365, 156–160. [Google Scholar] [CrossRef]
- Manz, B.N.; Tan, Y.X.; Courtney, A.H.; Rutaganira, F.; Palmer, E.; Shokat, K.M.; Weiss, A. Small molecule inhibition of Csk alters affinity recognition by T cells. Elife 2015. [Google Scholar] [CrossRef]
- Dobenecker, M.W.; Schmedt, C.; Okada, M.; Tarakhovsky, A. The ubiquitously expressed Csk adaptor protein Cbp is dispensable for embryogenesis and T-cell development and function. Mol. Cell. Biol. 2005, 25, 10533–10542. [Google Scholar] [CrossRef] [Green Version]
- Schmedt, C.; Saijo, K.; Niidome, T.; Kuhn, R.; Aizawa, S.; Tarakhovsky, A. Csk controls antigen receptor-mediated development and selection of T-lineage cells. Nature 1998, 394, 901–904. [Google Scholar] [CrossRef]
- Tan, Y.X.; Manz, B.N.; Freedman, T.S.; Zhang, C.; Shokat, K.M.; Weiss, A. Inhibition of the kinase Csk in thymocytes reveals a requirement for actin remodeling in the initiation of full TCR signaling. Nat. Immunol. 2014, 15, 186–194. [Google Scholar] [CrossRef]
- Xu, S.; Huo, J.; Tan, J.E.; Lam, K.P. Cbp deficiency alters Csk localization in lipid rafts but does not affect T-cell development. Mol. Cell. Biol. 2005, 25, 8486–8495. [Google Scholar] [CrossRef] [Green Version]
- Inderberg, E.M.; Mensali, N.; Oksvold, M.P.; Fallang, L.E.; Fane, A.; Skorstad, G.; Stenvik, G.E.; Progida, C.; Bakke, O.; Kvalheim, G.; et al. Human c-SRC kinase (CSK) overexpression makes T cells dummy. Cancer Immunol. Immunother. 2018, 67, 525–536. [Google Scholar] [CrossRef] [PubMed]
- Eichmann, T.O.; Lass, A. DAG tales: The multiple faces of diacylglycerol--stereochemistry, metabolism, and signaling. Cell Mol. Life Sci. 2015, 72, 3931–3952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishna, S.; Zhong, X. Role of diacylglycerol kinases in T cell development and function. Crit. Rev. Immunol. 2013, 33, 97–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gwack, Y.; Feske, S.; Srikanth, S.; Hogan, P.G.; Rao, A. Signalling to transcription: Store-operated Ca2+ entry and NFAT activation in lymphocytes. Cell Calcium 2007, 42, 145–156. [Google Scholar] [CrossRef]
- Kong, K.F.; Yokosuka, T.; Canonigo-Balancio, A.J.; Isakov, N.; Saito, T.; Altman, A. A motif in the V3 domain of the kinase PKC-theta determines its localization in the immunological synapse and functions in T cells via association with CD28. Nat. Immunol. 2011, 12, 1105–1112. [Google Scholar] [CrossRef] [Green Version]
- Krishna, S.; Zhong, X.P. Regulation of Lipid Signaling by Diacylglycerol Kinases during T Cell Development and Function. Front. Immunol. 2013, 4, 178. [Google Scholar] [CrossRef] [Green Version]
- Merida, I.; Arranz-Nicolas, J.; Rodriguez-Rodriguez, C.; Avila-Flores, A. Diacylglycerol kinase control of protein kinase C. Biochem. J. 2019, 476, 1205–1219. [Google Scholar] [CrossRef]
- Hogan, P.G.; Chen, L.; Nardone, J.; Rao, A. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev. 2003, 17, 2205–2232. [Google Scholar] [CrossRef] [Green Version]
- Sanjuan, M.A.; Jones, D.R.; Izquierdo, M.; Merida, I. Role of diacylglycerol kinase alpha in the attenuation of receptor signaling. J. Cell Biol. 2001, 153, 207–220. [Google Scholar] [CrossRef] [Green Version]
- Zhong, X.P.; Hainey, E.A.; Olenchock, B.A.; Jordan, M.S.; Maltzman, J.S.; Nichols, K.E.; Shen, H.; Koretzky, G.A. Enhanced T cell responses due to diacylglycerol kinase zeta deficiency. Nat. Immunol. 2003, 4, 882–890. [Google Scholar] [CrossRef]
- Merida, I.; Torres-Ayuso, P.; Avila-Flores, A.; Arranz-Nicolas, J.; Andrada, E.; Tello-Lafoz, M.; Liebana, R.; Arcos, R. Diacylglycerol kinases in cancer. Adv. Biol. Regul. 2017, 63, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Prinz, P.U.; Mendler, A.N.; Masouris, I.; Durner, L.; Oberneder, R.; Noessner, E. High DGK-alpha and disabled MAPK pathways cause dysfunction of human tumor-infiltrating CD8+ T cells that is reversible by pharmacologic intervention. J. Immunol. 2012, 188, 5990–6000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi, R.P.; Koretzky, G.A. Diacylglycerol kinases: Regulated controllers of T cell activation, function, and development. Int. J. Mol. Sci. 2013, 14, 6649–6673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldanzi, G.; Bettio, V.; Malacarne, V.; Graziani, A. Diacylglycerol Kinases: Shaping Diacylglycerol and Phosphatidic Acid Gradients to Control Cell Polarity. Front. Cell Dev. Biol. 2016, 4, 140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merida, I.; Andrada, E.; Gharbi, S.I.; Avila-Flores, A. Redundant and specialized roles for diacylglycerol kinases alpha and zeta in the control of T cell functions. Sci. Signal. 2015, 8, re6. [Google Scholar] [CrossRef]
- Cipres, A.; Carrasco, S.; Merino, E.; Diaz, E.; Krishna, U.M.; Falck, J.R.; Martinez, A.C.; Merida, I. Regulation of diacylglycerol kinase alpha by phosphoinositide 3-kinase lipid products. J. Biol. Chem. 2003, 278, 35629–35635. [Google Scholar] [CrossRef] [Green Version]
- Merino, E.; Avila-Flores, A.; Shirai, Y.; Moraga, I.; Saito, N.; Merida, I. Lck-dependent tyrosine phosphorylation of diacylglycerol kinase alpha regulates its membrane association in T cells. J. Immunol. 2008, 180, 5805–5815. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Moreno, M.; Garcia-Lievana, J.; Soutar, D.; Torres-Ayuso, P.; Andrada, E.; Zhong, X.P.; Koretzky, G.A.; Merida, I.; Avila-Flores, A. FoxO-dependent regulation of diacylglycerol kinase alpha gene expression. Mol. Cell. Biol. 2012, 32, 4168–4180. [Google Scholar] [CrossRef] [Green Version]
- Santos, T.; Carrasco, S.; Jones, D.R.; Merida, I.; Eguinoa, A. Dynamics of diacylglycerol kinase zeta translocation in living T-cells. Study of the structural domain requirements for translocation and activity. J. Biol. Chem. 2002, 277, 30300–30309. [Google Scholar] [CrossRef] [Green Version]
- Shin, J.; Xie, D.; Zhong, X.P. MicroRNA-34a enhances T cell activation by targeting diacylglycerol kinase zeta. PLoS ONE 2013, 8, e77983. [Google Scholar] [CrossRef] [Green Version]
- Avila-Flores, A.; Arranz-Nicolas, J.; Andrada, E.; Soutar, D.; Merida, I. Predominant contribution of DGKzeta over DGKalpha in the control of PKC/PDK-1-regulated functions in T cells. Immunol. Cell Biol. 2017, 95, 549–563. [Google Scholar] [CrossRef] [PubMed]
- Joshi, R.P.; Schmidt, A.M.; Das, J.; Pytel, D.; Riese, M.J.; Lester, M.; Diehl, J.A.; Behrens, E.M.; Kambayashi, T.; Koretzky, G.A. The zeta isoform of diacylglycerol kinase plays a predominant role in regulatory T cell development and TCR-mediated ras signaling. Sci. Signal. 2013, 6, ra102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, R.; Wan, C.K.; Carpenter, J.H.; Mousallem, T.; Boustany, R.M.; Kuan, C.T.; Burks, A.W.; Zhong, X.P. Synergistic control of T cell development and tumor suppression by diacylglycerol kinase alpha and zeta. Proc. Natl. Acad. Sci. USA 2008, 105, 11909–11914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, B.K.; Kambayashi, T. The Immunomodulatory Functions of Diacylglycerol Kinase zeta. Front. Cell Dev. Biol. 2016, 4, 96. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, P.; Krishna, S.; Wang, J.; Lin, X.; Huang, H.; Xie, D.; Gorentla, B.; Huang, R.; Gao, J.; et al. Unexpected positive control of NFkappaB and miR-155 by DGKalpha and zeta ensures effector and memory CD8+ T cell differentiation. Oncotarget 2016, 7, 33744–33764. [Google Scholar]
- Riese, M.J.; Wang, L.C.; Moon, E.K.; Joshi, R.P.; Ranganathan, A.; June, C.H.; Koretzky, G.A.; Albelda, S.M. Enhanced effector responses in activated CD8+ T cells deficient in diacylglycerol kinases. Cancer Res. 2013, 73, 3566–3577. [Google Scholar] [CrossRef] [Green Version]
- Dominguez, C.L.; Floyd, D.H.; Xiao, A.; Mullins, G.R.; Kefas, B.A.; Xin, W.; Yacur, M.N.; Abounader, R.; Lee, J.K.; Wilson, G.M.; et al. Diacylglycerol kinase alpha is a critical signaling node and novel therapeutic target in glioblastoma and other cancers. Cancer Discov. 2013, 3, 782–797. [Google Scholar] [CrossRef]
- Jung, I.Y.; Kim, Y.Y.; Yu, H.S.; Lee, M.; Kim, S.; Lee, J. CRISPR/Cas9-Mediated Knockout of DGK Improves Antitumor Activities of Human T Cells. Cancer Res. 2018, 78, 4692–4703. [Google Scholar] [CrossRef] [Green Version]
- Arranz-Nicolas, J.; Ogando, J.; Soutar, D.; Arcos-Perez, R.; Meraviglia-Crivelli, D.; Manes, S.; Merida, I.; Avila-Flores, A. Diacylglycerol kinase alpha inactivation is an integral component of the costimulatory pathway that amplifies TCR signals. Cancer Immunol. Immunother. 2018, 67, 965–980. [Google Scholar] [CrossRef]
- Liu, K.; Kunii, N.; Sakuma, M.; Yamaki, A.; Mizuno, S.; Sato, M.; Sakai, H.; Kado, S.; Kumagai, K.; Kojima, H.; et al. A novel diacylglycerol kinase alpha-selective inhibitor, CU-3, induces cancer cell apoptosis and enhances immune response. J. Lipid Res. 2016, 57, 368–379. [Google Scholar] [CrossRef] [Green Version]
- Wesley, E.M.; Xin, G.; McAllister, D.; Malarkannan, S.; Newman, D.K.; Dwinell, M.B.; Cui, W.; Johnson, B.D.; Riese, M.J. Diacylglycerol kinase zeta (DGKzeta) and Casitas b-lineage proto-oncogene b-deficient mice have similar functional outcomes in T cells but DGKzeta-deficient mice have increased T cell activation and tumor clearance. Immunohorizons 2018, 2, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Riese, M.J.; Moon, E.K.; Johnson, B.D.; Albelda, S.M. Diacylglycerol Kinases (DGKs): Novel Targets for Improving T Cell Activity in Cancer. Front. Cell Dev. Biol. 2016, 4, 108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeon, M.S.; Atfield, A.; Venuprasad, K.; Krawczyk, C.; Sarao, R.; Elly, C.; Yang, C.; Arya, S.; Bachmaier, K.; Su, L.; et al. Essential role of the E3 ubiquitin ligase Cbl-b in T cell anergy induction. Immunity 2004, 21, 167–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, S.R.; Bastin, D.; Chen, L.; Nguyen, A.; Storbeck, C.J.; Lefebvre, C.; Stojdl, D.; Bramson, J.L.; Bell, J.C.; Wan, Y. Type I IFN blockade uncouples immunotherapy-induced antitumor immunity and autoimmune toxicity. J. Clin. Investig. 2019, 129, 518–530. [Google Scholar] [CrossRef] [PubMed]
- Shang, W.; Jiang, Y.; Boettcher, M.; Ding, K.; Mollenauer, M.; Liu, Z.; Wen, X.; Liu, C.; Hao, P.; Zhao, S.; et al. Genome-wide CRISPR screen identifies FAM49B as a key regulator of actin dynamics and T cell activation. Proc. Natl. Acad. Sci. USA 2018, 115, E4051–E4060. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| Protein | Mechanism of Action | Demonstrated Anti-Tumor Activity | Evidence of Autoimmunity | Development of Small Molecule Inhibitors | Development in Clinical Trials |
|---|---|---|---|---|---|
| c-Cbl | -Degradation of critical TCR signaling components (e.g., TCRζ) -Establishment of anergy | No | Yes | − | No |
| Cbl-b | -Degradation of critical TCR signaling components (e.g., CD3ζ) -Establishment of anergy | Yes | Yes | + | Yes |
| GRAIL | -Degradation of critical TCR signaling components (e.g., TCRβ) -Establishment of anergy | No, but protein is over-expressed in TILs | Yes | − | No |
| NEDD4 | -Degradation of critical TCR signaling components (e.g., Bcl10) | Yes | No | − | No |
| Itch | -Degradation of critical TCR signaling components (e.g., Bcl10 and CD3ζ) -Establishment of anergy | No | Yes | ++ | No |
| Deltex1 | -Substrate undetermined -Establishment of anergy | No | Yes | − | No |
| TRAF6 | -Regulates PI3K-Akt pathway by altering protein localization or function by ubiquitination | Yes [39] | No | + | No |
| MDM2 | -Degradation of NFATc2 | Yes | No | +++ (not as cancer immunotherapy) | Yes |
| Peli1 | -Degradation of c-Rel and NIK | No | Yes | − | No |
| SOCS3 | -Substrate undetermined | Yes [40] | No | Yes (in neuron regeneration) | No |
| SOCS6 | -Degradation of Lck | No | No | No | No |
| Cish | -Degradation of PLCγ1 | No | No | No | Yes (NCT03538613) |
| Protein (Gene) | Mechanism of Action | Demonstrated Anti-Tumor Activity | Evidence of Autoimmunity | Development of Small Molecule Inhibitors | Development in Clinical Trials |
|---|---|---|---|---|---|
| SHP1 | -Dephosphorylation and inactivation of critical TCR components (e.g., Zap-70, CD3ζ, and TCRζ) | Yes | Yes | +++ | Yes |
| SHP2 | -Sequestered from dephosphorylating and activating Lck by PD1 binding -May facilitate TCR signaling in some instances | Yes (potentially only when inhibited in NK cells) | No | +++ | Yes |
| PTEN | -Inactivation of the PI3K pathway by dephosphorylating PIP3 | Yes (when inhibited in mature T cells) | Yes | − | No |
| SHIP | -Inactivation of the PI3K pathway by dephosphorylating PIP3 and IP4 | No | No | +++ | Yes |
| PEP | -Dephosphorylation and inactivation of the critical TCR kinases Lck and Fyn | Yes | Yes | + | No |
| PTP-PEST | -Dephosphorylation and inactivation of key signaling molecules involved in Ras activation and actin reorganization -Establishment of anergy | No | Yes | − | No |
| PTPH1 | -Dephosphorylation and inactivation of the critical TCR component CD3ζ | No | No | − | No |
| PP2A | -Dephosphorylation and inactivation of the critical TCR component Carma 1 | No | No | + | |
| CSK | -Phosphorylation and inactivation of the critical TCR kinases Lck and Fyn | No | No | − | No |
| DGKα | -Phosphorylation and inactivation of the critical TCR component DAG -Establishment of anergy | Yes | No | + ++ | No |
| DGKζ | -Phosphorylation and inactivation of the critical TCR component DAG -Establishment of anergy | Yes | No | ++ | No |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sitaram, P.; Uyemura, B.; Malarkannan, S.; Riese, M.J. Beyond the Cell Surface: Targeting Intracellular Negative Regulators to Enhance T cell Anti-Tumor Activity. Int. J. Mol. Sci. 2019, 20, 5821. https://doi.org/10.3390/ijms20235821
Sitaram P, Uyemura B, Malarkannan S, Riese MJ. Beyond the Cell Surface: Targeting Intracellular Negative Regulators to Enhance T cell Anti-Tumor Activity. International Journal of Molecular Sciences. 2019; 20(23):5821. https://doi.org/10.3390/ijms20235821
Chicago/Turabian StyleSitaram, Poojitha, Bradley Uyemura, Subramaniam Malarkannan, and Matthew J. Riese. 2019. "Beyond the Cell Surface: Targeting Intracellular Negative Regulators to Enhance T cell Anti-Tumor Activity" International Journal of Molecular Sciences 20, no. 23: 5821. https://doi.org/10.3390/ijms20235821
APA StyleSitaram, P., Uyemura, B., Malarkannan, S., & Riese, M. J. (2019). Beyond the Cell Surface: Targeting Intracellular Negative Regulators to Enhance T cell Anti-Tumor Activity. International Journal of Molecular Sciences, 20(23), 5821. https://doi.org/10.3390/ijms20235821